

Abstract
Exposure of cells to various stresses often leads to the induction of a group of proteins called heat shock proteins (HSPs, molecular chaperones)1,2. Hsp70 is one of the most highly inducible molecular chaperones, but its expression must be maintained at low levels under physiological conditions to permit constitutive cellular activities to proceed3,4. Heat shock transcription factor 1 (HSF1) is the transcriptional regulator of HSP gene expression5, but it remains poorly understood how newly synthesized HSPs return to basal levels when HSF1 activity is attenuated. CHIP (carboxy terminus of Hsp70-binding protein), a dual-function co-chaperone/ubiquitin ligase, targets a broad range of chaperone substrates for proteasomal degradation6–11. Here we show that CHIP not only enhances Hsp70 induction during acute stress but also mediates its turnover during the stress recovery process. Central to this dual-phase regulation is its substrate dependence: CHIP preferentially ubiquitinates chaperone-bound substrates, whereas degradation of Hsp70 by CHIP-dependent targeting to the ubiquitin–proteasome system occurs when misfolded substrates have been depleted. The sequential catalysis of the CHIP-associated chaperone adaptor and its bound substrate provides an elegant mechanism for maintaining homeostasis by tuning chaperone levels appropriately to reflect the status of protein folding within the cytoplasm.
The activation status of HSF1 is governed in large part by the balance between the amount of misfolded proteins and the chaper-one availability in the cell12,13. However, HSF1 also seems to have additional layers of regulation in the cell14. For example, our previous data indicate that CHIP elicits the transcriptional activation of HSF1, independently of its ability to facilitate the clearance of misfolded proteins in the cell15. To improve our understanding of the effects of CHIP on both HSF1 activity and chaperone availability, we manipulated CHIP expression levels in HEK-293 cells by plasmid-mediated overexpression or double-stranded RNA (dsRNA)-dependent knockdown. In concordance with our previous observations, CHIP overexpression triggered the activation of HSF1 in a dose-dependent manner as indicated by luciferase reporter assays (Fig. 1a). Immunoblotting demonstrated elevated levels of Hsp70 as expected. Conversely, cells depleted of CHIP by dsRNA had lower basal levels of HSF1 activity. To our surprise, however, no decrease in Hsp70 levels was observed in these cells, and instead we found a moderate but consistent induction of Hsp70 protein levels (Fig. 1b). To explore this observation further, we analysed Hsp70 expression in CHIP−/− and wild-type fibroblasts. Consistent with their reduced stress response in the absence of CHIP was our observation that CHIP−/− cells demonstrated much lower Hsp70 induction after heat shock (42 °C, 15 min; Fig. 1c). However, the basal Hsp70 level before heat shock was paradoxically higher in CHIP−/− than in CHIP+/+ cells. Taken together, these experiments imply that CHIP regulates Hsp70 expression through mechanisms that operate differentially under physiological and stressful conditions.
Figure 1. CHIP regulates Hsp70 availability.
