

Summary
Since the advent of glucocorticoid therapy for autoimmune disease in the 1940s, their widespread application has led to the concurrent therapy-limiting discovery of many adverse metabolic side effects. Unanticipated hyperglycemia associated with the initiation of glucocorticoids often leads to preventable hospital admissions, prolonged hospital stays, increased risks for infection and reduced graft function in solid organ transplant recipients. Challenges in managing steroid-induced diabetes stem from wide fluctuations in post-prandial hyperglycemia and the lack of clearly defined treatment protocols. The mainstay of treatment is insulin therapy coincident with meals.
This article aims to review the pathogenesis, risk factors, diagnosis and treatment principles unique to steroid-induced diabetes.
Keywords: steroid-induced diabetes, glucocorticoids, insulin resistance, new onset diabetes after transplant
Introduction
Glucocorticoids are extensively used in almost every subspecialty of medicine. Indications for short-term acute steroid therapy can be seen in exacerbation of chronic obstructive pulmonary disease, acute gout, chemotherapy protocols, bacterial meningitis and in pregnant women for fetal lung maturation, to name a few. Disease processes benefiting from chronic glucocorticoid use include the following: pulmonary diseases such as idiopathic interstitial pneumonia, hypersensitivity pneumonitis and sarcoidosis; autoimmune conditions; neurologic diseases such as myasthenia gravis and multiple sclerosis; and inflammatory bowel diseases. More recently, chronic glucocorticoid therapy plays an important role in modulating the immune system following solid organ transplantation. Although widely prescribed for their anti-inflammatory and immunosuppressive properties, glucocorticoids have various common metabolic side effects including hypertension, osteoporosis and diabetes. Steroid-induced diabetes mellitus (SIDM) has been recognized as a complication of glucocorticoid use for over 50 years [1].
Definition
Steroid-induced diabetes mellitus is defined as an abnormal increase in blood glucose associated with the use of glucocorticoids in a patient with or without a prior history of diabetes mellitus. The criteria for diagnosing diabetes by the American Diabetes Association [2] is an 8 h fasting blood glucose ≥ 7.0 mmol/L (126 mg/dL), 2 h post 75 g oral glucose tolerance test (OGTT) ≥ 11.1 mmol/L (200 mg/dL), HbA1c ≥ 6.5% or in patients with symptoms of hyperglycemic, a random plasma glucose of ≥ 11.1 mmol/L (200 mg/dL).
Prevalence
Given the widespread use of glucocorticoids in both the inpatient and ambulatory care setting, it is not surprising that at our 550-bed teaching hospital, approximately 40% of all inpatient consults to the Endocrinology Consult Service are for new onset steroid-induced diabetes or type 2 diabetes exacerbated by steroid use. This figure is consistent with the rate of 56% noted at other institutions [3].
The length of time on steroids, the relative potency of the glucocorticoid and the absolute dose all play a role in the occurrence of SIDM. In a retrospective study of 11 855 patients receiving various doses of glucocorticoids, Gurwitz et al. assessed the need of hypoglycemic therapy. The calculated odds ratio for patients receiving the equivalent of 50, 100 and greater than 120 mg of hydrocortisone daily were 3.02, 5.82 and 10.35, respectively, compared with controls [4]. In order to appreciate the magnitude of SIDM, one needs to consider that steroids cause predominantly post-prandial hyperglycemia and therefore, looking at impaired fasting glucose as the sole criteria, may underestimate the true incidence of SIDM.
Populations affected by chronic glucocorticoids
New onset diabetes after transplant (NODAT) is used to describe those patients in whom diabetes occurs for the first time in a post-transplant setting [5]. The incidence of NODAT is quite variable and likely underestimated because of lack of uniformity in the definition [6]. Varying immunosuppression protocols have caused discrepant incidence rates, although all agree that the incidence of NODAT is high in renal, liver, heart and lung transplant recipients (Table 1) [7–10]. In addition, the presence of NODAT has an adverse outcome on the survival of the transplanted organ as well as the health of the recipient [10].
Table 1. Examples of incidence of steroid-induced diabetes following solid organ transplantation.
| Organ | % with SIDM | Reference |
|---|---|---|
| Liver | 24% | [7] Anderson, et al. 2006 |
| Lung | 60% | [8] Belle-Van Meerkerk et al. 2012 |
| Heart | 29% | [9] Depczynski et al. 2000 |
| Kidney | 17% | [10] Yates et al. 2012 |
SIDM, steroid-induced diabetes mellitus.
