

Abstract
Gene expression can be altered by small molecules that target DNA; sequence as well as shape selectivities are both extremely important for DNA recognition by intercalating and groove-binding ligands. We have characterized a carbohydrate scaffold (1) exhibiting DNA “shape readout” properties. Thermodynamic studies with 1 and model duplex DNAs demonstrate the molecule's high affinity and selectivity towards B* form (continuous AT-rich) DNA. Isothermal Titration Calorimetry (ITC), Circular Dichroism (CD) titration, Ultraviolet (UV) thermal denaturation, and Differential Scanning Calorimetry were used to characterize the binding of 1 with a B* form AT-rich DNA duplex d[5′-G2A6T6C2-3′]. The binding constant was determined using ITC at various temperatures, salt concentrations, and pH. ITC titrations were fit using a two-binding site model. The first binding event was shown to have a 1:1 binding stoichiometry and was predominantly entropy driven with a binding constant of approximately 108 M−1. ITC-derived binding enthalpies were used to obtain the binding-induced change in heat capacity (ΔCp) of -225±19 cal/mol·K. The ionic strength dependence of the binding constant indicated a significant electrolytic contribution in ligand:DNA binding, with approximately four to five ion pairs involved in binding. Ligand 1 displayed a significantly higher affinity towards AT-tract DNA over sequences containing GC inserts, and binding experiments revealed the order of binding affinity for 1 with DNA duplexes: contiguous B* form AT-rich DNA (d[5′-G2A6T6C2-3′]) > B form alternate AT-rich DNA (d[5′-G2(AT)6C2-3′]) > A form GC-rich DNA (d[5′-A2G6C6T2-3′]), demonstrating the preference of ligand 1 for B* form DNA.
Keywords: DNA major groove, carbohydrate scaffold, shape recognition
Introduction
The binding of small molecules with duplex DNA has become an active research area in recent years because the complexes formed by small molecules and DNA can alter natural gene expression and eventually regulate cell growth.1, 2 As is becoming evident from studies of DNA-protein interactions, sequence as well as shape selectivities are both valuable in achieving DNA recognition. Therefore, design of small molecules that target DNA requires renewed attention towards DNA shape selectivity. In this regard, carbohydrate-DNA interactions can offer useful insight into recognition of DNA by shape-selective non-planar pharmacophores. DNA-binding ligands consist largely of planar aromatic units. Most DNA-binding ligands, even those having non-planar carbohydrate appendages, owe a large portion of their binding free energy to base-stacking interactions provided by aromatic rings. These scaffolds are observed in both intercalators and minor groove-binding molecules. Additionally, while a large emphasis has been placed on the study of carbohydrate-protein interactions in the last few decades, the study of DNA-carbohydrate interactions is still in its infancy. This study aims to advance our understanding of shape-selective recognition of DNA by carbohydrates.
While structural knowledge of DNA-ligand complex is important in rational drug design, thermodynamic parameters point to the principal driving forces for ligand-DNA interaction. Thermodynamic characterization provides quantitative information about the ligand-DNA interaction, helping identify specific interactions that contribute to the free energy components.3-7
Aminoglycosides, particularly neomycin, have been shown to stabilize numerous nucleic acid structures that possess narrow grooves characteristic of A-form DNA major grooves.8-12 These include B-form DNA minor grooves 13, 14, and grooves of RNA triplexes 15-18, DNA triplexes, 19, DNA/RNA hybrid duplexes/triplexes, 20, 21 and quadruplexes.22 To explore the potential of aminoglycosides to specifically recognize wider grooves, such as the B-form DNA major groove, we have synthesized neomycin dimers by covalently attaching two neomycin units via a linker.9, 12, 23 We have shown previously that one of these neomycin dimers has high affinity and specificity for the major groove of B-form DNA, particularly in AT tracts consisting of ∼10−12 base pairs.9, 24 In an effort to optimize the shape and sequence-selectivity of aminosugar-DNA interactions, here we have characterized the thermodynamics of the binding interaction between neomycin dimer 1 (Figure 1) and continuous AT (B* form DNA with the sequence d[G2A6T6C2]), alternating AT B form DNA with the sequence d[G2(AT)6C2] and GC-rich A form DNA with the sequence d[A2G6C6T2].
Figure 1.

Structure of the ligands used in this study.
Our results show that dimer 1 has a binding preference that changes with DNA shape and sequence, as indicated by comparison of the thermodynamic fingerprints of the complexes. The DNA duplexes: continuous AT, alternate AT, and GC-rich, have different minor and major groove dimensions. The contiguous AT-rich DNA is a model B* form conformation with a narrower minor groove, and a rigid structure when compared to a B form DNA helix.25-28 The major groove of B form DNA is wider and shallower than B* DNA. In this work we find that binding of 1 with the B* form DNA is an entropy driven process, as is expected, largely due to the liberation of ordered water from the major groove of the DNA to the bulk solvent. Similar binding patterns have been observed between small molecule intercalators and AT rich DNA.
The DNA-ligand 1 interaction has been characterized at various buffer conditions (salt and pH) and temperature. The salt dependent studies indicate that ∼4–5 ion pairs form during the binding interaction between neomycin dimer and AT-rich DNA duplex. The predominant contributions to the binding free energy derive from polyelectrolytic free energy and the hydrophobic free energy. Our findings explore the mechanism of binding between a carbohydrate scaffold, neomycin dimer 1, with nucleic acids of different conformations using a battery of thermodynamic experiments.
Materials and Methods
Oligonucleotides synthesis and their characterization
All DNA oligonucleotides except for the 12-mer duplex d[5′-A12-x-T12-3′] oligonucleotide were purchased from Eurofins MWG operon (Huntsville, AL). The 12-mer duplex d[5′-A12-x-T12-3′] oligonucleotide was synthesized in-house on an Applied Biosystem 8890 DNA synthesizer using standard phosphoramidite chemistry and purified by HPLC on a Gen-Pak FAX (4.6 × 100 mm) ion exchange column, eluting with buffer A (25 mM Tris–HCl, 1 mM EDTA, 10 % CH3CN, pH 8.0) from 98% to 50% and buffer B (25 mM Tris–HCl, 1 mM EDTA, 1 M NaClO4, 10 % CH3CN, pH 8.0) from 2% to 50% in 15 min. The concentrations of the DNA sample solutions were determined by measuring UV absorbance at 90°C, and for commercially purchased oligomers, extinction coefficients provided by the company were used to calculate concentration: The following extinction coefficients (ε), in units of mol of nucleotide/L−1 cm−1, were used: ε260= 163,500 for d[5′-A8T8-3′], ε260 = 160,700 for d[5′-GA7T7C-3′], ε260 = 157,900 for d[5′-G2A6T6C2-3′], ε260 = 155,100 for d[5′-G3A5T5C3-3′] ε260= 152,300 for d [5′-G4A4T4C4-3′], ε260 = 147,300 for d[5′-G5A3T3C5-3′], ε260 = 145,000 for d[5′-G6A2T2C6-3′], ε260 = 259,844 for d[5′-A12-x-T12-3′], ε260 = 112,500 for d[5′-CGCA3T3GCG-3′], ε260 = 148,300 for d[5′-A3G5C5T3-3′], ε260 = 187,000 for d[5′-G2A12T12G2-3′], ε260 = 126,200 for d[5′-C2 T12C2-3′], ε260 = 147,300 for [5′-G4CA3T3GC4-3′], ε260 = 144,500 for [5′-G4C2A2T2G2C4-3′], ε260 = 147,300 [5′-CGA4T4CG-3′], ε260 = 117,500 for [5′-G4CA3T3GC4-3′],. All commercially obtained nucleic acids were used without further purification; in-house synthesized oligomers were purified by anion exchange HPLC. Equimolar amounts of duplex strands for each DNA complex were then left to incubate at 4°C overnight for annealing.
Chemicals
Synthesis of 1 has been reported elsewhere (9). Neomycin was purchased from MP Biomedicals (Solon, OH) and all other chemicals were purchased from Sigma Aldrich. All chemicals were used without further purification.