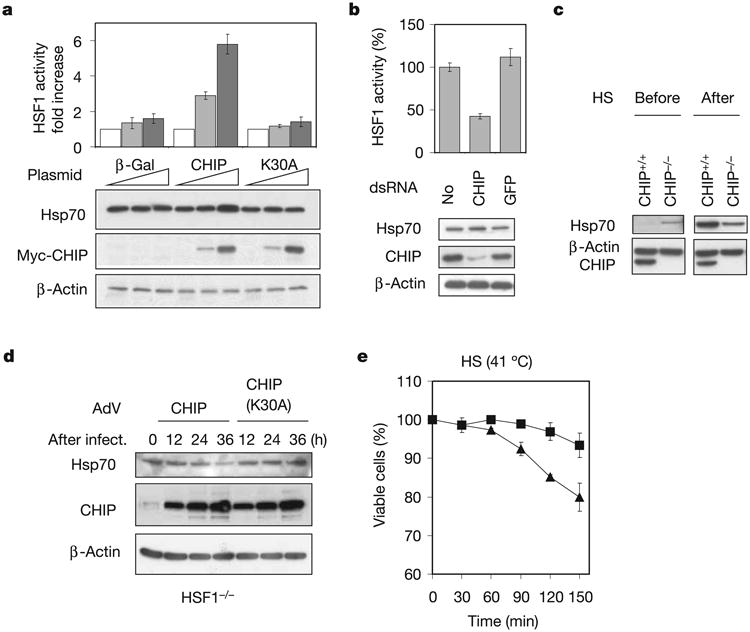
a, HEK-293 cells were transfected with plasmids as indicated with 0 μg (white bars), 0.1 μg (grey bars) and 0.5 μg (black bars) per well in a six-well plate. HSF1 activity was measured by dual luciferase reporter assay. Hsp70 levels were determined by immunoblotting. b, HEK-293 cells were transfected with dsRNA as indicated. Both HSF1 activity and Hsp70 levels were determined. c, Immortalized CHIP+/+ and CHIP−/− cell lines were heat shocked for 15 min at 42 °C (HS) and left to recover for 4 h at 37 °C. Cell lysates from before and after heat shock were immunoblotted with Hsp70 antibody. d, HSF1−/− cells were infected with AdV expressing CHIP or CHIP(K30A). Hsp70 levels were determined at different times after infection by immunoblotting of whole cell lysates. e, AdV-infected HSF1−/− cells were incubated at 41 °C for various durations and cell viability was measured. Triangles, AdV/CHIP; squares, AdV/CHIP(K30A). Values are means ± s.d.
To assess the effects of CHIP on Hsp70 availability under conditions in which its ability to activate HSF1 is inoperative, we used an immortalized fibroblast cell line derived from HSF1−/− mice. Recombinant adenovirus (AdV)-mediated CHIP overexpression markedly decreased Hsp70 in these cells, whereas no such effect was observed by overexpressing an inactive mutant of CHIP, CHIP(K30A), that does not dock with chaperones16 (Fig. 1d). Previous studies have indicated that HSF1−/− cells are sensitive to stress because they lack an appropriate heat shock response17. We predicted that the diminished levels of Hsp70 after CHIP over-expression should render these cells more susceptible to stresses such as heat shock. As expected, HSF1−/− cells were less viable when CHIP was overexpressed during incubation at 41 °C than were cells overexpressing CHIP(K30A) (Fig. 1e). Taken together, these observations indicate a role for CHIP in multiple steps of the stress response: while activating HSF1 and consequently inducing HSP expression, CHIP also negatively regulates steady-state levels of Hsp70 through an HSF1-independent mechanism.
As a co-chaperone ubiquitin ligase, CHIP is capable of ubiquitinating its associated chaperone partners in vivo and in vitro18, although it has not been apparent that this leads to their proteasomal degradation, nor have physiological roles for CHIP-dependent chaperone ubiquitination been assigned. To explore this relationship further, we examined turnover of HSPs in CHIP−/− cells after reintroducing either CHIP, CHIP(K30A) or green fluorescent protein (GFP) alone as an additional control. Because the basal level of Hsp70 in mouse fibroblasts is very low, the infected cells were transiently exposed to 42 °C for 30 min and recovered at 37 °C for 6 h. The turnover of stress-induced Hsp70 was examined by incubating cells in the presence of the protein synthesis inhibitor cycloheximide. Hsp70 was remarkably stable (t1/2 ≫ 8 h) in the absence of CHIP (Fig. 2a). However, adding back CHIP resulted in much shorter t1/2 for Hsp70 (less than 4 h), whereas little change in t1/2 was observed by adding back CHIP(K30A). In addition to Hsp70, CHIP interacts with Hsc70 and Hsp90, and CHIP also affected the stability of these proteins (Fig. 2a). After subtraction of their different turnover rates in the absence of CHIP, the most prominent net effect of CHIP was found on Hsp70 (Fig. 2a). The stability of Grp78, an endoplasmic reticulum-localized chaperone that does not interact with CHIP, was not affected by the manipulation of CHIP levels. The differential effects of CHIP on Hsc70 and Hsp70 were further confirmed in a pulse–chase analysis followed by immunoprecipitation (Fig. 2b).