The population of patients following solid organ transplant is not the only population treated with glucocorticoids who develop SIDM: 12.7% of lupus patients [11], 14.7% of patients with respiratory ailments [10] and 23.5% of leprosy patients [12] developed diabetes following treatment with glucocorticoids. In addition, endogenous overproduction of glucocorticoids resulting in Cushing's syndrome often translates to central obesity, muscle wasting, hepatic steatosis, hypertension and insulin resistance. In either overt or ‘subclinical’ Cushing's 53% and 45% of subjects had either frank diabetes or impaired glucose tolerance, respectively [13].
Pathophysiology
The effect of glucocorticoids on glucose metabolism is likely the result of impairment of multiple pathways including beta cell dysfunction (sensitivity to glucose and ability to release insulin) and insulin resistance in other tissue.
Clinical studies
The role of beta cell function and other tissues' sensitivity to insulin may be different depending on whether the glucocorticoid effect is acute or chronic. One study compared an acute single dose of prednisolone (75 mg) with 30 mg of prednisolone daily for 15 days. The acute treatment inhibited several parameters of beta cell function. Conversely, prolonged glucocorticoid exposure showed partial recovery of beta cell function but similarly impaired glucose tolerance, suggesting additional factors are important in SIDM other than beta cell dysfunction [14].
In addition to timing, the ‘glucocorticoid potency’ is a factor in the severity of post-glucocorticoid hyperglycemia. Yasuda et al. demonstrated that hydrocortisone, dexamethasone and prednisone result in varying degrees of insulin resistance based on decreased binding affinity of insulin rather than a decrease in receptor number [15].
Furthermore, normoglycemic men given a bolus of either cortisol or corticotropin releasing hormone (which causes an increase in endogenous cortisol) resulted in the expected elevation of plasma cortisol but caused an abrupt inhibition of insulin secretion even before there was a change in glucose concentration. Insulin resistance, measured by insulin secretion rate, developed 4–6 h after cortisol elevation and persisted for > 16 h [16].
In vitro studies
Further evidence for a direct effect of glucocorticoids on beta cell function has been from cultured rat insulinoma insulin-secreting, INS-1E cells [17]. Measurement of impaired insulin release in response to a glucose challenge was seen in prednisone-treated INS-1E cells. The inhibition was reversed in the presence of prednisone with the glucocorticoid receptor antagonist, RU486 [17]. The authors suggest that the defect may be due to impaired endoplasmic reticulum homeostasis, which in turn may lead to beta cell death.
Glyceroneogenesis
One of the etiologies of SIDM is based on the profound and reciprocal effect glucocorticoids have on glyceroneogenesis in liver and adipose tissue (Figure 1). In adipose tissue, glyceroneogenesis controls the rate of fatty acid release in the blood, while in the liver glyceroneogenesis is responsible for the synthesis of triacylglyerol from fatty acids and glycerol 3-phosphate [18]. The regulation of this process in both liver and adipose is via the enzyme phosphoenylpyruvate carboxykinase (PEPCK). In the presence of glucocorticoids, PEPCK gene expression in adipose tissue is suppressed, inhibiting glyceroneogenesis. In contrast, PEPCK in liver stimulates glycerol production and fatty acid concentration in the blood increased by the action of lipoprotein lipase [19]. The net result of glucocorticoids, therefore, is to increase the amount of fatty acids released into the blood. An increase in fatty acids interferes with glucose utilization and results in insulin resistance, especially in skeletal muscle. Thiazolidinediones promote expression of adipose and skeletal muscle PEPCK and reduce serum levels of fatty acids therefore reducing insulin resistance [20].
Figure 1.