Circular Dichroism (CD) spectroscopy
Circular Dichroism (CD) experiments were conducted at 25°C using a Jasco J-810 spectropolarimeter with a thermo-electrically controlled cell holder. A quartz cell with a 1 cm path length was used in all CD studies. CD spectra were recorded as an average of three scans from 300 nm to 200 nm. Scanning experiments were carried out in 1.8 mL of 4.0 μM/duplex nucleic acid solutions in buffer without the presence of ligand. In CD titration experiments, small aliquots of concentrated ligand solutions were added to the 1.8 mL of 4.0 μM/duplex nucleic acid solutions in buffer and allowed to equilibrate for at least 5 minutes prior to scanning. The resulting scans were plotted for CD signal changes with respect to wavelength and Kaleidagraph 3.5 software (Synergy software) was used to process the data.
Isothermal Titration Calorimetry
In a typical experiment, an aliquot (9 μL) of 1 (125 μM in 10 mM Sodium Cacodylate (SC), 100 mM KCl, 0.5 mM EDTA, and pH 6.8) was injected at 25°C into an isothermal sample chamber containing an oligonucleotide duplex solution (1.42 mL, 4 μM/duplex in 10 mM SC, 100 mM KCl, 0.5 mM EDTA, and pH 6.8) via a 296 μL rotary syringe (rotating at 300 rpm). The interval time between each injection was 300 s, the duration of each injection was 10 s, and the initial delay prior to the first injection was 60 s. Injection of 1 at the same concentration into the pH 6.8 buffer solution at 25°C was used as a blank. Each injection generated a heat burst curve measured in microcalories per second and the area under each curve was determined by integration using Origin (Version 5.0, MicroCal, Northampton, MA) software to quantitate the heat associated with that injection. The associated heat from drug-buffer injection was then subtracted from the corresponding heat associated with each drug-DNA injection to yield the heat of drug binding for that injection. The final corrected injection heats were plotted as a function of molar ratio ([drug]/[DNA]) and subsequently fitted with either a one binding site model or with a two independent binding site model curve.
Heat Capacity Measurements
The binding enthalpies obtained from isothermal titration calorimetry (ITC) experiments were used to calculate the change in heat capacity (ΔCp) measured as the temperature dependence of the binding enthalpy using the relationship given below:
| (1) |
Ultra Voilet (UV) Spectroscopy for Thermal Melting
All UV spectra were obtained using a 12-cell holder Cary 1E UV-Vis spectrophotometer with a temperature controller attached. Quartz cells with 1 cm path length were used for all experiments. The DNA duplex melting was monitored at wavelength of 260 nm and the DNA samples were heated from 20.0°C to 98.0°C at a rate of 0.3°C/min. Buffer conditions were: 100 mM KCl, 10 mM Sodium Cacodylate (SC), 0.5 mM EDTA, and pH 5.5, with [DNA] = 1 μM/duplex for the salt-dependent studies, and 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 6.8 for the thermal melt comparisons with Poly(dA)·Poly(dT). All resulting temperature-absorbance profiles were plotted using Kaleidagraph 3.5 software (Synergy software). For Tm determination, first derivative analysis was used.
Fluorescent Intercalator Displacement (FID) Assay
The single point fluorescent intercalator displacement (FID) assay between DNA oligomers and neomycin dimer (1) was conducted in a 96-well plate. A solution containing DNA (1 μM/duplex) and Thiazole Orange (TO) (0.5 μM/base pair) was prepared by incubating DNA and intercalator for 30 min in buffer (100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 6.8) prior to the measurements. Each well of the 96-well plate (flat bottom, black) was loaded with DNA+TO solution (200 μL). A 1 μL aliquot of the 200 μM stock solution of 1 (for the desired final concentration of 1 μM to give an equimolar ratio of DNA:1) was added and the fluorescence was measured in triplicate after incubation for 5 min. Fluorescence readings were reported as percentage fluorescence relative to control wells, with the reference fluorescence normalized such that [TO+DNA] defines 100% fluorescence, and [TO] defines 0% fluorescence.
Differential Scanning Calorimetry (DSC) Experiments
Differential scanning calorimetry (DSC) measurements were performed using a VP-DSC Microcalorimeter from Microcal. The DSC experiments compared sample and reference cells. Both cells were first loaded with 0.511 mL of buffer solution, equilibrated at 5°C for 15 min, and scanned from 5 to 90°C at a scan rate of 60°C/h. Extreme care was taken to minimize the presence of air bubbles in loading of the sample cell. The data were recorded every 2 s. The solutions were brought back to 5°C at the rate of 60°C/h and incubated for 15 min. The above procedure was repeated three times in order to check for reversibility and reproducibility. Then, the sample cell was emptied, rinsed, and loaded with 0.511 mL of a DNA duplex solution (40 μM/base pair) and scanned under the same conditions. The net DSC scan was analyzed for thermodynamic parameters using Origin 5.0 software (MicroCal). The pre- and post-transitional baselines were determined from a least squares fits of straight lines to the data points, respectively, below the onset of the transition peak and following the return of the transition peak to the baseline. A sigmoidal baseline was determined under the transition peak by extrapolating the pre- and post-transitional baselines and employing the profile of the transition peak. The difference in the extrapolated baselines at the transition temperature divided by the number of moles of duplex is the measured heat capacity change.
Results and discussion
Validating the Binding Stoichiometry: ITC Studies
Results from a 512 sequence FID screen previously performed in our laboratory demonstrated that neomycin dimer 1 (Figure 1) binds with AT-rich DNA duplexes with high affinity and to a binding region of 10−12 base pairs/molecule.9, 24 For the purposes of the work presented here, a series of AT- and GC-rich DNA duplexes were designed and studied with 1 using ITC. In the design of these sequences, the length of the AT stretch was varied to obtain optimal binding stoichiometry of 1:1 with neomycin dimer 1 and the compositions of the base pairs were also varied in order to explore a range of different DNA conformations from B* to A. The AT stretches in DNA duplexes were varied from 6 to 16 base pairs. The GC-rich DNA sequences usually exhibit an A-form conformation, depending upon buffer conditions. Duplexes with three or more contiguous Adenines exhibit B* form structure, while duplexes with alternate AT stretches exhibit B form structure (27). The ITC titrations and the thermodynamic parameters of a select few duplexes are summarized in Supporting Information (Table S1 and S2 and Figure S1 in Supporting Information). Ligand 1 illustrated high binding affinity towards AT-rich DNA duplexes. Additionally, most of the ITC titrations displayed two binding sites. The first binding event is attributed to a specific binding event whereas the second binding is largely a non-specific electrostatic interaction as reflected by the binding stoichiometries associated with the ITC binding profiles. The ITC profile for two DNA duplexes could not be fit to either a one- or two-site model and hence no thermodynamic parameters could be derived for the d[5′-G4C2A3T3G2C4-3′] and d[5′-CGA4T4CG-3′] duplexes (Table S1 and S2, Supporting Information).
To further examine the thermodynamics of interaction of 1 with a B* form AT-rich DNA duplexes, we chose a DNA duplex that displayed a 1:1 binding stoichiometry with 1. The DNA duplex d[5′-G2A6T6C2-3′], which contains 12 base pair of continuous AT base pairs is expected to confer the requisite B* confirmation to this duplex.
Effect of GC Base Pair Insert(s) in the A Tract
Our previous results show that 1 binds with high affinity towards AT-rich DNA duplexes (10). To observe the selectivity of 1 towards AT-rich DNA, DNA duplexes were designed and studied using both FID (Fluorescent Intercalator Displacement) assay 29 and ITC (this work). The DNA duplexes with mismatches contain a GC base pair to break the stretches of AT base pairs. The ITC experiments were conducted with the DNA sequences (Results summarized in Figure S2, Supporting Information) and the ITC data of the mismatch sequences were unable to be fit with either one- or two-site binding isotherms, therefore, no direct binding affinity comparisons can be made between the continuous AT-rich sequence and the mismatch sequences. However, as is indicative from the shapes of the isotherms (Figure S2, Supporting Information), specific binding sites (Figure S1 and S2) are absent in all three of the AT-rich sequences that contain the GC inserts. Nevertheless, the binding enthalpies were determined using excess site titrations (Table S3 in Supporting Information). The binding enthalpies are much higher (−5.12 to −6.81 kcal/mol) for continuous AT-rich DNA, than for DNA sequences containing the GC inserts (−2.78 to −3.68 kcal/mol). FID studies were then conducted with three GC-containing sequences (Table S4, Suppoting Information). 1 shows a clear preference towards DNA duplexes with continuous AT-rich stretches such as d[5′-G2A6T6C2-3′] and d[5′-A8T8-3′] as seen from the % change in thiazole orange (TO) fluorescence upon addition of one molar equivalent of 1 to a solution of DNA+TO. A smaller TO displacement is seen with sequences containing GC inserts, such as d[G2A5GT6C2:G2A6CT5C2], d[G2A5CT6C2:G2A6GT5C2], and d[5′-A7GCT7-3′] (Table S4 in Supporting Information). Ligand 1 displayed a higher affinity for the contiguous AT-rich duplex than for the noncontiguous DNA duplex, reflected by a higher percentage TO displacement by 1.