Figure 2. Selective effects of CHIP on Hsp70 in vivo and in vitro.

a, CHIP−/− cells were infected with AdV expressing GFP, CHIP or CHIP(K30A). Infected cells were incubated for 30 min at 42 °C and left to recover for 6 h at 37 °C to induce Hsp expression. Cycloheximide (CHX) was then added and cell aliquots were collected at the times indicated. Whole cell lysates were immunoblotted with a panel of antibodies. The effects of CHIP on Hsp levels were quantified by subtracting their turnover in the presence of GFP. Squares, Hsp70; diamonds, Hsp90; triangles, Hsc70; circles, Grp78. Values are means ± s.d. b, AdV-infected CHIP−/− cells were radiolabelled with [35S]methionine for 30 min at 3 h after heat shock and chased with unlabelled methionine for up to 8 h. Immunoprecipitations were performed with monoclonal antibody against Hsp70 or Hsc70. c, In vitro ubiquitination assays were performed by incubating equal amounts of Hsc70 and Hsp70 in reactions containing E1, E2 (UbcH5a), CHIP, ubiquitin (Ub) and ATP. Purified 26S proteasomes were included in the reaction mixture for the coupled degradation assay. Levels of Hsc70 (squares) and Hsp70 (diamonds) were determined by immunoblotting and quantified by densitometry. Values are means ± s.d. d, In vitro ubiquitination assays were performed with ubiquitin mutants bearing a single lysine as indicated, and the reaction was stopped after 120 min incubation. WT, wild-type.
We next examined whether the selective effects of CHIP on Hsp70 in vivo can be recapitulated in vitro with a reconstituted ubiquitination and degradation system. Equal amounts of Hsc70 and Hsp70 were incubated in a reaction containing E1, E2 (UbcH5a), CHIP, ubiquitin and ATP. Although CHIP is capable of targeting both chaperones for ubiquitination, Hsp70 was much more susceptible (Fig. 2c). The addition of purified 26S proteasomes resulted in rapid degradation of Hsp70, whereas only modest proteasome-dependent degradation was observed for Hsc70. No proteasomal degradation of Hsp70 was observed in the absence of CHIP (Fig. 2c, bottom panel). The degradative effects of CHIP on stress-inducible Hsp70 were further investigated by using ubiquitin mutants bearing a single lysine. We found that CHIP was able to initiate both canonical and non-canonical ubiquitin chains on both Hsc70 and Hsp70. However, Lys 48-linked high-molecular-weight species were selectively observed in reactions containing Hsp70 but not in those containing Hsc70 (Fig. 2d), which probably accounts for their differential susceptibility to proteasome-mediated degradation. Because Hsc70 and Hsp70 have nearly identical amino-acid sequences except for residues near the C-terminal region that are responsible for CHIP binding, it is possible that divergent residues flanking the binding sequence EEVD contribute partly to the differential activity of CHIP. Consistent with this notion is the observation that in vitro binding assays have shown different binding affinities of Hsc70 and Hsp70 for CHIP16.
When engaged by CHIP, Hsp70 functions as an adaptor for CHIP to access chaperone clients. Thus, it is unlikely that CHIP could negatively regulate Hsp70 availability while simultaneously facilitating the degradation of chaperone clients. We therefore postulated that a competitive relationship might exist in CHIP-mediated ubiquitination between adaptor and substrate. To test this hypothesis in vivo, we first examined the turnover of Hsp70 in the enforced presence of unfoldable cytoplasmic protein. Because proteotoxic insults such as heat shock have various cellular effects, we performed these experiments with cytoplasmically expressed bovine serum albumin (cBSA). This protein is incapable of acquiring its normal conformation in the reducing cytosolic environment19. Its degradation by the proteasome can be further facilitated by CHIP over-expression (Supplementary Fig. 1). We found that expression of cBSA significantly decreased the proteasome-dependent turnover of Hsp70 (as demonstrated through proteasome inhibition with MG132) in comparison with the expression of β-galactosidase (β-Gal), a stably folded protein that is not a CHIP substrate (Fig. 3a).