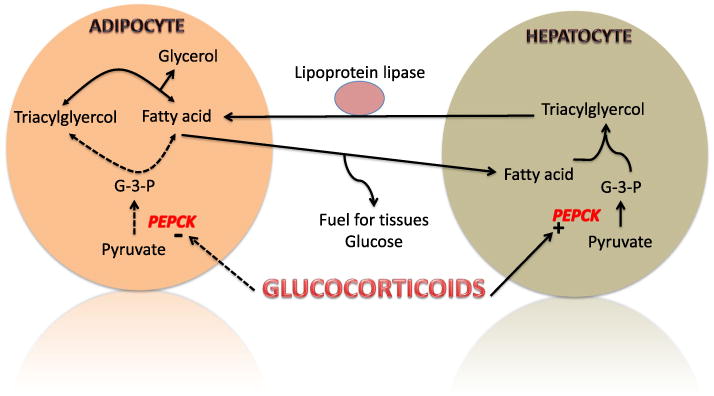
The effect of glucocorticoids on glycerneogenesis in adipose tissue and liver. Phosphoenylpyruvate carboxykinase (PEPCK) is reciprocally upregulated in liver and downregulated in adipose by glucocorticoids. This results in a buildup of free fatty acids in the blood, which in turn result in insulin resistance and increase gluconeogenesis
Molecular basis of glucocorticoid action on glucose regulation
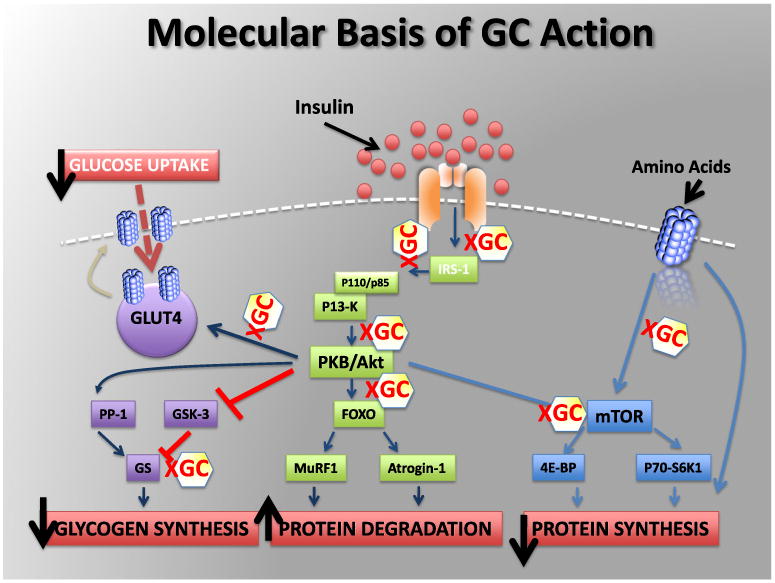
The insulin-mediated pathways of glycogen synthesis and protein degradation and synthesis are directly influenced by glucocorticoids (Figure 2). Skeletal muscle is responsible for the majority of insulin-mediated glucose uptake. Insulin recruits GLUT4 glucose transporters to the cell surface enabling glucose uptake into cell. Glucocorticoids impair insulin-mediated glucose uptake by directly interfering with components of the insulin signalling cascade, such as glycogen synthase kinase-3, glycogen synthase and GLUT4 translocation [21,22]. An increase in protein degradation and decrease in protein synthesis is due to glucocorticoid inhibition of post-insulin receptor cascades involving PKB/Akt and mTOR pathways.
Figure 2. Molecular basis of glucocorticoid (GC) action. See text for details. After van Raalte et al. [21].
Risk factors for steroid-induced diabetes mellitus
Proposed risk factors for steroid-induced diabetes beyond cumulative dose and longer duration of steroid course include traditional risk factors for type 2 diabetes: older age, family history, high body mass index and impaired glucose tolerance [23]. The association with family history of diabetes is not well defined. Simmons et al. compared the demographics and clinical characteristics of patients with new onset SIDM with those with type 2 diabetes with and without steroid treatment. Those individuals who developed new onset SIDM had significantly less family history of diabetes when compared with individuals with type 2 diabetes mellitus and glucocorticoid treatment [24].
Concurrent immunosuppression
Other immunosuppressive agents can also affect glycemic control through other mechanisms, thus confounding impact of glucocorticoid therapy. In transplant patients, the use of calcineurin inhibitors (particularly tacrolimus) contributes to glucose intolerance by suppressing insulin production [24]. In those patients with systemic lupus erythematosis, patients on high-dose steroid therapy, development of diabetes was associated with concurrent use of mycophenalate mofetil, possibly attributed to impaired insulin secretion from increased beta cell stress [11,25].