The binding preference of 1 for AT-rich over GC-rich sequences can be explained with consideration of these DNA duplex conformations. AT-rich DNA duplexes, known as “A-tract” adopt a conformation referred to as B* DNA. B* DNA differs from B DNA in numerous ways: (1) a narrower minor groove, (2) an ordered spine of hydration, featuring a higher order hydration shell in the DNA grooves, (3) a negative propeller twist, and (4) higher rigidity 25, 31. The B* DNA contains contiguous ApA or ApT steps from 5′ towards 3′. Any discontinuity in such a sequence pattern can lead to the disruption of the “A-tract” (e.g. GC base pairs or TpA step from 5′ to 3′)25, 30. Neomycin dimer 1 binds with high affinity towards continuous AT-rich DNA duplexes. The first binding event is entropically driven because of the destabilization of the spine of hydration that causes liberation of water molecules from DNA to the bulk solvent. However, the sequences in which a GC base pair was introduced to disrupt the A tract behave differently. The ITC titrations of the “GC insert” sequences with 1 were not fittable by our one- and two-site models (Figure S2, Supporting Information). The ITC titrations between 1 and the GC–rich DNA (A-form DNA) were primarily enthalpically driven, and demonstrated that 1 had 100- to 500-fold lower binding affinity A DNA than it demonstrated for B* DNA9. The alternate AT-rich DNA sequences, with a B DNA conformation, also bind 1 with two orders of magnitude lower affinity than the continuous AT-rich B* DNA 9. Both comparisons indicate that neomycin dimer 1 has higher binding affinity towards DNA duplexes adopting a B* conformation.
CD Spectroscopy and ITC Investigation of Binding Stoichiometry and DNA Conformation in AT-Rich vs. GC-Rich DNA Duplexes
CD titration was employed to determine effect of 1 on DNA conformation and to determine the binding stoichiometry between 1 and the DNA duplex d[5′-G2A6T6C2-3′] (Figure 2 and Figure S3 in Supporting Information). The CD spectrum of d[5′-G2A6T6C2-3′]shows a positive band around 280 nm and 220 nm and a negative band of the same magnitude near 248 nm (Figure 2A). The continuous addition of the drug induces a small change in the CD spectrum suggesting the occurrence of a binding event 31-33. The drug was continually added until no more changes were observed in the CD spectrum (Figure 2A). A plot between CD intensity and ratio of 1 to DNA results in an inflection point indicating the apparent number of drug molecules associating with the DNA duplex. For d[5′-G2A6T6C2-3′], there is approximately one molecule of 1 binding to each DNA duplex (Figure 2B; Figure S3, Supporting Information). A CD titration was also performed with a GC-rich DNA duplex, d[5′-A2G6C6T2-3′]. The GC-rich DNA duplex possesses an A-form DNA conformation as it exhibits a strong positive band around 260 nm and two negative bands around 211 and 240 nm (Figure 2C). The continuous addition of 1 results in an alteration in the CD spectrum (Figure 2C). The inflection point demonstrated that approximately two molecules of 1 bind with each d[5′-A2G6C6T2-3′] GC rich duplex (Figure 2D).
Figure 2.
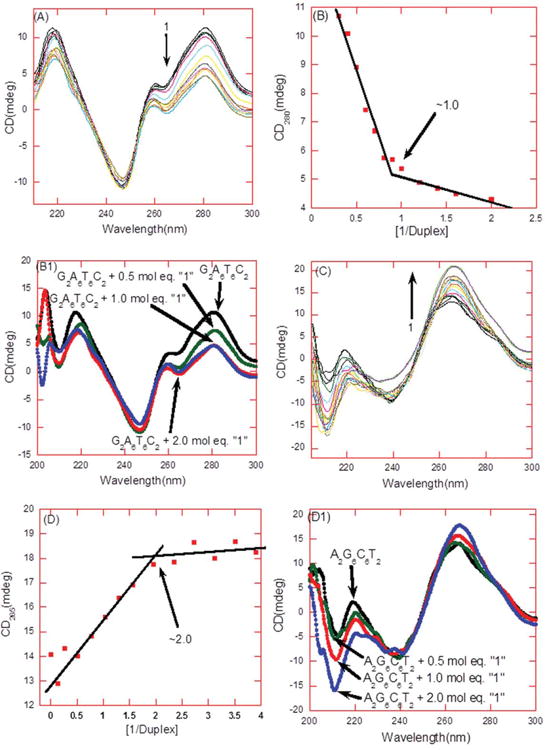
(A) CD titration of d[5′-G2A6T6C2-3′] in the presence of 1. From top to bottom at 280 nm the CD intensity decreased with an increasing amount of 1. (B) The plot between changes in CD intensity (at 280 nm) as a function of the molar ratio of 1 to the DNA duplex. The continuous lines in the plot reflect the linear least squares fit of each apparent linear domain of the experimental data (filled squares) before and after the apparent inflection point. The inflection point corresponds to the binding site size. (B1) CD scans display a change in the CD intensity at stoichiometry ratio (drug/DNA) of 0, 0.5, 1.0, and 2.0. (C) CD titration of d[5′-G2A6T6C2-3′] in the presence of 1. From bottom to top at 265 nm the CD intensity increased with an increasing amount of 1. (D) The plot between changes in CD intensity (at 265 nm) as a function of the molar ratio of 1 to the DNA duplex. The continuous lines in the plot reflect the linear least squares fit of each apparent linear domain of the experimental data (filled squares) before and after the apparent inflection point. The inflection point corresponds to the binding site size. (D1) CD scans display a change in the CD intensity at stoichiometry ratio (drug/DNA) of 0, 0.5, 1.0, and 2.0. Buffer condition: 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH =5.5. T = 25°C. [DNA] = 4 :M/duplex.
We have shown through previous work, combined with these studies, that the monomer neomycin binds to A form DNA duplexes (unpublished works), whereas 1 binds to the wider groove of B and B* form DNA duplexes (ITC-derived thermodynamic parameters for the aforementioned DNA duplexes are summarized in Table S1 and S2, Supporting Information). Neomycin dimer 1 displayed a binding constant of (1.29±0.34)×108 M−1 with the AT-rich DNA duplex, d[5′-G2A6T6C2-3′], that is approximately 200-fold higher than the binding constant measured for the GC-rich DNA duplex, (d[5′-A2G6C6T2-3′, Ka = (6.82±1.31)×105 M−1]). ITC and CD studies of the GC-rich duplex d[5′-A2G6C6T2-3′] demonstrated two binding sites for the monomer neomycin, with similar affinity as observed for 1. This suggests that only half of the dimer unit (one monomer of neomycin) is involved in binding to the A form duplex, providing one explanation for the observed lower affinity of ligand 1 for A form as compared with B or B* form DNA.
Ligand 1 Increases the UV Thermal Denaturation Temperature of DNA Duplexes
The salt-dependent UV melting experiments were performed in the presence and absence of 1 and neomycin (as control) to monitor the effect of these ligands on the thermal melting of the DNA duplex d [5′-G2A6T6C2-3′] at pH 5.5 (Figure S4 in Supporting Information). Our data show that (1) 1 stabilizes the DNA duplex by ∼8.5°C (at rdd = 1, where rdd =ratio of drug/duplex) which is higher in comparison to neomycin-induced stabilization (0.7°C, at rdd = 2) at 100 mM KCl, pH 5.5 (Figure S4 and Table S5, Supporting Information), and that (2) the thermal stability decreases with an increase in salt concentration (Figure S5, Supporting Information), which indicates the role of ammonium groups of 1 in the binding interaction. These data were collected at pH 5.5 instead of pH 6.8 to ensure virtually full protonation of the ammonium groups.