Figure 3. Substrate-regulated Hsp70 turnover in vivo and in vitro.
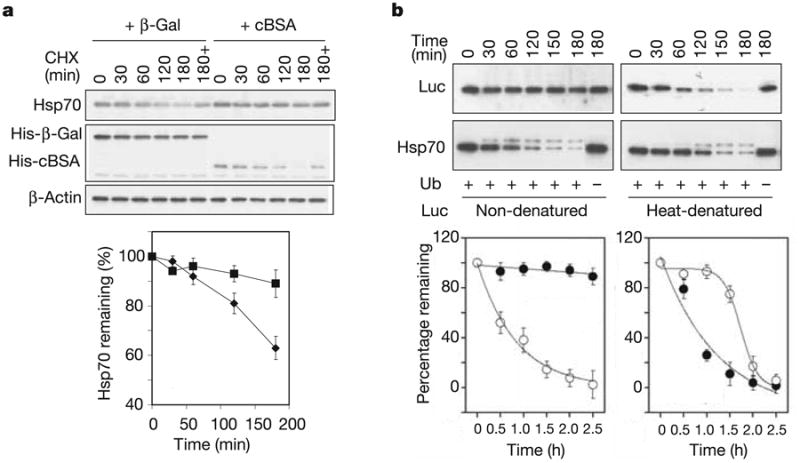
a, HEK-293 cells were co-transfected with plasmids expressing CHIP, Flag-Hsp70 and cBSA (squares) orβ-Gal (diamonds). The turnover of Hsp70 was determined with the use of a cycloheximide (CHX) chase followed by immunoblotting. ‘180 + ’ indicates the presence of 20 μM MG132 during the chase. Values are means ± s.d. b, In vitro degradation assays were performed with Hsp70 (open circles) and luciferase (Luc; filled circles) as substrates. For heat-denatured luciferase, luciferase was heated at 43 °C for 10 min with Hsp70 and Hdj2 (ref. 20). Both the luciferase and Hsp70 levels were determined in the same samples by immunoblotting and quantified by densitometry. Values are means ± s.d.
The data in Fig. 3a imply that misfolded proteins compete with Hsp70 for the ubiquitin ligase activity of CHIP. However, there is no mutual competition between Hsp70 turnover and substrate degradation because Hsp70 overexpression does not interfere with the degradation of cBSA (Supplementary Fig. 2). This indicates that CHIP might preferentially ubiquitinate chaperone-bound substrates, whereas chaperone ubiquitination occurs after substrate abundance is depleted. To test this hypothesis directly, we reconstituted CHIP-dependent degradation by means of the proteasome. Purified Hsp70, in the presence of native firefly luciferase or thermally denatured luciferase (a known CHIP substrate20), was incubated with E1, E2(UbcH5a) and CHIP in reaction buffer containing ubiquitin, ATP and 26S proteasome. Non-denatured luciferase was minimally degraded even after incubation for 3 h, whereas Hsp70 was degraded with linear kinetics in the same sample (Fig. 3b). In contrast, heat-denatured luciferase underwent rapid degradation (t1/2 ≈ 30 min). Remarkably, under these conditions the steady-state level of Hsp70 was stable until most denatured luciferase had been degraded. The biphasic kinetics of Hsp70 degradation under these conditions is markedly different from the kinetics in the absence of denatured substrate, supporting the concept of a hierarchy in the CHIP-targeted degradation of its chaperone adaptors and their associated substrates.
On the basis of these observations, we further predicted that stress-induced Hsp70 undergoes CHIP-mediated autoregulation during stress recovery in which most misfolded proteins are degraded or refolded21. To examine the physiological kinetics of stress-induced Hsp70 expression in vivo, we measured total Hsp70 levels in normal mouse fibroblasts during the acute phase of the stress response and the chronic phase of stress recovery. Hsp70 was rapidly induced to its peak level about 4 h after heat shock (42 °C, 10 min) and maintained its steady-state level for about 8 h, followed by a decrease (Fig. 4a). This biphasic pattern echoes the substrate-dependent Hsp70 turnover observed in reconstitution assays in vitro (Fig. 3b). The degradation of misfolded proteins approximates first-order kinetics (Fig. 3a), whereas the stability of Hsp70 is maintained until substrate concentrations fall below a threshold, at which time Hsp70 degradation accelerates rapidly (Figs 3b and 4a). Further supporting the model that threshold levels of misfolded substrates determine when the clearance of excess Hsp70 begins, there is a close correlation between the intensities of applied stress and the sustained period of Hsp70 expression (Supplementary Fig. 3).