Hypomagnesemia
Numerous studies have reported an inverse relationship between glycemic control and serum magnesium levels. Van Laecke et al. conducted a single-centre retrospective analysis consisting of 254 renal transplant recipients demonstrating that hypomagnesemia during the first month post-transplant was associated with the development of NODAT [26].
Hepatitis C virus
Liver disease contributes to impaired glucose tolerance, but there is evidence that chronic hepatitis C virus (HCV) infection itself is an independent risk factor for the development of diabetes in the general population and in liver transplant recipients [27,28]. A meta-analysis by Fabrizi et al. associated HCV seropositivity with a significantly increased risk of NODAT in kidney transplantation [29]. Baid et al. also demonstrated that the prevalence of NODAT was significantly higher in HCV positive patients (64% versus 28%) who underwent liver transplant [27].
Clinical course
The tendency for patients to develop new hyperglycemia in the setting of initiating glucocorticoid therapy is often not anticipated. In the elderly, without close follow-up or monitoring of blood sugars, there is a risk of precipitating hyperglycemic hyperosmolar states [30], which would require admission to the hospital for aggressive hydration and insulin therapy. It is generally thought that glucocorticoids result mainly in an increase in post-prandial blood glucose [31,32]. Use of continuous blood glucose monitor in COPD patients treated with prednisolone demonstrated that hyperglycemia predominately occurs in the afternoon and evening, indicating that this would be the most appropriate time to screen for SIDM as well as the period of time to direct specific treatment [33].
Early detection
Strategies are needed to detect those at risk for developing steroid-induced diabetes before starting chronic therapy. The 2004 updated international consensus guidelines for NODAT suggest that pre-transplant evaluation include fasting plasma glucose, and when this is normal, an OGTT [22]. When OGTTs were performed in a cohort of renal transplant patients 1 week before and approximately 1 year after transplant, 23.7% of the patients had NODAT, which was associated with higher fasting and 2 h plasma glucose in the pre-transplant setting. Additionally, the ratio of proinsulin to insulin was higher at baseline in the patients that developed NODAT group [34].
Oral glucose tolerance testing should be performed as early as possible in post-transplant patients to detect diabetes in those deemed to be at risk [35]. In a cohort of renal transplant recipients 3 months post-transplant, Valderhaug et al. used the criteria of a fasting plasma glucose 95–124 mg/dL or an HbA1c greater than or equal to 5.8% to identify a subset that should undergo further diagnostic testing such as an OGTT [35].
Comorbidities
New onset diabetes after transplant is a strong predictor of graft failure in the transplant population. Roth et al. reported 12-year graft survival in diabetic patients to be 48% versus 70% in controls [33]. Graft failure in the renal transplant population who develop NODAT is attributed to ongoing hyperglycemia leading to recurrent diabetic nephropathy [36].
One of the largest barriers to tapering glucocorticoids or switching to steroid-sparing immunosuppression to improve glucose control is the risk of allograft rejection [36], which itself is associated with increased risk for NODAT.
Diabetes is also associated with increased risk of cardiovascular events and a myriad of microvascular complications. The literature suggests that kidney transplant recipients who develop NODAT are at a twofold–threefold increased risk of fatal and non-fatal cardiovascular disease events as compared with non-diabetic patients [37].
Treatment
Optimal treatment of SIDM warrants a different management strategy than non-steroid-induced diabetes. For instance, metformin, which is often used first-line in type 2 diabetes, is not recommended for SIDM because of its many relative or absolute contraindications, which include nausea/vomiting, hypoxia and liver or kidney disease. As noted previously in the discussion about glyceroneogenesis, the role of thiazolidinediones is yet to be fully explored.
Non-pharmacologic intervention
As with all types of diabetes, initial steps to improve glycemic control include lifestyle modification which includes exercise and dietary counselling to provide options that can perhaps lessen post-prandial hyperglycemia.
Insulin
Because initiation of glucocorticoids can cause post-prandial hyperglycemia and the tapering of glucocorticoids can lead to normalization of glycemic control, current guidelines may insufficiently address this. Basal bolus insulin therapy remains the most flexible option for patients and includes three components: basal insulin, prandial insulin and a supplemental correction factor insulin [38]. Conventional use of long-acting basal insulin with traditional weight-based dosing may cause nocturnal hypoglycemia [33]. More studies exploring dose titration of insulin in patients on glucocorticoids possibly utilizing technology like continuous glucose monitoring system are needed. In general, however, timing of glucocorticoids, to a midday or an evening meal with concomitant administration of intermediate acting insulin, is judicious.