Comparison of 1:d[5′-G2A6T6C2-3′] Binding with 1:poly(dA).poly(dT) Binding Using McGhee's Model for Helix-Coil Transition of DNA
The binding constant calculated using ITC titrations between 1 and oligomeric A tract DNA was compared to binding affinities derived using McGhee fits34 of the UV melting transition of a polymeric A tract [poly(dA).poly(dT)] DNA in the presence of 1. The binding parameters for [poly(dA).poly(dT)] duplex binding to 1 were also calculated independently using ITC and DSC measurements and entered in the van't Hoff equation to determine the binding constant (Table I).
Table I. Thermodynamic Profile of the poly(dA).poly(dT) Interaction with 1 in 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 6.8.
| ΔHwc (kcal/mol) | 4.5±0.2 |
| Tm (°C) | 69.0 |
| Tm (°C) | 84.8 |
| n (base pair/drug) | 16.0 |
| ΔH (kcal/mol) | 11.2±2.1 |
| KT(25°C) (M−1) | (4.0±0.1)×108 |
The binding constant was calculated using the van't Hoff equation. ΔHwc (kcal/mol) = the melting enthalpy for DNA duplex, Tm (°C) = UV melting temperature in the absence of ligand, Tm(°C) = UV melting temperature in the presence of ligand, n = binding site size (base pair/ligand), ΔH (kcal/mol) = heat of enthalpy during the DNA-ligand interaction, KT(25°C)(M−1) = binding constant calculated using the van't Hoff equation.
The parameters for binding of 1 to poly(dA).poly(dT) were determined independently by fitting experimental UV thermal denaturation curves to McGhee model for DNA melting transition in the presence of ligands (Table I).34 All the binding parameters remained unchanged from the previous calculation of the binding constant, in which the McGhee model was used for the UV thermal melting of DNA in the presence of ligands. Both the UV thermal denaturation data and the best fit using McGhee model were plotted, compared, and summarized (Figure 3 and Table II).
Figure 3.
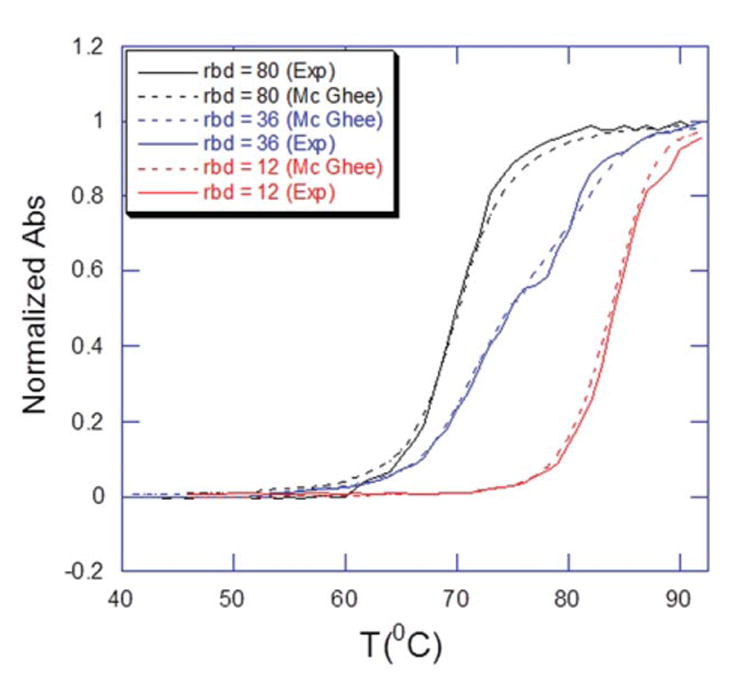
Overlay of the normalized UV thermal denaturation profiles of poly(dA).poly(dT) in the presence of “1” at indicated rbd’s (ratio of base pair to drug) using experiment (thick line) and theoritical (McGhee fit, dotted line) data.
Table II. Binding Parameters Used in the McGhee Model for Determining the UV Melting Transition of DNA in the Presence of Ligands.
| [Poly(dA).poly(dT)] (M bp) | [1] (M) | [1]sim (M) | K (M−1) | n (bp/drug) | σ |
|---|---|---|---|---|---|
| 15.0×10−6 | 0.2×10−6 | 0.2×10−6 | 2.0×108 | 80 | 1.0×10−3 |
| 15.0×10−6 | 0.4×10−6 | 0.4×10−6 | 2.0×108 | 36 | 1.0×10−3 |
| 15.0×10−6 | 1.3×10−6 | 1.3×10−6 | 2.0×108 | 12 | 1.0×10−3 |
[1] is the concentration of the neomycin dimer and [1]sim=the concentration of neomycin dimer used in simulation. σ=the nucleation parameter used in the simulation. n=the DNA-binding site size, K=the binding constant of neomycin dimer with the DNA at 25°C. Some parameters were constraint in the simulation. The melting enthalpy of the DNA (ΔHwc) and the binding enthalpy (ΔHb) between DNA-ligand interactions are 4.5±0.2 kcal mol−1 and −11.2±2.1 kcal mol−1, respectively.
The UV thermal denaturation experiments were conducted at three different concentrations of 1. The UV melting plots were generated using Mc Ghee's model with the following parameters: enthalpy for DNA melting in the absence of ligand (ΔHwc) = 4.54±0.2 kcal mol−1, enthalpy for ligand binding to the helical base pairs (ΔHb) = −11.2±2.1 kcal mol−1, and the cooperative parameter for the ligand binding to helical base-pairs (ωh) = 1.0. The other parameters were adjusted to generate the best fit.35-37 Both the normalized UV thermal melting plots (experimental and from McGhee plot) were overlaid, resulting in a fair agreement of curves (Figure 3). The experimentally-derived binding constant was 4.0×108 (M−1), and was slightly higher as compared with the theoretically calculated binding constant of 2×108 (M−1). This small discrepancy may be explained by slight overestimation of the binding affinities at pH 6.8 due to binding induced heats of drug protonation.
ITC-derived Binding Affinities Used to Characterize the Binding of 1 with Duplex DNA: Differences Between Binding Sites in Alternating vs. Continuous AT-Rich Duplexes
We have employed ITC to characterize the binding affinity between 1 and DNA duplex, and performed ITC titrations to study the effect of pH, temperature, and ionic strength on the binding interaction (Figure 4) a typical ITC profile of interaction between 1 and DNA duplex d[5′-G2A6T6C2-3′]). Most of the isotherms (at different pH, temperature and ionic strength) exhibited two binding events as reflected by the biphasic titration plot. The first entropically driven binding event results in a binding stoichiometry ratio of 1:1 between 1 and DNA duplex while the second binding event is enthalpy driven and involves 2-4 molecules of ligand binding. Previous reports have characterized similar ITC profiles in ligand-DNA interaction.9, 38-40
Figure 4.
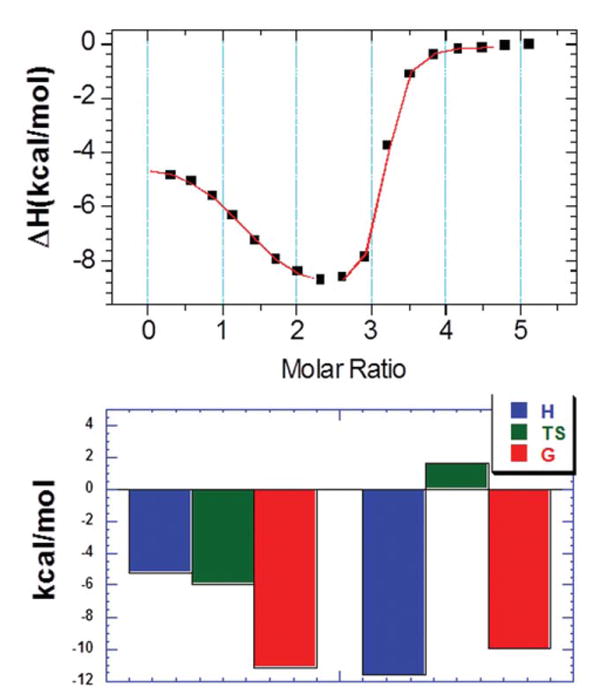
ITC profile (top) and thermodynamic contribution (bottom) of binding interaction between 1 and d[5′-G2A6T6C2-3′]. The ITC profile was fit using the two sets of sites binding model where first binding site was predominately entropy driven while second binding site was enthalpy driven.