Figure 4. CHIP orchestrates both stress response and recovery processes.
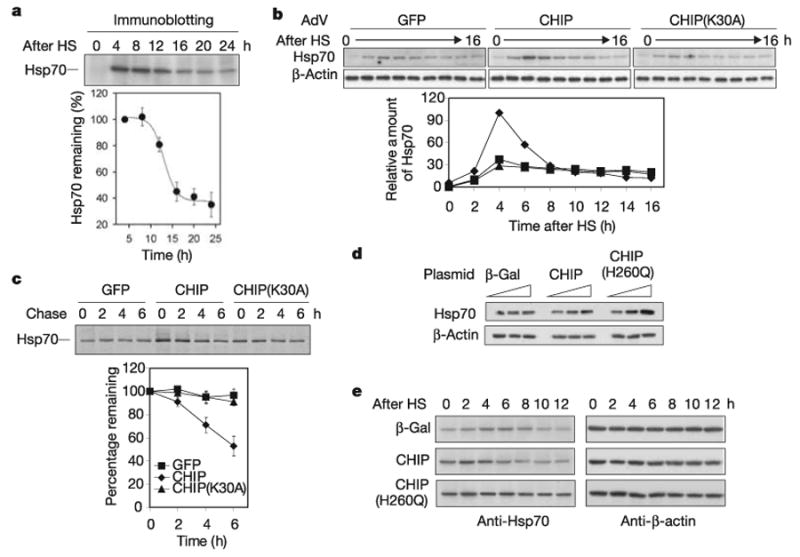
a, Mouse fibroblasts were heat shocked (HS) at 42 °C for 10 min, and aliquots were collected every 4 h during recovery at 37 °C. Hsp70 levels were determined by immunoblotting. Values are means ± s.d. b, CHIP−/− cells were infected with AdV expressing GFP (squares), CHIP (diamonds) or CHIP(K30A) (triangles). Infected cells were heat shocked at 42 °C for 10 min, and aliquots were collected every 2 h during recovery at 37 °C. Hsp70 levels were determined by immunoblotting and quantified by densitometry. c, The same AdV-infected CHIP−/− cells as in b were heat shocked at 42 °C for 10 min, and radiolabelled with [35S]methionine for 30 min at 3 h after HS and chased with unlabelled methionine for up to 6 h. Immunoprecipitation was performed with a Hsp70-specific antibody and quantified with a PhosphorImager. Symbols are as in b. Values are means ± s.d. d, HEK-293 cells were transfected with plasmids as indicated with 0, 0.1 and 0.5 μg per well in a six-well plate. Hsp70 levels were determined by immunoblotting. e, Transfected HEK-293 cells were heat shocked 42 °C for 5 min, and aliquots were collected every 2 h during recovery at 37 °C. Hsp70 levels were determined by immunoblotting.
The results presented here show a previously unknown mechanism to regulate inducible HSP expression and resolution of the stress response. Our observations indicate that the effect of CHIP on Hsp70 might not counteract its active role in the clearance of misfolded proteins and that both functions might instead be integrally related during the stress response and recovery process. To define the role of CHIP in orchestrating both phases, we examined the behaviour of Hsp70 in CHIP−/− cells during acute stress and the chronic recovery phase before and after reintroducing CHIP with the use of AdV. As expected from previous observations, adding back CHIP, but not CHIP(K30A), resulted in enhanced Hsp70 induction after heat shock, which was consistent with the role of CHIP in activating HSF1 (Fig. 4b). During the recovery phase, however, a rapid decrease in steady-state levels of Hsp70 was observed only in the presence of wild-type CHIP. In contrast, cells infected with AdV expressing GFP or CHIP(K30A) showed a prolonged plateau of Hsp70 levels. The role of CHIP in the regulation of Hsp70 availability was further confirmed by pulse–chase analysis followed by immunoprecipitation, which indicated that the t1/2 of Hsp70 was much shorter during the stress recovery phase when CHIP was reintroduced into CHIP−/−cells (Fig. 4c). Thus, the presence of CHIP actively maintains cellular homeostasis not only by enhancing Hsp70 induction during the acute phase of stress but also by removing excess Hsp70 during the recovery process.