Secretagogues
Oral secretagogues such as sulfonylurea therapy do not specifically target post-prandial hyperglycemia and thus long-acting agents may be associated with hypoglycemia if the patient does not eat meals regularly. For patients with mild hyperglycemia who are unable or unwilling to give injections of insulin, a trial of short-acting secretagogues such as nateglinide or repaglinide taken before meals could be considered [38].
Incretin mimetics
Incretin-based therapy with GLP-1 receptor agonists and DPP-4 inhibitors control glucose levels by stimulating insulin and inhibiting glucagon secretion in the fasting and post-prandial setting. A single dose of exenatide was able to improve glucose intolerance and insulin resistance in mice [39]. In humans, a randomized, double-blind, placebo-controlled trial was performed in which subjects on prednisone therapy received either the GLP-1 receptor agonist exenatide or saline [40]. Exenatide prevented prednisone-induced glucose intolerance and islet cell dysfunction primarily by decreasing glucagon and decreasing gastric emptying (Table 2) [40]. In the post-transplant setting, as more studies will be conducted with these and other agents, attention to drug–drug interactions is essential.
Table 2. Effect of prednisolone with and without exenatide following a mixed meal [40].
| Molecule | Prednisolone only | Prednisolone + exenatide |
|---|---|---|
| Glucose | Increase | Decrease |
| Insulin | No change | Decrease |
| C-peptide | Decrease | Decrease |
| Glucagon | Increase | Decrease |
Conclusion
As the therapeutic benefits of glucocorticoids continue to expand across medical specialties, the incidence of steroid-induced or steroid-exacerbated diabetes will continue to rise. Similar to non-steroid-related diabetes, the principles of early detection and risk factor modification apply. Diagnosing impaired fasting glucose or impaired glucose tolerance prior to the initiation of chronic glucocorticoids will better identify those who would benefit from steroid-sparing treatment, or if this is not an option, blood glucose monitoring while starting therapy. Further investigation into the precise mechanism of steroid-induced insulin resistance will provide insight into future diabetes prevention efforts and targeted therapies.
Footnotes
Conflict of interest: The authors have no conflicts of interest.
References
- 1.Conn JW, Fajans SS. Influence of adrenal cortical steroids on carbohydrate metabolism in man. Metabolism. 1956;5:114–127. [PubMed] [Google Scholar]
- 2.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donihi AC, Raval D, Saul M, Korytkowski MT, DeVita MA. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr Pract. 2006;12:358–362. doi: 10.4158/EP.12.4.358. [DOI] [PubMed] [Google Scholar]
- 4.Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med. 1994;154:97–101. [PubMed] [Google Scholar]
- 5.Starzl TE. Experience in Renal Transplantation. Saunders; Philadelphia: 1964. [Google Scholar]
- 6.Balla A, Chobanian M. New-onset diabetes after transplantation: a review of recent literature. Curr Opin Organ Transplant. 2009;14:375–379. doi: 10.1097/MOT.0b013e32832dbb98. [DOI] [PubMed] [Google Scholar]
- 7.Anderson AL, Lewis DA, Steinke DT, Ranjan D, Smith KM, Clifford TM. Effects of hyperglycemia on the development of new-onset diabetes after liver transplantation. Prog Transplant. 2009;19:298–303. doi: 10.1177/152692480901900403. [DOI] [PubMed] [Google Scholar]
- 8.Belle-van Meerkerk G, van de Graaf EA, Kwakkel-van Erp JM, et al. Diabetes before and after lung transplantation in patients with cystic fibrosis and other lung diseases. Diabet Med. 2012;29:e159–162. doi: 10.1111/j.1464-5491.2012.03676.x. [DOI] [PubMed] [Google Scholar]
