

Summary
Background and Purpose
Ischemic stroke (IS) and coronary artery disease (CAD) share several risk factors and each have a substantial heritability. We conducted a genome-wide analysis to evaluate the extent of shared genetic determination of the two diseases.
Methods
Genome-wide association data were obtained from the METASTROKE, CARDIoGRAM, and C4D consortia. We first analyzed common variants reaching a nominal threshold of significance (p<0.01) for CAD for their association with IS and vice versa. We then examined specific overlap across phenotypes for variants that reached a high threshold of significance. Finally, we conducted a joint meta-analysis on the combined phenotype of IS or CAD. Corresponding analyses were performed restricted to the 2,167 individuals with the ischemic large artery stroke (LAS) subtype.
Results
Common variants associated with CAD at p<0.01 were associated with a significant excess risk for IS and for LAS and vice versa. Among the 42 known genome-wide significant loci for CAD, three and five loci were significantly associated with IS and LAS, respectively. In the joint meta-analyses, 15 loci passed genome-wide significance (p<5×10-8) for the combined phenotype of IS or CAD and 17 loci passed genome-wide significance for LAS or CAD. Since these loci had prior evidence for genome-wide significance for CAD we specifically analyzed the respective signals for IS and LAS and found evidence for association at chr12q24/SH2B3 (pIS=1.62×10-07) and ABO (pIS =2.6×10-4) as well as at HDAC9 (pLAS=2.32×10-12), 9p21 (pLAS =3.70×10-6), RAI1-PEMT-RASD1 (pLAS =2.69×10-5), EDNRA (pLAS =7.29×10-4), and CYP17A1-CNNM2-NT5C2 (pLAS =4.9×10-4).
Conclusions
Our results demonstrate substantial overlap in the genetic risk of ischemic stroke and particularly the large artery stroke subtype with coronary artery disease.
Introduction
Stroke and coronary artery disease (CAD) are among the most common causes of premature death and loss of disability-adjusted life years worldwide.1, 2 Both conditions are risk factors for one another 3, 4 and in combination they are used for assessment of risk or as a therapeutic target in clinical trials. Stroke and CAD share several risk factors and many aspects of their underlying pathophysiology. This shared biology applies to ischemic stroke (IS) and particularly to the sub-type of atherosclerotic stroke (large artery stroke, LAS).4, 5 Twin and family studies have demonstrated that both IS and CAD are highly heritable6, 7 with some evidence of a shared heritability for both diseases.8
Recent genome-wide association studies (GWAS) have identified some common genetic variants that are associated with IS 9-11 and multiple loci that are associated with CAD.12, 13 Interestingly, some of the variants that were originally found to affect CAD risk also associate with LAS 14, 15 suggesting a shared genetic architecture. However, there has been no systematic study assessing shared genetic susceptibility to both IS and CAD or to LAS and CAD on a genome-wide level in large datasets.
Combining genome-wide data from the METASTROKE, CARDIoGRAM, and C4D consortia we examined whether IS and its subtype LAS share genetic risk with CAD with respect to common genetic variation. We further explored the most robustly associated variants for CAD for their association with both IS and LAS and vice versa. Finally, we conducted a joint meta-analysis of IS and CAD to search for variants that are associated with the combined and thus broader vascular phenotype.
Methods
Participating studies and study design
The study sample consisted of GWAS case-control samples from the METASTROKE 9, CARDIoGRAM 12, and C4D 16 consortia (Supplementary Table I). All participating studies used a case-control or nested case-control design. Most participating studies were cross-sectional, whereas some were prospective, population-based studies.