The U-box is required for CHIP ubiquitin ligase activity but is dispensable in the stimulation of HSF1 activity15. We therefore predicted that inactivation of the CHIP U-box would lead to sustained high levels of Hsp70 in the cell after heat shock. CHIP(H260Q), a U-box mutant that does not bind E2, shows dominant-negative effects in HEK-293 cells. Indeed, we observed a much higher basal level of Hsp70 in cells transfected with CHIP(H260Q) (Fig. 4d). After a mild heat shock, the amount of Hsp70 was largely sustained in cells expressing CHIP(H260Q), whereas overexpression of CHIP accelerated both the elevation and restoration of Hsp70 levels (Fig. 4e). These results confirm that the ubiquitin ligase activity of CHIP is required for Hsp70 degradation during stress recovery in vivo.
Several principles emerge from our results. First, CHIP-mediated Hsp70 turnover represents a novel regulatory mechanism for Hsp70 in addition to HSF1-mediated transcriptional regulation. Second, these data indicate that there are multiple coordinated steps by which the chaperone and ubiquitin–proteasome systems interact to regulate the stress response. Without appropriate CHIP function, there is impaired ability to induce Hsp70 during stress and concomitantly a deficient buffering of misfolded proteins. At the same time, CHIP is required for proteasome-dependent clearance of unfoldable proteins and the recovery from the stress response by means of Hsp70 degradation. Finally, CHIP-mediated sequential catalysis of the chaperone adaptor and substrate indicates an elegant and simple mechanism to regulate the target specificity for a single ubiquitin ligase. In the multisubunit ubiquitin ligases such as Skp1–Cdc53/ Cul1–F-box (SCF), the F-box adaptor targets a distinct array of substrates for ubiquitination and proteasomal degradation22. Intriguingly, most F-box adaptors in the SCF complex are labile proteins as a result of autocatalysis by the intrinsic E3 activity23–25. In the case of CHIP, Hsp70 acts as an adaptor, or ‘F-box equivalent’, for CHIP to ubiquitinate chaperone clients; degradation of Hsp70 may therefore be a substrate-dependent variation on this autocatalytic mechanism.
Methods
Cell lines, plasmids and viruses
HEK-293 cells were maintained in DMEM medium containing 10% FBS. CHIP+/+ and CHIP−/− cell lines were established from fibroblasts isolated from adult lungs of CHIP+/+ and CHIP−/− mice by transfection with SV40 large Tantigen (kindly provided by D. Ron). The HSF1−/− murine embryonic fibroblast cell line was provided by I. Benjamin. Mammalian expression vectors for β-Gal, cBSA, Hsp70, CHIP and CHIP(K30A) are based on pcDNA3.1. Recombinant adenoviruses expressing CHIP, CHIP(K30A) or GFP have been described previously15.
Antibodies and short interfering RNAs
Monoclonal anti-Hsp70 (SPA810), anti-Hsp90α (SPA845) and anti-Hsc70 (SPA815) were purchased from Stressgen. Monoclonal anti-Grp78 antibody was from Santa Cruz. Monoclonal anti-His antibody was from Qiagen. Monoclonal anti-β-actin was from Sigma. Polyclonal anti-luciferase was from Promega. Double-stranded RNA for CHIP (target sequence 5′-GGAGCAGGGCAAUCGUCUG-3′) and GFP (target sequence 5′-AACGAGAAGCGCGAUCACAUG-3′) were synthesized by Dharmacon.