The temperature-dependent ITC titrations, (shown in Figure S6, Supporting Information), were performed at pH 6.8 with the ITC profiles and corrected integrated heat (after subtracting the buffer heat). The thermodynamic parameters resulting from these ITC profiles are summarized as well (Table S6, Supporting Information). From these data we may discern the following conclusions: (1) the binding affinity of the first binding event is substantially higher (∼50 times) than the second; and that (2) the first binding event is entropy driven while the second is enthalpy driven. As explained in the previous sections, the origin of this binding is explained by the conformation of the DNA duplex used in the study.
The DNA duplex d[5′-G2A6T6C2-3′] has a continuous stretch of purine or pyrimidine bases which gives rise to a B* form conformation. We undertook a thermodynamic comparison with two alternating versions of AT-rich duplex d[5′-G2(AT)6C2-3′] and d[5′-(AT)8-3′] that exhibit a B form DNA conformation, and the results are summarized in Figure S1 and Table S1 and S2, in Supporting Information. The ITC profiles of both duplexes with 1 is similar in shape to d[5′-G2A6T6C2-3′] but differs in the respective contributions of enthalpy and entropy to the overall binding. Under similar experimental conditions, the binding of 1 with {d[5′-(AT)8-3′]} is overwhelmingly enthalpy driven. The ITC titration between 1 and {d[5′-G2(AT)6C2-3′]} displayed one binding site with a binding constant of (3.16±0.74)×106 M−1. The predominant contribution to binding comes from the binding enthalpy, as opposed to the binding interaction between d[5′-G2A6T6C2-3′] and 1 in which the binding interaction is predominantly entropy driven. The ITC-derived binding affinity of 1 is 50-fold higher for d[5′-G2A6T6C2-3′] than d[5′-G2(AT)6C2-3′].
As previously explained, the high positive entropy contribution in the binding of 1 to duplex d[5′-G2A6T6C2-3′] likely arises from the disruption of the primary hydration sphere of the DNA resulting in the release of water molecules into the bulk solvent.3, 41, 42
Probing the Contribution of Protonation During the Complexation of 1 with DNA
Ligand 1, synthesized from two units of neomycin, is expected to exhibit a similar range of pKa values for its amine groups as the monomer neomycin does (between 5.92 to 9.51).43 In short, we expected that most of the amino groups on 1 would be close to being fully protonated at pH 5.5. The ITC titrations were therefore performed at pH 5.5 (Figure S7, and the results are summarized in Table S7, Supporting Information). The binding enthalpies at both the pH's (5.5 and 6.8) differed markedly. Such divergence clearly suggests a contribution of drug protonation heats during the drug-DNA interaction, as observed by ITC.
There was a slight decrease in both the binding affinity and enthalpy at a pH of 5.5 at 25°C. As discussed previously, the first binding event is entropy driven with a small contribution from enthalpy hence the binding affinity is not affected despite the great modulation in the enthalpy. To ensure the protonation of unbound drug at pH 5.5, an ITC experiment was conducted in a 2-(N-morpholino)ethanesulfonic acid (MES) buffer. MES and SC buffers exhibit different heats of ionization, -0.47 and 3.71 kcal/mol, respectively. Ligand protonation during DNA binding is expected to result in a difference in the observed enthalpy (ΔHobs) (Table S8, supporting information). The ΔHobs are −5.36±0.3 and −5.21±0.1 kcal/mole for MES and sodium cacodylate buffer respectively, suggesting little to no protonation or deprotonation effects during the complexation of 1 with duplex DNA at a pH of 5.5.
Calculation of Heat Capacity Change Associated with the Binding Interaction
The heat capacity change induced during the complexation of 1 and DNA can be obtained using the relationship given as,
| (2) |
Where ΔHobs is the binding enthalpy calculated by direct ITC titration in a temperature range (Figure S7, Supporting Information the data points of which are summarized in the Table S7). Note that the binding heat enthalpy becomes increasingly exothermic with an increase in temperature. A plot (Figure 5) of ΔHobs as a function of temperature allows us to derive the heat capacity change. The slope of the data points yields a ΔCp value of −225±19 cal/mol·K. The value of ΔCp falls within the range of −100 to -500 cal/mol·K, which is typically observed for ligand-nucleic acid and nucleic acid-protein interactions.44-47 More than one factor may impact ΔCp, such as the reduction in the solvent accessible surface area. During the complexation of a ligand with DNA, the negative heat capacity change has been suggested to indicate the burial of the nonpolar surface area, while the positive heat capacity change ascribes the burial of the polar surface area 45, 46, 48-50. Ligand 1 binding with DNA duplex is associated with a negative heat capacity, suggesting that there is a reduction in the solvent accessible area during the binding interaction. Another such contributing factor could be a change in conformation induced by the drug-DNA interaction. Indeed, a substantial number of DNA-ligand interactions studied thermodynamically thus far, among these the minor groove binders 45, ligands with dual recognition, intercalation 4, 36, 51, 52 or bisintercalation 35, lead to changes in DNA conformation. While the CD titration of 1 with DNA shows changes in the DNA CD spectrum, there is little change at ligand to DNA ratio of 1:1.
Figure 5.
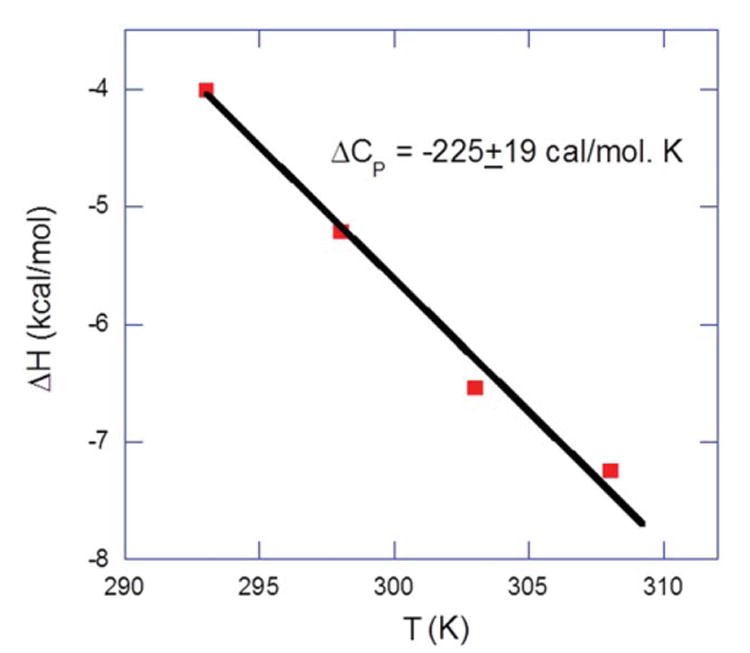
Temperature dependence of the binding enthalpy of 1-DNA interactions. The data points were determined experimentally (derived by direct ITC titration between 1 and DNA) within a temperature range of 20-35°C at 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 5.5. The slope of the linear least-square fit of the data gives a δ Cp value of −225±19 cal mol−1 K−1.
Salt Dependence of the 1-to-DNA Interaction
As 1 is a positively charged ligand, the binding interaction with negatively charged DNA is expected to be influenced by the salt concentration used in this study. The ITC experiments were conducted at 60, 87.5, 100, and 125 mM KCl concentrations (Figure S8, Supporting Information) to analyze the ionic strength dependence of the binding of 1 with DNA (the ITC-derived binding parameters are provided in Table S9 and S10). The binding affinity of 1 with DNA duplex decreases with a corresponding increase in the salt concentration. The salt-dependent binding parameters were used to calculate the number of condensed counter ions liberated or the formation of ion pairs during the binding interaction between 1 and DNA duplex using condensation theory developed by Record et al (Eq. 3).53
| (3) |
In Eq. 2, Z denotes the ion-pairs formed between ligand-DNA, φ is the number of the counter-ions associate with each phosphate group on nucleic acid, a value normally used as 0.88 for polynucleotide DNA. The φ value varies with different nucleic acids and with the length of the nucleic acids. For oligonucleotides, this value is expected to be smaller than the polynucleotides because of a reduced charge density and a reduced release of counter ions per bound cation during complexation;54-56 hence a φ value of 0.75, as previously estimated, is used here. K0 is the binding constant at 1 M [K+] and the binding affinity at 1 M salt concentration is considered to be free from electrostatic interactions. Using Eq. 2, we estimated the number of ammonium groups participating in electrostatic interactions with the duplex DNA. Fitting the experimental points using linear regression yielded a slope of −3.25 (Zφ) (Figure 12A). Using a φ value of 0.75, we find that 1 uses ∼4–5 ammonium groups out of twelve at pH 5.5 in its binding with DNA duplex (Z=3.25/0.75). Hence, even though 1 is likely present as a dodeca-cation at pH 5.5, it binds to the DNA duplex via formation of four to five ion-pairs. Such a significant contribution from electrostatic interactions requires the use of the Record et al.'s 53 polyelectrolyte theory, with which we can dissect the total free energy into its components including electrolytic (ΔGpe) and non-electrolytic (ΔGnon-pe) contributions (Eq. 4):
| (4) |
Similarly, the polyelectrolyte contribution can be calculated by using Eq. 5 53:
| (5) |
The ITC-derived binding energy and contributions of its components (electrolyte and non-electrolyte) were summarized (Table S11, Supporting Information) and K0 at 1.0 M [K+] was then estimated by extrapolating from the graph in Figure 6A, and was found to be 1.77×105 M−1.
