

Abstract
Compounds that stabilize the G-quadruplexes formed by human telomeres can inhibit the telomerase activity and are potential cancer therapies. We have developed an assay for the screening of compounds with high affinity for human telomeric G-quadruplexes (HTG). The assay uses a thiazole orange fluorescent reporter molecule conjugated to the aminoglycoside, neomycin, as a probe in a fluorescence displacement assay. The conjugation of the planar base stacking thiazole orange with the groove binding neomycin results in high affinity probe that can determine the relative binding affinity of high affinity HTG binding drugs in a high throughput format. The robust assay is applicable for the determination of the binding affinity of HTG in the presence of K+ or Na+.
Keywords: G-quadruplex, Telomere, Aminoglycoside, TO-neo, High throughput screen
Over the last few decades, a number of different telomeric ends have been identified including the human telomeres.1 The human telomeric ends are non-coding DNA rich in tandem repeats of (TTAGGG). In addition to single and double stranded DNA, the guanine rich telomeres of chromosomal DNA can adopt unique structures formed by the stacking of G-quartets called G-quadruplexes (Fig. 1).2 Recent work on human telomeres has shown that the human telomeric G-quadruplex (HTG) may be a target for emerging cancer therapies. The interaction of telomeres with the enzyme telomerase has been linked to cancer proliferation.3–5 However, the formation of G-quadruplexes inhibits the activity of telomerase.5 Thus, work is under way to find compounds that can stop cancer proliferation by stabilizing G-quadruplexes.
Figure 1.
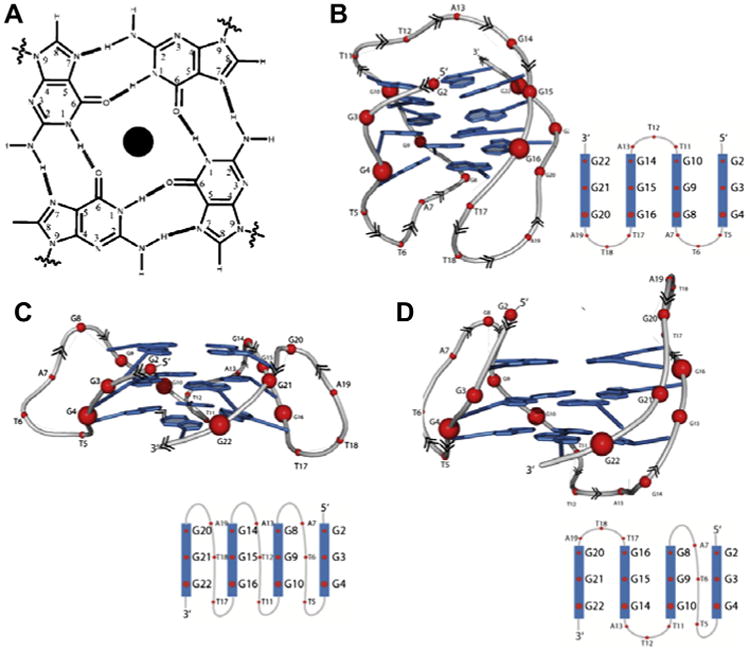
(A) A diagram showing the arrangement of guanines in a G-quartet. Polymorphism of HTG in the presence of sodium and potassium ions (B) a Basket-type conformation in the presence of Na+ (PDB: 143D) (C) a Propellar-type conformation in the presence of K+ with parallel strand topology (PDB: 3SC8), and (D) a Hybrid (3 + 1)-type conformation with three parallel and one antiparallel strands (PDB: 2GKU).
The study of compounds that stabilize HTG is currently lacking in two related areas. First, the number of compounds that bind with high affinity and high specificity is limited. While ligands that interact with different G-quadruplexes have been discovered in the past decade, most of the G-quadruplex ligands contain planar aromatic moieties and stack to G-quadruplexes with moderate binding affinities.6–11 In order to increase the specificity of compounds for HTG the approach of identifying ligands with high selectivity for G-quadruplex grooves is needed.6–9
Aminoglycosides have been shown to bind in the major groove of a variety of nucleic acid targets.11–16 There is also strong evidence that aminoglycosides bind in the grooves of G-quadruplex DNA.17 The ability of a drug to bind in the groove of quadruplexes offers the potential for greater selectivity due to the ability to ‘sense’ differences in groove width as well as interactions with the functional groups in the groove. Therefore, a higher selectivity for G-quadruplex structures can be envisioned by targeting the grooves HTG.