Dual luciferase assay
A firefly luciferase gene under the control of the strictly stress-regulated human Hsp70B promoter was used to assess HSF1 transcriptional activity. To control for the differences in transfection efficiency and other effects, a Renilla luciferase gene driven by a cytomegalovirus promoter was included in all transfections26. Reporter activity was determined with a dual-luciferase reporter assay (Promega).
In vitro reconstitution assay
His-tagged CHIP, and UbcH5a, were produced in Escherichia coli BL21(DE3) followed by purification with Ni2+-nitrilotriacetate agarose. In vitro ubiquitination assays were performed in the presence of 0.1 μM purified rabbit E1 (BioMol), 2 μM UbcH5a, 3 μM CHIP, 50 μM ubiquitin (Sigma), 1 mM dithiothreitol, 2 mM MgCl2 and 4 mM ATP. For in vitro degradation assays, 50nM purified 26S proteasomes (BioMol) was included in the reaction mixture. Reactions were performed at 37 °C, and samples were analysed by SDS–PAGE and immunoblotting with appropriate antibodies. Heat treatment of firefly luciferase was described elsewhere21. In brief, 0.5 μM luciferase was heated at 43 °C for 10 min with 1 μM Hsp70 and 1 μM Hdj2 on the presence of 4mM ATP in 50mM Tris-HCl pH7.5 containing 2mM MgCl2. After being heated, samples were quickly chilled in an ice bath. Aliquots were incubated in the reaction mixture as indicated above.
Pulse–chase analysis
Cells were radiolabelled with [35S]Met (100 μCi) for the indicated durations. After washing with PBS containing excess unlabelled Met (1 mg ml−1), cells were chased in the complete DMEM for the indicated duration. Whole cell lysates were made in Tris-buffered saline pH7.5 containing 1% Triton X-100 and 10 Uml−1 DNase A (Roche). Lysates were either directly resolved by SDS–PAGE or used for immunoprecipitation with Protein G beads coated in monoclonal anti-Hsp70 (SPA810).
Supplementary Material
Acknowledgments
We thank D. Thiele for critical reading of the manuscript. This work was supported by grants from the National Institutes of Health, the American Heart Association and the Burroughs Wellcome Fund.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information: Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
The authors declare no competing financial interests.
References
- 1.Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 2.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 3.Feder JH, Rossi JM, Solomon J, Solomon N, Lindquist S. The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 1992;6:1402–1413. doi: 10.1101/gad.6.8.1402. [DOI] [PubMed] [Google Scholar]
- 4.Volloch VZ, Sherman MY. Oncogenic potential of Hsp72. Oncogene. 1999;18:3648–3651. doi: 10.1038/sj.onc.1202525. [DOI] [PubMed] [Google Scholar]
- 5.Wu C. Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol. 1995;11:441–469. doi: 10.1146/annurev.cb.11.110195.002301. [DOI] [PubMed] [Google Scholar]
- 6.Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- 7.Connell P, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nature Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 8.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nature Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 9.Demand J, Alberti S, Patterson C, Hohfeld J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr Biol. 2001;11:1569–1577. doi: 10.1016/s0960-9822(01)00487-0. [DOI] [PubMed] [Google Scholar]
- 10.Xu W, et al. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA. 2002;99:12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 14.Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai Q, et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballinger CA, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, et al. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y, et al. Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem. 2001;276:45791–45799. doi: 10.1074/jbc.M105931200. [DOI] [PubMed] [Google Scholar]
- 20.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weibezahn J, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 22.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nature Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 23.Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci USA. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou P, Howley PM. Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol Cell. 1998;2:571–580. doi: 10.1016/s1097-2765(00)80156-2. [DOI] [PubMed] [Google Scholar]
- 25.Wee S, Geyer RK, Toda T, Wolf DA. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nature Cell Biol. 2005;7:387–391. doi: 10.1038/ncb1241. [DOI] [PubMed] [Google Scholar]
- 26.Boellmann F, et al. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc Natl Acad Sci USA. 2004;101:4100–4105. doi: 10.1073/pnas.0304768101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.