The METASTROKE consortium included 15 GWA studies involving 12,389 IS cases and 62,004 controls. Among them were 2,167 LAS cases and 49,159 LAS controls, and 2,365 cardioembolic stroke (CES) cases and 56,140 CES controls. Genotyping in individual cohorts was carried out using Affymetrix or Illumina platforms, and approximately 2.5 million imputed genotypes were generated. Individual METASTROKE results of the association analyses from every center were analyzed using a fixed-effects inverse-variance weighted model with METAL.9, 17 All data were quality-controlled as previously described.9
The CARDIoGRAM consortium included 14 GWA studies involving 22,233 CAD cases and 64,762 controls. The genotyping platforms used and imputation approach was similar to METASTROKE. The C4D consortium included 3 studies involving a total of 11,165 CAD cases and 10,964 controls. Genotyping was carried out using Illumina arrays containing a common set of about 575,000 genotyped SNPs.16 The meta-analysis of all CAD studies was carried out using a fixed-effects or random-effects model depending on the extent of heterogeneity as described previously.18 All data were quality controlled as previously described.16, 18
Phenotype definitions of stroke and CAD are described in the original reports.9, 12, 16 In brief, stroke was defined as a typical clinical syndrome with radiological confirmation. Stroke subtyping was done using the Trial of Org 10172 in Acute Stroke Treatment (TOAST) classification system. Definitions for CAD slightly varied between cohorts but usually included myocardial infarction, symptoms of angina pectoris, and/or >50% coronary artery stenosis. Participating studies were approved by relevant institutional review boards, and all participants provided written or oral consent for genetic research using protocols approved by the relevant institutional body.
Statistical analysis
For the analysis of variants showing a nominal threshold of significance (p<0.01) for IS, LAS, or CAD, data were taken from the METASTROKE (for IS and LAS) and in the combined CARDIoGRAM and C4D (for CAD) sample. Variants with a p-value<0.01 for a given phenotype were then tested for association with the alternate phenotype(s) to determine whether the observed distribution of p-values significantly deviated from the expected distribution. To ensure that only independent loci are incorporated in the analysis, we performed LD-based pruning with an r2- cut off of 0.3 retaining the SNP with the lowest p-value in the original study for each locus. QQ plots were drawn to plot –log (p-values) where SNPs with effects in opposite directions were plotted separately from SNPs with effects in the same direction. To determine the deviation of the p-value distribution shown in the QQ plots, p-values were z-transformed. Under the null hypothesis, the z-transformed effects follow a standard normal distribution. One-tailed significance was determined by comparing the absolute values of the z-scores to a random normal one-tailed distribution using a standard t-test 2×2 contingency tables were constructed for different p-values and r2-cutoffs and Fisher’s exact test was used to evaluate the significance of the contingency tables.
Directionality of effects (OR associated with the minor allele) of top variants for the three phenotypes (IS, LAS, CAD) in other phenotypes (CAD for IS and LAS variants; IS and LAS for CAD variants) were examined by calculating the proportion of effects going in the same direction and comparing this proportion to that expected by chance (50%). For this, an exact binomial test was performed. The analysis was repeated on the LD-pruned data to ensure independence of tested SNPs. Bonferroni correction was applied to determine study-wide significance.
To rule out that the agreement in p-values at individual risk loci is limited to single variants we calculated the correlation of p-values using Spearman’s rank correlation for defined genomic regions (consistent drop of p-values <0.05) for each potentially shared risk locus. This allows to quantify the agreement between the p-value distributions of the different phenotypes using Spearman’s rho as a read out, where rho=1 is defined as a perfect positive correlation and rho=-1 as a perfect inverse correlation.
Meta-Analysis methods
We performed meta-analyses of the combined data from CARDIoGRAM (for CAD) and METASTROKE (for IS and LAS) using two methods. First, we performed subtype-specific meta-analyses using the protocol published by Mägi and colleagues.19, 20 This method was originally developed for sex-specific genome-wide association studies but can also be applied to other dichotomous covariates. The algorithm is implemented in the GWAMA software 19, 20 and accounts for possible heterogeneity between study subgroups by formally allowing for interaction between genotypes and subgroups under an additive model. Here a subgroup-differentiated p-value below individual p-values for individual subgroups is indicative of an association with both sub-phenotypes. To evaluate whether the resulting meta-analysis p-values are significant after correcting for multiple testing, we evaluated the false discovery rate (FDR) of these p-values. The R package “fdrtool” was used to estimate q-values, a direct measure of the proportion of false positive results in the presence of a statistically significant result.