Our approach for increasing the affinity and specificity of HTG binding ligands is the conjugation of aminoglycoside molecules with various planar molecules. When the selectivity of groove binding aminoglycosides is combined with the base stacking surface areas of intercalating molecules with a HTG, ligands with much a higher selectivity18 and a higher affinity19 for the G-quadruplex can be designed.
A related limiting factor in the discovery of HTG binding ligands is the lack of a high throughput method of screening molecules that bind in the groove of HTG. In order to develop a screen to determine compound's affinity for HTG, we have synthesized a fluorescent probe by covalently conjugating thiazole orange with neomycin (TO-neo) as shown in Scheme 1. The synthesis of TO-neo (5) was achieved by coupling a thiazole orange derivative (3) with Boc protected neomycin amine (4). The modified thiazole orange derivative (3) was synthesized in three steps using a similar procedure reported in literature.20,21 As displayed in Scheme 1, 3-methyl-2-(methylthio)benzo[d]thiazol-3-ium (1) was prepared in one step by reacting 3-methylbenzo[d]thiazole-2(3H)-thione with methyl iodide. In a separate reaction 1-(4-carboxybutyl)-4-methylquinolinium (2) was synthesized by reacting 4-methylquinoline with 5-bromovaleric acid. The quaternary salts 1 and 2 were reacted in the presence of triethyl amine to afford compound 3 which bears a carboxylic acid functional group. The Boc protected neomycin amine (4) was synthesized using previously reported procedures.22,23 The coupling of 3 and 4 was achieved using O-(benzotriazol-1-yl)-N,N,N′,N″ tetramethyluronium tetrafluoroborate (TBTU) as the coupling agent to afford Boc protected thiazole orange-neomycin conjugate (5a). The Boc protecting groups in 5a were deprotected using trifluoroacetic acid to afford the desired probe TO-neo (5) in 80% yield.
Scheme 1.

Reagents and conditions: (a) CH3I, 4 h, 50 °C, 84%; (b) 5-bromovaleric acid, 3 h, 110 °C, 38%; (c) Et3N, 50 °C for 2 h, then 1 h at room temperature, 24%; (d) DMF, TBTU, DIPEA, room temperature, 65%; (e) TFA, DCM, 3 h, rt, 80%.
Structural information on HTG has been obtained with solution studies by NMR2 and in the crystalline state using the sequence d[AGGG(TTAGGG)3].24 HTG is polymorphic structures and can exist as a parallel strand G-quadruplex25 or various mixtures of parallel and antiparallel strand G-quadruplex (Fig. 1). The difference in the structure of HTG is highly dependent on the cation present. When formed in 100 mM sodium chloride and 10 mM sodium cacodylate, HTG is dominated by a single basket type parallel/antiparallel structure consisting of both lateral and diagonal loops (Fig. 1B).2 The dominant structure of the Na+ of HTG is monitored by its signature CD signal, with a maximum at 295 nm and minimum at 260 nm, characteristic of alternating anti and syn glycosidic conformations along each DNA strand (Supplementary Fig. S1).
When the 22 base HTG is formed in the buffer containing 10 mM HEPES and 100 mM potassium chloride at pH 7.0, G-quadruplex exists as a complex mixture likely to contain some ensemble of structures such as basket type structure, the propeller type parallel structure (Fig. 1C) and 3 + 1 hybrid structure (Fig. 1D).26,27 The K+ form of HTG has a broader CD spectrum in the 240–295 nm wavelength range with signature positive peaks at 295 and 265 nm and a negative peak at 235 nm (Supplementary Fig. S1).