Figure 6.
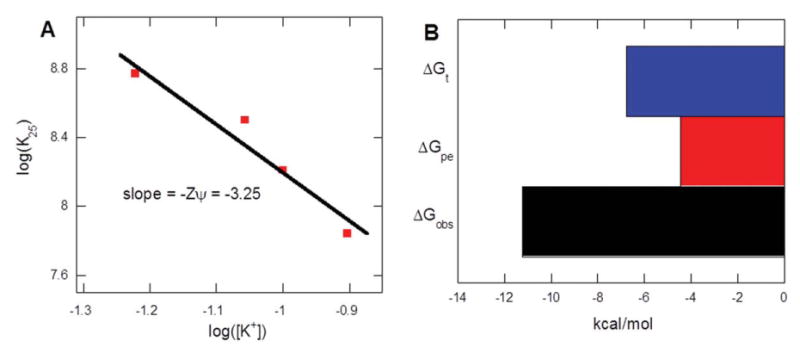
Salt dependence of direct ITC derived binding constants at 25°C. The experimental data points were fit by using linear regression yielding a slope of −3.25, indicating the participation of approximately ∼4-5 NH3 + groups of 1 in electrostatic interactions with the host DNA. Buffer conditions: 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 5.5.
Dissection of the Free Energy of Binding into Various Components
The binding free energy of drug-DNA interaction can be determined by using the standard Gibbs free energy equation (Eq. 6):
| (6) |
Where ΔGobs is the Gibbs free energy of ligand-DNA interaction and can be directly calculated from the binding constant (Kobs). The observed binding constant Kobs is derived from the ITC experiment (Table III). To dissect the free energy of binding (ΔGobs) into its various components, we followed the method outlined by Chaires and coworkers4, 5, 57 as represented in Eq. 7:
Table III. Temperature Dependence of the Binding Constant of 1 with DNA Oligonucleotide d[5′-G2A6T6C2-3′].
| T (°C) | N1 | K1 (M−1) × 108 | ΔGobs(kcal/mol) | ΔH1(kcal/mol) | ΔS1(cal/mol K) |
|---|---|---|---|---|---|
| 20 | 1.1±0.1 | 2.0±0.5 | −11.1 | −4.0±0.2 | 24.3 |
| 25 | 1.1±0.0 | 1.6±0.3 | −11.2 | −5.2±0.1 | 20.1 |
| 30 | 1.1±0.1 | 1.1±0.4 | −11.1 | −6.5±0.5 | 15.1 |
| 35 | 1.1±0.1 | 1.0±0.3 | −11.3 | −7.3±0.6 | 13.0 |
ΔG1=the binding-free energy calculated from binding constant (K1) using ΔGobs = - RT ln K1. Buffer conditions: 100 mM KCl, 10 mM SC, 0.5 mM EDTA, and pH 5.5.
| (7) |
ΔGconf is the free energy resulting from the change in conformation during the formation of the complex, ΔGhyd is the free energy contribution from the burial of the hydrophobic surface area of the drug from the solution into the DNA, ΔGt+r is the free energy contribution resulting from the loss of the rotational and translational degrees of freedom during the complex formation between drug and DNA, and ΔGpe is the free energy contribution from electrostatic interactions that cause the release of ions in solution (Figure 7). The free energy contribution from the electrostatic, van der Waals interactions and hydrogen bonding between ligand and DNA are estimated by ΔGmol (Figure 7).
Figure 7.
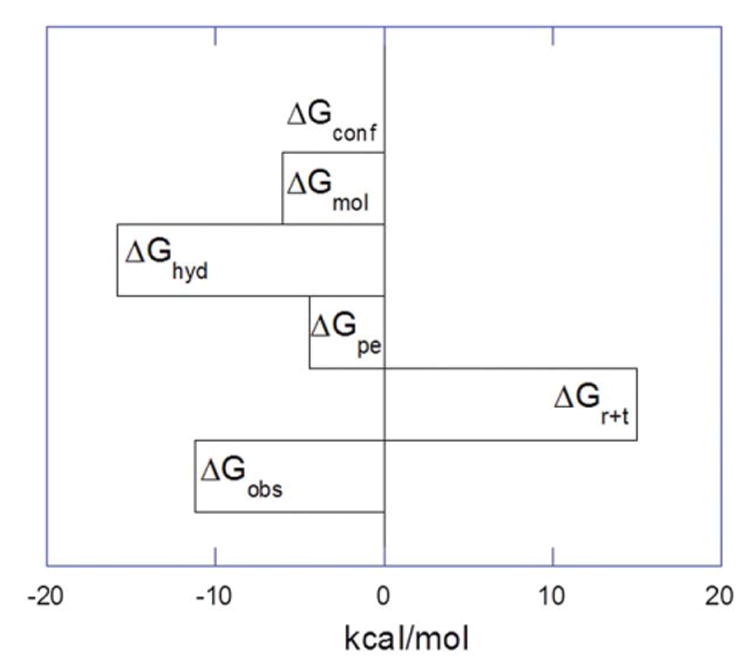
Dissection of the components of free energy of 1 binding with d[5′-G2A6T6C2-3′] at pH = 5.5 (in the absence of drug protonation during the binding event) at 25 °C. The contribution from +Gconf is considered to be zero for the interaction of 1 with DNA. The +Ghyd is the average, without considering the error value (±10).
In this section, we discuss the origin of each component of the free energy for the interaction of 1 and DNA (as summarized in Table S12). ΔGpe, the free energy contribution from the release of ions in the solution contributes significantly to the overall free energy because of the interaction of positively charged 1 with the DNA duplex. The polyelectrolytic contribution of −4.43 kcal/mol was determined via the following equation:
| (8) |
Zφ = 3.25, R = 0.019 kcal/mol·K, T = 298 K, and [KCl] = 0.100 M
ITC-derived binding enthalpies were used to calculate the heat capacity change (−225±19 cal/mol·K), which in turn was then used to calculate the free energy contribution from the transfer of hydrophobic surface of 1 from the solution into the DNA. This contribution (ΔGhyd) was derived via the commonly used relationship for a wide range of DNA-binding drugs and DNA sequences58:
| (9) |
The free energy contribution calculated using this equation has a range of −17.5 to −22.5 kcal/mol, and represents a significant contribution to the magnitude of the overall free energy of binding for such as polycationic ligand. Nevertheless, the range of free energy seems reasonable in comparison to other hydrophobic ligands such as Hoechst 33258 (ΔCp = −330 cal/mol·K, ΔGhyd = ∼ −26.4 kcal/mol),45 and Chartreusin (ΔCp = −391 cal/mol·K, ΔGhyd = ∼−30 kcal/mol),51 where a much more negative heat capacity change leads to a more substantial free energy of interaction. As discussed earlier, the salient contribution of the induced heat capacity change (ΔCp) is derived from the burial of the highly polar surface of 1 from the solution into the duplex DNA. These values are significantly larger than the total free energy of binding, helping counterbalance the entropic costs of dimer 1: DNA binding. Though we lack the structural information, negligible changes in CD of the DNA upon drug binding at pH 5.5 and pH 6.8 (Supporting Information) allow us to estimate an insignificant contribution from DNA conformational changes during the formation of the complex; hence the contribution from ΔGconf is estimated here as zero. The considerable loss of translational and rotational degrees of freedom during the complex formation between ligand and DNA contributes unfavorably to the total free energy and has been empirically calculated by Spolar and Record.50 We used the previously estimated value of ΔGt+r of +15 kcal/mol (20% error), with its well-recognized limitations. From these estimates, the value of ΔGmol, the free energy contribution from intermolecular interactions, including van der Waals forces, and hydrogen binding is calculated as −6.02 kcal/mol:
The free energy contribution from the molecular interaction of forces is minimal in comparison to the overall binding free energy. Recall that the polyelectrolytic contribution (ΔGpe) contributed −4.43 kcal/mol of the total −11.20 kcal/mol for ΔGobs suggesting that for the 1:DNA interaction characterized here, approximately ∼40% of the free energy of binding is derived from ionic interactions. In the interaction of 1 with DNA, the first binding site being evaluated here is largely entropy-driven, resulting in the displacement of water molecules from the DNA groove. Hence, the limited van der Waals and H-bonding interactions implied here by the ΔGmol value of −6.02 kcal/mol are somewhat surprising, but can be easily explained by the entropic fingerprint of binding. Similar observations have been noticed during the interaction of Chartreusin binding to duplex DNA51 as well as the binding of dye Hoechst 33258 with the DNA duplex.45 These findings lend credence to the role of DNA shapes and conformational preference (shape readout) in ligand:DNA interactions, as opposed to focusing exclusively on the role of “direct readout” mechanisms in designing macromolecule specific small molecules, in which intermolecular interactions such as hydrogen bonds, van der Waals contacts are used to make specific interactions to DNA bases.