Second, we used the method of Zaykin and Kozbur 21, which is similar to the method by Lin and Sullivan22 to account for overlap of an estimated ~38,000 controls between the CARDIoGRAM and METASTROKE samples from the KORA, WTCCC2, CHARGE and deCODE studies (the exact number of overlapping controls could not be determined in the absence of individualized data) which may lead to the inflation of meta-analysis p-values. This program compensates for this lack of independence in test statistics created by the use of the same controls by computing the correlation between studies and using this measure for correction of p-values obtained from a standard meta-analysis. In the absence of exact numbers for overlapping controls we simulated different scenarios of overlapping controls.
Results
Analysis of variants meeting a low threshold of significance of association with CAD, IS, and LAS
We first tested whether single nucleotide polymorphisms (SNPs) with some evidence for association with CAD also associate with IS, and vice versa. Specifically, we constructed a QQ plot in the IS GWAS meta-analysis using variants that displayed a p-value of <0.01 for CAD in CARDIoGRAM/C4D.12, 16 We next constructed a QQ plot in the CAD GWAS meta-analysis using variants that displayed a p-value of <0.01 for IS in METASTROKE.9 For both analyses, deviation of the observed from the expected distribution was highly significant with p <10-82 (Figure 1A).
Figure 1.
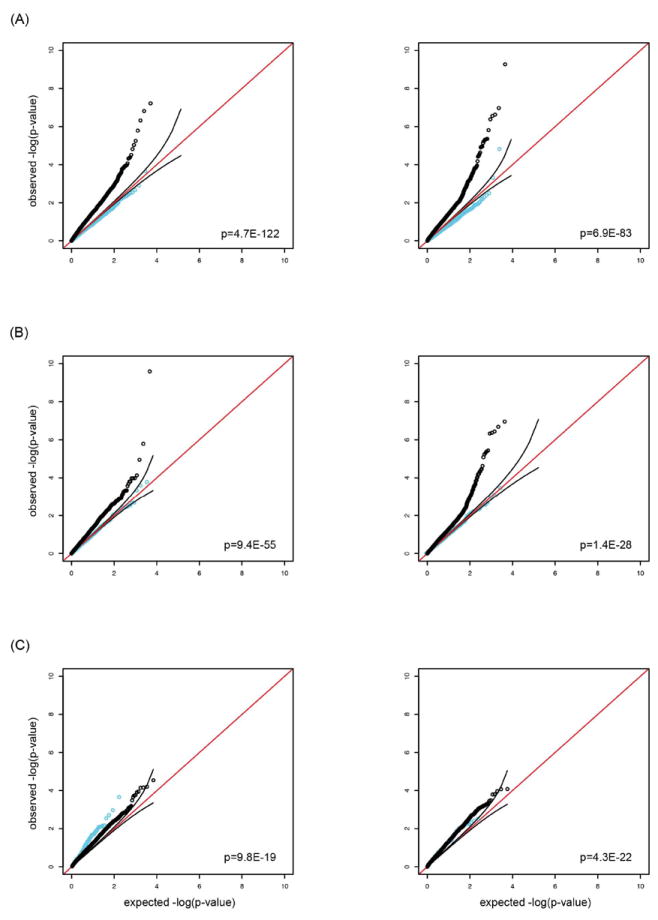
QQ plots for individual vascular phenotypes considering variants reaching a low threshold of significance (p<0.01) in alternate vascular phenotypes: CAD variants in all IS (left) and all IS variants in CAD (right) (A), CAD variants in LAS (left) and LAS variants in CAD (right) (B); CAD variants in CES (left) and CES variants in CAD (right) (C). SNPs with effects going into the same direction in the respective samples are shown in black. SNPs with effects going into opposite directions in the respective samples are shown in light blue. Data were drawn from METASTROKE, CARDIoGRAM and C4D. Red line: expected line corresponding to a normal distribution; black lines represent 95% confidence intervals of the expected distribution. For display purposes variants from the 9p21 locus are omitted from the figure. p-values correspond to the analysis of directionally consistent SNPs (black line).
Next, we generated corresponding QQ plots for LAS and CAD. Again, deviation of the observed from the expected distribution was highly significant in both analyses (p <10-27)(Figure 1B). Corresponding QQ plots for cardioembolic stroke (CES) and CAD also showed some deviation of the observed from the expected distribution (Figure 1C). However, the deviation was less pronounced than for LAS and CAD. Focusing on variants with a p-value of <0.0001 revealed a significant excess of shared signals between IS and CAD (3.75×10-8) and between LAS and CAD (p=3.4 × 10-3) but not between CES and CAD (p=1.0) (Supplementary Figure I).