The TO-neo binds with high affinity to both the Na+ and K+ forms of HTG. A titration of the 22 base model of HTG into TO-neo results in an increase in fluorescence as a function of HTG concentration for both the Na+ and K+ forms (Fig. 2). The increase in the fluorescence can be attributed to stacking interactions of the thiazole orange of TO-neo with the DNA bases of the G-quadruplex.
Figure 2.
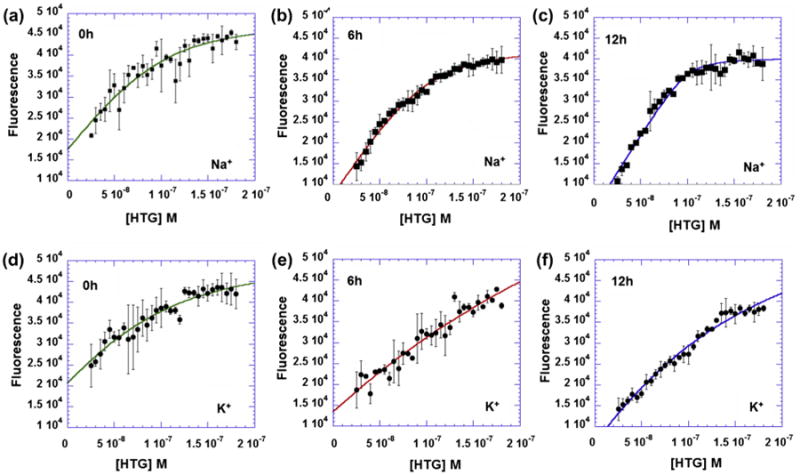
The HTG:TO-neo binding isotherms. The HTG was titrated (25–180 nM) into 100 nM TO-neo in 10 mM sodium cacodylate, 0.5 mM EDTA, 100 mM NaCl at pH 7.0 after incubating for (a) 0 h (b) 6 h (c) 12 h or in 10 mM HEPES, 100 mM KCl at pH 7.0 after incubating for (d) 0 h (e) 6 h (f) 12 h. Titrations were performed in a 96-well round bottom Greneir black plate with an excitation wavelength of 485 nm and emission fluorescence measured at 535 nm from 100 reads, and all points of the isotherms were the average of two wells. Time trials were determined from separate plates to insure minimal photo bleaching resulting from multiple measurements.
While the isotherm of the Na+ and K+ forms of HTG are similar, significant differences are present in the interaction of TO-neo with the different structures. The binding ratio of HTG to TO-neo is similar for the Na+ and K+ structures, with the Na+ having a HTG to TO-neo binding ratio of 0.86 and the K+ with 1.15 ratio of HTG to TO-neo. While these values appear similar, the difference in the ratios is likely indicative of real differences in the interaction of TO-neo with HTG in the presence of Na+ and K+ (see below).
In order to determine the binding affinity of HTG for TO-neo, the binding isotherm was fit to a two state model at a 1:1 binding ratio as previously described.28 The fitting was performed for titrations measured at various time intervals for both the Na+ and K+ conditions to determine if a stable equilibrium had been reached. In the presence of both Na+ and K+, the equilibrium of HTG binding to TO-neo is slow and the titration curve indicates multiple states are initially present. The initial measurements of the Na+ form have large deviations for all concentrations of HTG and a modest fit of the isotherm (Fig. 2a, R = 0.94). At 6 h, the deviations between measurements at each concentration is significantly reduced and the fit of the isotherm is significantly improved (Fig. 2b, R = 0.99). The errors in measurements and fit of the isotherm remains constant after 6 h for the Na+ form of HTG, indicating that the binding equilibrium had been reached.
Similar to the Na+ conditions, the initial measurements in the presence of potassium ions for each HTG titration point into TO-neo were associated with large deviations and the fit of the binding isotherm was less than ideal (Fig. 2d, R = 0.95). However, the K+ structure does not reach equilibrium until 12 h (Fig. 2e, R = 0.99).
The most significant difference in the Na+ and K+ forms of HTG is binding affinity for TO-neo. Analysis of the isotherm indicates that in the presence of Na+ only, the binding affinity is 9.6 × 107 M−1. The binding affinity in the presence of K+ is 9.3 × 106 M−1. Thus the TO-neo has a 10-fold greater affinity for the Na+ form of HTG than the K+ form of HTG.