An Energy Compensation Comparison of Ligand 1 Known DNA-Binding Ligands
A dataset that contains the energy compensation plot of 26 drug-DNA interactions was reproduced from the plot by Chaires,59, which was derived from the original plot presented by Jacobson that illustrated the protein-DNA energy compensation.60 The binding free energy of 1: DNA duplex d[5′-G2A6T6C2-3′] was dissected into its discrete enthalpy and entropy components and plotted as shown in Figure 8. The enthalpy-entropy compensation plot provides us with a pattern that distinguishes intercalative binding from groove binding. The intercalator-DNA interactions are generally enthalpically favored, whereas the groove binder-DNA interactions are entropically favored. The energy compensation data of 1 was entered in the plot at both the pH values (5.5 and 6.8) and monitored relative to their positions in the pattern (Figure 8).
Figure 8.
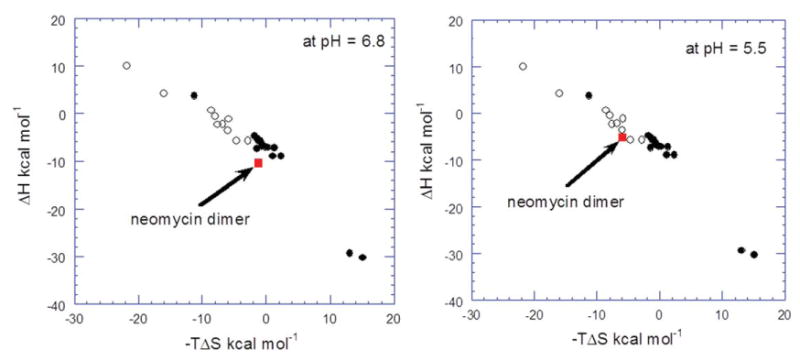
The energy compensation plot of DNA-drug interaction. The filled squares illustrate the energy compensation plot of intercalators and the open squares represent the data for groove binders. 1 is represented by the red square for comparison. The energy compensation data of 1 with the DNA duplex d[5′-G2A6T6C2-3′] compared at both pH readings. The figure is adapted and modified from the work of Chaires (59).
At pH 5.5, 1 clearly falls within the subgroup of groove binders (Figure 8). The thermodynamic data (from ITC) also support the claim that 1-DNA interaction is an entropy- favored binding event. At a pH of 6.8, however, the Figure 8 shows that the enhanced enthalpic signature of binding of 1 to DNA positions it among intercalative ligands, which is clearly not possible for a non-planar ligand such as 1. It is important to note that at pH 6.8, the heat of binding induced drug protonation adds to the binding enthalpy and the overall enthalpy of the 1-DNA interaction is thus over estimated. Therefore, the data points in Chaires' compensation plot of 1-DNA interaction do not fall within the expected groove binding pattern of the energy compensation dataset at the pH of 6.8. Caution must be used in using ligand-DNA enthalpies in the Chaires' plot. Intrinsic enthalpies of ligand-DNA interactions are needed for analysis and any binding-induced protonation contributions to enthalpies of interaction can lead to erroneous results.
In summary, dimeric neomycin 1, is one of the first “all carbohydrate” ligands, with no planar or intercalative moiety present, to bind to duplex DNA with extremely high affinities (>108 M−1). Ligand 1 shows a conformational preference in DNA binding, with the highest affinity for B* DNA containing at least two AT tracts d[5′-G2A6T6C2-3′]. Binding of 1 to DNA is largely driven by the release of ordered waters from the DNA groove and a small intrinsic free energy of molecular interaction. Ligand 1 binds to DNA as a hexa-cation at pH 5.5, as seen in the salt dependence of its binding to DNA. Recognition of DNA shapes and conformations 61, 62 by groove binding small molecules such as 1 is essential for a comprehensive approach to DNA recognition. Our studies outline a first step in the development of such molecules with “shape readout” properties.
Supplementary Material
Acknowledgments
Contract grant sponsor: National Science Foundation
Contract grant number: CHE/MCB-0134972
Contract grant sponsor: National Institutes of Health
Contract grant number: R15CA125724
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Dervan PB. Bioorg Med Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 2.Neidle S. Nat Prod Rep. 2001;18:291–309. doi: 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]
- 3.Marky LA, Snyder JG, Remeta DP, Breslauer KJ. J Biomol Struct Dyn. 1983;1:487–507. doi: 10.1080/07391102.1983.10507457. [DOI] [PubMed] [Google Scholar]
- 4.Chaires JB, Satyanarayana S, Suh D, Fokt I, Przewloka T, Priebe W. Biochemistry. 1996;35:2047–2053. doi: 10.1021/bi952812r. [DOI] [PubMed] [Google Scholar]
- 5.Chaires JB. Biopolymers. 1997;44:201–215. doi: 10.1002/(SICI)1097-0282(1997)44:3<201::AID-BIP2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 6.Lane AN, Jenkins TC. Q Rev Biophys. 2000;33:255–306. doi: 10.1017/s0033583500003632. [DOI] [PubMed] [Google Scholar]
- 7.Haq I. Arch Biochem Biophys. 2002;403:1–15. doi: 10.1016/S0003-9861(02)00202-3. [DOI] [PubMed] [Google Scholar]
- 8.Arya DP. Acc Chem Res. 2010;44:134–146. doi: 10.1021/ar100113q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, Xue L, Arya DP. J Am Chem Soc. 2011;133:7361–7375. doi: 10.1021/ja108118v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willis B, Arya DP. Biochemistry. 2010;49:452–469. doi: 10.1021/bi9016796. [DOI] [PubMed] [Google Scholar]
- 11.Xue L, Ranjan N, Arya DP. Biochemistry. 2011;50:2838–2849. doi: 10.1021/bi1017304. [DOI] [PubMed] [Google Scholar]
- 12.Kumar S, Arya DP. Bioorg Med Chem Lett. 2011;21:4788–4792. doi: 10.1016/j.bmcl.2011.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willis B, Arya DP. Biochemistry. 2006;45:10217–10232. doi: 10.1021/bi0609265. [DOI] [PubMed] [Google Scholar]
- 14.Willis B, Arya DP. Curr Org Chem. 2006;10:663–673. [Google Scholar]
- 15.Arya DP, Micovic L, Charles I, Coffee RL, Jr, Willis B, Xue L. J Am Chem Soc. 2003;125:3733–3744. doi: 10.1021/ja027765m. [DOI] [PubMed] [Google Scholar]
- 16.Arya DP, Coffee RL, Jr, Willis B, Abramovitch AI. J Am Chem Soc. 2001;123:5385–5395. doi: 10.1021/ja003052x. [DOI] [PubMed] [Google Scholar]
- 17.Arya DP, Coffee RL, Jr, Charles I. J Am Chem Soc. 2001;123:11093–11094. doi: 10.1021/ja016481j. [DOI] [PubMed] [Google Scholar]
- 18.Arya DP, Coffee RL., Jr Bioorg Med Chem Lett. 2000;10:1897–1899. doi: 10.1016/s0960-894x(00)00372-3. [DOI] [PubMed] [Google Scholar]
- 19.Xi H, Kumar S, Dosen-Micovic L, Arya DP. Biochimie. 2010;92:514–529. doi: 10.1016/j.biochi.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw NN, Xi H, Arya DP. Bioorg Med Chem Lett. 2008;18:4142–4145. doi: 10.1016/j.bmcl.2008.05.090. [DOI] [PubMed] [Google Scholar]