- 9.Depczynski B, Daly B, Campbell LV, Chisholm DJ, Keogh A. Predicting the occurrence of diabetes mellitus in recipients of heart transplants. Diabet Med. 2000;17:15–19. doi: 10.1046/j.1464-5491.2000.00206.x. [DOI] [PubMed] [Google Scholar]
- 10.Yates CJ, Fourlanos S, Hjelmesaeth J, Colman PG, Cohney SJ. New-onset diabetes after kidney transplantation-changes and challenges. Am J Transplant. 2012;12:820–828. doi: 10.1111/j.1600-6143.2011.03855.x. [DOI] [PubMed] [Google Scholar]
- 11.Ha Y, Lee KH, Jung S, Lee SW, Lee SK, Park YB. Glucocorticoid-induced diabetes mellitus in patients with systemic lupus erythematosus treated with high-dose glucocorticoid therapy. Lupus. 2011;20:1027–1034. doi: 10.1177/0961203311402246. [DOI] [PubMed] [Google Scholar]
- 12.Papang R, John AS, Abraham S, Rao PS. A study of steroid-induced diabetes mellitus in leprosy. Indian J Lepr. 2009;81:125–129. [PubMed] [Google Scholar]
- 13.Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab. 2011;22:499–506. doi: 10.1016/j.tem.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 14.van Raalte DH, Nofrate V, Bunck MC, et al. Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur J Endocrinol. 2010;162:729–735. doi: 10.1530/EJE-09-1034. [DOI] [PubMed] [Google Scholar]
- 15.Yasuda K, Hines E, 3rd, Kitabchi AE. Hypercortisolism and insulin resistance: comparative effects of prednisone, hydrocortisone, and dexamethasone on insulin binding of human erythrocytes. J Clin Endocrinol Metab. 1982;55:910–915. doi: 10.1210/jcem-55-5-910. [DOI] [PubMed] [Google Scholar]
- 16.Plat L, Byrne MM, Sturis J, et al. Effects of morning cortisol elevation on insulin secretion and glucose regulation in humans. Am J Physiol. 1996;270:E36–42. doi: 10.1152/ajpendo.1996.270.1.E36. [DOI] [PubMed] [Google Scholar]
- 17.Linssen MM, van Raalte DH, Toonen EJ, et al. Prednisolone-induced beta cell dysfunction is associated with impaired endoplasmic reticulum homeostasis in INS-1E cells. Cell Signal. 2011;23:1708–1715. doi: 10.1016/j.cellsig.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Cadoudal T, Leroyer S, Reis AF, et al. Proposed involvement of adipocyte glyceroneogenesis and phosphoenolpyruvate carboxykinase in the metabolic syndrome. Biochimie. 2005;87:27–32. doi: 10.1016/j.biochi.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Franckhauser S, Antras-Ferry J, Robin P, Robin D, Granner DK, Forest C. Expression of the phosphoenolpyruvate carboxykinase gene in 3T3-F442A adipose cells: opposite effects of dexamethasone and isoprenaline on transcription. Biochem J. 1995;305(Pt 1):65–71. doi: 10.1042/bj3050065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadoudal T, Blouin JM, Collinet M, et al. Acute and selective regulation of glyceroneogenesis and cytosolic phosphoenolpyruvate carboxykinase in adipose tissue by thiazolidinediones in type 2 diabetes. Diabetologia. 2007;50:666–675. doi: 10.1007/s00125-006-0560-5. [DOI] [PubMed] [Google Scholar]
- 21.van Raalte DH, Ouwens DM, Diamant M. Novel insights into glucocorticoid-mediated diabetogenic effects: towards expansion of therapeutic options? Eur J Clin Invest. 2009;39:81–93. doi: 10.1111/j.1365-2362.2008.02067.x. [DOI] [PubMed] [Google Scholar]
- 22.Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism. 1998;47:3–6. doi: 10.1016/s0026-0495(98)90184-6. [DOI] [PubMed] [Google Scholar]
- 23.Kim SY, Yoo CG, Lee CT, et al. Incidence and risk factors of steroid-induced diabetes in patients with respiratory disease. J Korean Med Sci. 2011;26:264–267. doi: 10.3346/jkms.2011.26.2.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simmons LR, Molyneaux L, Yue DK, Chua EL. Steroid-induced diabetes: is it just unmasking of type 2 diabetes? ISRN Endocrinol. 2012;2012:910905. doi: 10.5402/2012/910905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzantini M, Torre C, Miccoli M, et al. Adverse events during longterm low-dose glucocorticoid treatment of polymyalgia rheumatica a retrospective study. J Rheumatol. 2012;39:552–557. doi: 10.3899/jrheum.110851. [DOI] [PubMed] [Google Scholar]