Despite the difference in the HTG structure in the presence of Na+ and K+, TO-neo binds tightly to either structures and gives a large change in fluorescence upon binding. In both the Na+ and K+ conditions, the TO-neo binds with higher affinity than that of thiazole orange or neomycin alone under similar conditions.17,29 Additionally, neomycin appears to dictate the binding ratio of 1:1 in potassium as opposed to the 2:1 binding ratio typically observed between thiazole orange and G-quadruplexes.29 Thus, it was proposed that TO-neo could be used to probe both structures of HTG.
In order to establish a high throughput assay for screening HTG binding drugs a compound library of neomycin–anthraquinone conjugated molecules were screened to identify the compound with greatest affinity for HTG. The neomycin–anthraquinone compounds have been previously identified as quadruplex binding compounds,19 and compound DPA561 was identified from the screening of a small compound library (12 compounds) as the most effective compound at displacing TO-neo (Fig. 3). DPA561 is a neomycin–anthraquinone conjugate and a similar neomycin–anthraquinone conjugate has been shown to bind with an affinity of 1.25 × 107 M−1.19 As predicted, and as seen with the conjugation of TO with neomycin, the conjugation of the groove binding neomycin with the planar anthraquinone results in a compound with much greater affinity for HTG than either compound alone.19 Thus, DPA561 was used as the positive standard to develop a high throughput assay for high affinity HTG compounds.
Figure 3.
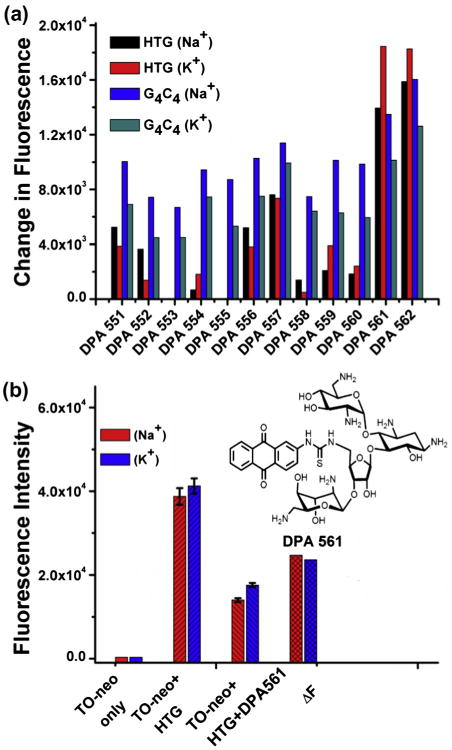
(a) Fluorescence change of TO-neo bound with HTG quadruplex or G4C4duplex in the presence of Na+/K+ upon addition of neomycin-anthraquinone conjugates (DPA 551–562). (b) Optimization of Z-factor using one of the best binders DPA 561. Fluorescent TO-neo Displacement from HTG was performed in the presence of Na+ (red) and K+ (blue). The displacement measurements were performed in a 96-well round bottom Greneir black plate with an excitation wavelength of 485 nm and emission fluorescence measured at 535 nm from 100 reads. All values were the average of 48 wells. The change in fluorescence (ΔF) was calculated by subtracting the average of fluorescence measurement from 100 nM of HTG:TO-neo complex from the average measurement of 100 nM complex with 200 nM DPA561 (insert). (For interpretation of the reference to color in this figure legend, the reader is referred to the web version of this article.)
Another important aspect of compound screening for HTG binding is the discrimination between duplex DNA and quadruplex DNA. To further develop the screening process we assessed the ability of TO-Neo to bind to the duplex DNA sequence d(G4C4)2. We found that a significant increase in the fluorescence intensity occurs upon the addition of TO-neo (data not shown). Additionally, the screening of the same compound library used in the HTG screen shows a significant decrease in the fluorescence with the addition of the compounds. Thus TO-neo, can be used to screen compounds that bind to duplex DNA and quadruplex DNA, allowing for a direct approach to determine compounds that discriminate between the two structures.