- 21.Shaw NN, Arya DP. Biochimie. 2008;90:1026–1039. doi: 10.1016/j.biochi.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 22.Ranjan N, Andreasen KF, Kumar S, Hyde-Volpe D, Arya DP. Biochemistry. 2010;49:9891–9903. doi: 10.1021/bi101517e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael K, Wang H, Tor Y. Bioorg Med Chem. 1999;7:1361–1371. doi: 10.1016/s0968-0896(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 24.Arya DP, Coffee RL, Jr, Xue L. Bioorg Med Chem Lett. 2004;14:4643–4646. doi: 10.1016/j.bmcl.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Haran TE, Mohanty U. Q Rev Biophys. 2009;42:41. doi: 10.1017/S0033583509004752. [DOI] [PubMed] [Google Scholar]
- 26.Hud NV, Plavec J. Biopolymers. 2003;69:144–158. doi: 10.1002/bip.10364. [DOI] [PubMed] [Google Scholar]
- 27.Breslauer KJ, Remeta DP, Chou WY, Ferrante R, Curry J, Zaunczkowski D, Snyder JG, Marky LA. Proc Natl Acad Sci U S A. 1987;84:8922–8926. doi: 10.1073/pnas.84.24.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexeev DG, Lipanov AA, Ya Skuratovskii I. Nature. 1987;325:821–823. doi: 10.1038/325821a0. [DOI] [PubMed] [Google Scholar]
- 29.Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. J Am Chem Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- 30.Watkins D, Hsiao C, Woods KK, Koudelka GB, Williams LD. Biochemistry (N Y) 2008;47:2325–2338. doi: 10.1021/bi701826f. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov VI, Minchenkova LE, Schyolkina AK, Poletayev AI. Biopolymers. 1973;12:89–110. doi: 10.1002/bip.1973.360120109. [DOI] [PubMed] [Google Scholar]
- 32.Gray DM, Ratliff RL. Biopolymers. 1975;14:487–498. doi: 10.1002/bip.1975.360140305. [DOI] [PubMed] [Google Scholar]
- 33.Gray DM, Ratliff RL, Vaughan MR. Meth Enzymol. 1992;211:389–406. doi: 10.1016/0076-6879(92)11021-a. [DOI] [PubMed] [Google Scholar]
- 34.McGhee JD. Biopolymers. 1976;15:1345–1375. doi: 10.1002/bip.1976.360150710. [DOI] [PubMed] [Google Scholar]
- 35.Leng F, Priebe W, Chaires JB. Biochemistry (N Y) 1998;37:1743–1753. doi: 10.1021/bi9720742. [DOI] [PubMed] [Google Scholar]
- 36.Leng F, Chaires JB, Waring MJ. Nucleic Acids Research. 2003;31:6191–6197. doi: 10.1093/nar/gkg826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui T, Wei S, Brew K, Leng F. J Mol Biol. 2005;352:629–645. doi: 10.1016/j.jmb.2005.07.048. [DOI] [PubMed] [Google Scholar]
- 38.Freyer MW, Buscaglia R, Nguyen B, David Wilson W, Lewis EA. Anal Biochem. 2006;355:259–266. doi: 10.1016/j.ab.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 39.Freyer MW, Buscaglia R, Hollingsworth A, Ramos J, Blynn M, Pratt R, Wilson WD, Lewis EA. Biophys J. 2007;92:2516–2522. doi: 10.1529/biophysj.106.098723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freyer MW, Buscaglia R, Cashman D, Hyslop S, Wilson WD, Chaires JB, Lewis EA. Biophys Chem. 2007;126:186–196. doi: 10.1016/j.bpc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 41.Marky LA, Breslauer KJ. Proc Natl Acad Sci U S A. 1987;84:4359–4363. doi: 10.1073/pnas.84.13.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marky LA, Blumenfeld KS, Breslauer KJ. Nucleic Acids Res. 1983;11:2857–2870. doi: 10.1093/nar/11.9.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaul M, Barbieri CM, Kerrigan JE, Pilch DS. J Mol Biol. 2003;326:1373–1387. doi: 10.1016/s0022-2836(02)01452-3. [DOI] [PubMed] [Google Scholar]
- 44.Fisher HF, Singh N. Methods in Enzymology. 1995;259:194–221. doi: 10.1016/0076-6879(95)59045-5. [DOI] [PubMed] [Google Scholar]
- 45.Haq I, Ladbury JE, Chowdhry BZ, Jenkins TC, Chaires JB. J Mol Biol. 1997;271:244–257. doi: 10.1006/jmbi.1997.1170. [DOI] [PubMed] [Google Scholar]
- 46.Mazur S, Tanious FA, Ding D, Kumar A, Boykin DW, Simpson IJ, Neidle S, Wilson WD. J Mol Biol. 2000;300:321–337. doi: 10.1006/jmbi.2000.3869. [DOI] [PubMed] [Google Scholar]
- 47.Barbieri CM, Srinivasan AR, Pilch DS. J Am Chem Soc. 2004;126:14380–14388. doi: 10.1021/ja0457516. [DOI] [PubMed] [Google Scholar]
- 48.Haq I, Jenkins TC, Chowdhry BZ, Ren J, Chaires JB. Methods Enzymol. 2000;323:373–405. doi: 10.1016/s0076-6879(00)23374-0. [DOI] [PubMed] [Google Scholar]
- 49.Haq I, Ladbury J. J Mol Recognit. 2000;13:188–197. doi: 10.1002/1099-1352(200007/08)13:4<188::AID-JMR503>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 50.Spolar RS, Livingstone JR, Record MT. Biochemistry (N Y) 1992;31:3947–3955. doi: 10.1021/bi00131a009. [DOI] [PubMed] [Google Scholar]
- 51.Barceló F, Capó D, Portugal J. Nucleic Acids Res. 2002;30:4567–4573. doi: 10.1093/nar/gkf558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barceló F, Scotta C, Ortiz-Lombardía M, Méndez C, Salas JA, Portugal J. Nucleic Acids Res. 2007;35:2215–2226. doi: 10.1093/nar/gkm037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Record MT, Jr, Anderson CF, Lohman TM. Q Rev Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 54.Olmsted MC, Bond JP, Anderson CF, Record MT., Jr Biophys J. 1995;68:634–647. doi: 10.1016/S0006-3495(95)80224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang W, Ni H, Capp MW, Anderson CF, Lohman TM, Record MT., Jr Biophys J. 1999;76:1008–1017. doi: 10.1016/S0006-3495(99)77265-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen B, Stanek J, Wilson WD. Biophys J. 2006;90:1319–1328. doi: 10.1529/biophysj.105.071381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chaires JB. Curr Opin Struct Biol. 1998;8:314–320. doi: 10.1016/s0959-440x(98)80064-x. [DOI] [PubMed] [Google Scholar]
- 58.Record MT, Jr, Ha JH, Fisher MA. Methods Enzymol. 1991;208:291–343. doi: 10.1016/0076-6879(91)08018-d. [DOI] [PubMed] [Google Scholar]
- 59.Chaires JB. Arch Biochem Biophys. 2006;453:26–31. doi: 10.1016/j.abb.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 60.Jen-Jacobson L, Engler LE, Jacobson LA. Structure. 2000;8:1015–1023. doi: 10.1016/s0969-2126(00)00501-3. [DOI] [PubMed] [Google Scholar]
- 61.Hamilton PL, Arya DP. Nat Prod Rep. 2012;29:134–143. doi: 10.1039/c1np00054c. [DOI] [PubMed] [Google Scholar]
- 62.Xi H, Davis E, Ranjan N, Xue L, Hyde-Volpe D, Arya DP. Biochemistry. 2011;50:9088–9113. doi: 10.1021/bi201077h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.