- 26.Van Laecke S, Van Biesen W, Verbeke F, De Bacquer D, Peeters P, Vanholder R. Posttransplantation hypomagnesemia and its relation with immunosuppression as predictors of new-onset diabetes after transplantation. Am J Transplant. 2009;9:2140–2149. doi: 10.1111/j.1600-6143.2009.02752.x. [DOI] [PubMed] [Google Scholar]
- 27.Baid S, Cosimi AB, Farrell ML, et al. Posttransplant diabetes mellitus in liver transplant recipients: risk factors, temporal relationship with hepatitis C virus allograft hepatitis, and impact on mortality. Transplantation. 2001;72:1066–1072. doi: 10.1097/00007890-200109270-00015. [DOI] [PubMed] [Google Scholar]
- 28.Bahtiyar G, Shin JJ, Aytaman A, Sowers JR, McFarlane SI. Association of diabetes and hepatitis C infection: epidemiologic evidence and pathophysiologic insights. Curr Diab Rep. 2004;4:194–198. doi: 10.1007/s11892-004-0023-7. [DOI] [PubMed] [Google Scholar]
- 29.Fabrizi F, Martin P, Dixit V, Bunnapradist S, Kanwal F, Dulai G. Post-transplant diabetes mellitus and HCV seropositive status after renal transplantation: meta-analysis of clinical studies. Am J Transplant. 2005;5:2433–2440. doi: 10.1111/j.1600-6143.2005.01040.x. [DOI] [PubMed] [Google Scholar]
- 30.Baldwin D, Apel J. Management of hyperglycemia in hospitalized patients with renal insufficiency or steroid-induced diabetes. Curr Diab Rep. 2013;13(1):114–120. doi: 10.1007/s11892-012-0339-7. [DOI] [PubMed] [Google Scholar]
- 31.Oyer DS, Shah A, Bettenhausen S. How to manage steroid diabetes in the patient with cancer. J Support Oncol. 2006;4:479–483. [PubMed] [Google Scholar]
- 32.Clore JN, Thurby-Hay L. Glucocorticoid-induced hyperglycemia. Endocr Pract. 2009;15:469–474. doi: 10.4158/EP08331.RAR. [DOI] [PubMed] [Google Scholar]
- 33.Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab. 2011;96:1789–1796. doi: 10.1210/jc.2010-2729. [DOI] [PubMed] [Google Scholar]
- 34.Nam JH, Mun JI, Kim SI, et al. Beta-cell dysfunction rather than insulin resistance is the main contributing factor for the development of postrenal transplantation diabetes mellitus. Transplantation. 2001;71:1417–1423. doi: 10.1097/00007890-200105270-00011. [DOI] [PubMed] [Google Scholar]
- 35.Valderhaug TG, Jenssen T, Hartmann A, et al. Fasting plasma glucose and glycosylated hemoglobin in the screening for diabetes mellitus after renal transplantation. Transplantation. 2009;88:429–434. doi: 10.1097/TP.0b013e3181af1f53. [DOI] [PubMed] [Google Scholar]
- 36.Guerra G, Ilahe A, Ciancio G. Diabetes and kidney transplantation: past, present, and future. Curr Diab Rep. 2012;12:597–603. doi: 10.1007/s11892-012-0306-3. [DOI] [PubMed] [Google Scholar]
- 37.Hjelmesaeth J, Hartmann A, Leivestad T, et al. The impact of early-diagnosed new-onset post-transplantation diabetes mellitus on survival and major cardiac events. Kidney Int. 2006;69:588–595. doi: 10.1038/sj.ki.5000116. [DOI] [PubMed] [Google Scholar]
- 38.Oyer DS, Shah A, Bettenhausen S. How to manage steroid diabetes in the patient with cancer. J Support Oncol. 2006;4(9):479–482. [PubMed] [Google Scholar]
- 39.Zhao R, Fuentes-Mattei E, Velazquez-Torres G, et al. Exenatide improves glucocorticoid-induced glucose intolerance in mice. Diabetes Metab Syndr Obes. 2011;4:61–65. doi: 10.2147/DMSO.S15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Raalte DH, van Genugten RE, Linssen MM, Ouwens DM, Diamant M. Glucagon-like peptide-1 receptor agonist treatment prevents glucocorticoid-induced glucose intolerance and islet-cell dysfunction in humans. Diabetes Care. 2011;34:412–417. doi: 10.2337/dc10-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]