The TO-neo screen was standardized for HTG using DPA561. The screen was performed using a fluorescence plate reader in a 96-well plate format. The concentration of TO-neo and HTG was 0.1 μM in all wells, and experiments were performed with in 10 mM HEPES, 100 mM KCl at pH 7.0 or 10 mM sodium cacodylate, 0.5 mM EDTA, 100 mM NaCl at pH 7.0, with excitation wavelength of 485 nm and an emission wavelength of 535 nm. The displacement of TO-neo by DPA561 was measured as greater than a twofold decrease in the fluorescence intensity at a 2:1 ratio of DPA561 to TO-neo (Fig. 3).
The TO moiety provides a strong fluorescent signal when bound to HTG, and can be competitively displaced by HTG binding molecules, resulting in a decrease in fluorescence. The fluorescent based assay provides a platform that is readily adaptable to a high throughput format to be used to identify compounds that bind within the groove of the G-quadruplex, and determine the relative binding affinity of compounds with high affinity for HTG. Several fluorescent based assays have been developed to probe aminoglycoside binding30 to a variety of nucleic acid structures, and recently a fluorescent intercalator displacement assay using thiazole orange (TO)31,32 was developed to screen for compounds that bind to G-quadruplexes. However, these assays are limited in their ability to discriminate against compounds that bind with high affinity characteristic of a dual binding mode.
The quality of the TO-neo displacement assay was determined by the calculation of a Z′-factor using Eq. (1) for the displacement of TO-neo from HTG by DPA561.
| (1) |
The final assay results were obtained using the average (μn) and standard deviation (σn) from 48 wells of 0.1 μM TO-neo:HTG complex as the negative control and from the average (μp) and standard deviation (σp) 48 wells of 0.1 μM complex mixed with 0.2 μM DPA561 as the positive control. A Z′-factor of 1 is ideal. Z′-factor between 0.5 and 1 is considered excellent.33
Our results indicate that the assay is suitable for the detection of compounds that bind to HTG by the displacement of the TO-neo probe in a high throughput format and is a functional assay for both the Na+ and K+ structures. The difference in cations present does not have an effect on the displacement assay of TO-neo from HTG by DPA561. The Z′-factor for the Na+ using DPA561 as a standard was 0.70, which was almost identical to the 0.71 Z′-factor determined for the assay in the presence of the K+.
Because the K+ is a more biologically relevant cation in the vicinity of the telomeres, the formation of HTG in the presence of K+ may be the more relevant form. However, because HTG is highly polymorphic and the conformation is dependent on other factors such as flanking sequence25 and crowding effects,34 the presence of the Na+ form of HTG in biological systems cannot be ignored. The ability to detect compounds with moderate to high binding affinity for multiple forms is highly desirable. Therefore, the TO-neo fluorescence based assay is a high throughput capable screen for detecting high affinity dual mode binding ligands for polymorphic HTG.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health, NIH (GM097917) for financial support. We also thank Gregory Jones, Clemson University, for his assistance with HPLC studies.
Footnotes
Supplementary data: Supplementary data (experimental procedures and synthesis details of 5 are provided) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmcl.2013.10.030.
References and notes
- 1.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Nucleic Acids Res. 2006;34:5402. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Patel DJ. Structure. 1993;1:263. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 3.Autexier C, Lue NF. Annu Rev Biochem. 2006;75:493. doi: 10.1146/annurev.biochem.75.103004.142412. [DOI] [PubMed] [Google Scholar]
- 4.Blackburn EH. Cell. 2001;106:661. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 5.Zahler AM, Williamson JR, Cech TR, Prescott DM. Nature. 1991;350:718. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 6.Shin-ya K, Wierzba K, Matsuo K, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. J Am Chem Soc. 2001;123:1262. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 7.Hounsou C, Guittat L, Monchaud D, Jourdan M, Saettel N, Mergny JL, Teulade-Fichou MP. ChemMedChem. 2007;2:655. doi: 10.1002/cmdc.200600286. [DOI] [PubMed] [Google Scholar]
- 8.Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, Neidle S. Cancer Research. 2005;65:1489. doi: 10.1158/0008-5472.CAN-04-2910. [DOI] [PubMed] [Google Scholar]
- 9.Campbell NH, Patel M, Tofa AB, Ghosh R, Parkinson GN, Neidle S. Biochemistry (NY) 2009;48:1675. doi: 10.1021/bi802233v. [DOI] [PubMed] [Google Scholar]
- 10.Dixon IM, Lopez F, EstÃv̈e J, Blasco MA, Pratviel G, Meunier B. J Am Chem Soc. 2007;129:1502. doi: 10.1021/ja065591t. [DOI] [PubMed] [Google Scholar]
- 11.Xi H, Davis E, Ranjan N, Xue L, Hyde-Volpe D, Arya DP. Biochemistry. 2011;50:9088. doi: 10.1021/bi201077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamilton PL, Arya DP. Nat Prod Rep. 2012;29:134. doi: 10.1039/c1np00054c. [DOI] [PubMed] [Google Scholar]
- 13.Arya DP. Acc Chem Res. 2011;44:134. doi: 10.1021/ar100113q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis B, Arya DP. Curr Org Chem. 2006;10:663. [Google Scholar]
- 15.Willis B, Arya DP. Adv Carbohydr Chem Biochem. 2006;60:251. doi: 10.1016/S0065-2318(06)60006-1. [DOI] [PubMed] [Google Scholar]
- 16.Arya DP. Top Curr Chem. 2005;253:149. [Google Scholar]
- 17.Ranjan N, Andreasen KF, Kumar S, Hyde-Volpe D, Arya DP. Biochemistry. 2010;49:9891. doi: 10.1021/bi101517e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue L, Ranjan N, Arya DP. Biochemistry. 2011;50:2838. doi: 10.1021/bi1017304. [DOI] [PubMed] [Google Scholar]
- 19.Ranjan N, Davis E, Xue L, Arya DP. Chem Commun. 2013:5796. doi: 10.1039/c3cc42721h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lartia R, Asseline U. Chem: Eur J. 2006;12:2270. doi: 10.1002/chem.200500908. [DOI] [PubMed] [Google Scholar]
- 21.Yang P, DeCian A, Teulade-Fichou M, Mergny J, Monchaud D. Angew Chem, Int Ed. 2009;48:2188. doi: 10.1002/anie.200805613. [DOI] [PubMed] [Google Scholar]
- 22.Kirk SR, Luedtke NW, Tor Y. J Am Chem Soc. 2000;122:980. [Google Scholar]
- 23.Arya DP, Xue L, Tennant P. J Am Chem Soc. 2003;125:8070. doi: 10.1021/ja034241t. [DOI] [PubMed] [Google Scholar]
- 24.Parkinson GN, Lee MPH, Neidle S. Nature. 2002;417:876. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 25.Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D. Nucleic Acids Res. 2006;34:2723. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renciuk D, Kejnovská I, Školáková P, Bednárová K, Motlová J, Vorlícková M. Nucleic Acids Research. 2009;37:6625. doi: 10.1093/nar/gkp701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai J, Carver M, Yang D. Biochimie. 2008;90:1172. doi: 10.1016/j.biochi.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiti S, Chaudhury NK, Chowdhury S. Biochem Biophys Res Commun. 2003;310:505. doi: 10.1016/j.bbrc.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 29.Monchaud D, Allain C, Teulade-Fichou MP. Nucleosides Nucleotides Nucleic Acids. 2007;26:1585. doi: 10.1080/15257770701548212. [DOI] [PubMed] [Google Scholar]
- 30.Watkins D, Norris FA, Kumar S, Arya DP. Anal Biochem. 2013;434:300. doi: 10.1016/j.ab.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monchaud D, Allain C, Teulade-Fichou M. Bioorg Med Chem Lett. 2006;16:4842. doi: 10.1016/j.bmcl.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 32.Largy E, Hamon F, Teulade-Fichou M. Anal Bioanal Chem. 2011;400:3419. doi: 10.1007/s00216-011-5018-z. [DOI] [PubMed] [Google Scholar]
- 33.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 34.Heddi B, Phan AT. J Am Chem Soc. 2011;133:9824. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.