Cross-analysis of robustly associated variants for CAD and IS
We next analyzed directionality of effects (OR associated with the minor allele) for all variants that have previously shown genome-wide significance for association with CAD in CARDIoGRAMplusC4D 13 in the METASTROKE GWAS for IS and LAS. Among 46 CAD variants from 42 loci, 33 variants [72%] from 31 loci showed point estimates for IS that were directionally consistent for CAD (p=0.0045; exact binomial test, two-sided; Supplementary Table II). Three variants from three loci were significantly associated with IS (Table 1) at the 95% confidence level following Bonferroni correction (p<0.00108 for testing of 46 variants). The effects for IS were in the same direction as for CAD for all three of the variants. Corresponding results for LAS were 34 variants from 31 loci (74%; p=0.0016) and 5 variants from 5 loci with study-wide significance (Table 1 and Supplementary Table II). When considering LD-pruned SNPs (r2<0.3) the results were very similar with only two SNPs (rs11203042 and rs3217992) at two loci being removed from analysis. Among 44 CAD variants from 42 loci, 31 variants from 31 loci [70%] showed point estimates for IS that were directionally consistent (p=0.0096). Corresponding results for LAS were 32 variants from 31 loci (73%; p=0.0037).
Table 1. Association signals and directional consistency of effects of top variants for coronary artery disease, ischemic stroke, and large artery stroke.
Shown are variants that were significantly associated with both CAD and IS, or both CAD and LAS, or all three phenotypes (study-wide level of significance: p<0.00108 for CAD, p<0.008 for IS and p<0.0045 for LAS). Results are shown for the CARDIoGRAM 12* and METASTROKE9samples.
| Coronary Artery Disease
|
Ischemic Stroke | Large Artery Stroke
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lead SNPs | Band | Gene in region | Minor Allele |
p-value | Odds ratio (95%CI) |
Effects go in same direction1 |
p-value | Odds ratio (95%CI) |
Effects go in same direction2 |
p-value | Odds ratio (95%CI) |
Effects go in same direction2 |
| Top variants (known loci) for CAD/MI in the CARDIoGRAMplusC4D sample 13 | ||||||||||||
| rs12413409 | 10q24.32 | CYP17A1/CNNM2/NT5C2 | A | 1.24E-06 | 0.89 [0.84-0.93] | - | 0.0603 | 0.95 [0.89-1.00] | yes | 0.00049 | 0.80 [0.70-0.90] | yes |
| rs12936587 | 17p11.2 | RAI1-PEMT-RASD1 | A | 1.98E-07 | 0.93 [0.90-0.96] | - | 0.0051 | 0.95 [0.92-0.98] | yes | 2.69E-05 | 0.86 [0.80-0.92] | yes |
| rs3184504** | 12q24.12 | chr12q24/SH2B3 | T | 9.33E-07 | 1.07 [1.04-1.11] | - | 1.01E-06 | 1.08 [1.05-1.12] | yes | 0.00015 | 1.14 [1.06-1.22] | yes |
| rs2023938 | 7p21.1 | HDAC9 | C | 2.10E-03 | 1.08 [1.03-1.13] | - | 1.65E-06 | 1.14 [1.08-1.20] | yes | 2.33E-09 | 1.38 [1.24-1.53] | yes |
| rs579459 | 9q34.2 | ABO | C | 2.14E-07 | 1.10 [1.06-1.14] | - | 0.00026 | 1.08 [1.04-1.12] | yes | 0.0054 | 1.13 [1.04-1.22] | yes |
| rs1333049 | 9p21.3 | CDKN2BAS | C | 2.96E-56 | 1.24 [1.21-1.28] | - | 0.0053 | 1.05 [1.01-1.09] | yes | 3.70E-06 | 1.19 [1.11-1.28] | yes |
|
| ||||||||||||
| Top variants (p<10-5) for all ischemic stroke in the METASTROKE sample | ||||||||||||
| rs17696736* | 12q24.13 | chr12q24/SH2B3 | G | 6.56E-08 | 1.07 [1.04-1.10] | yes | 5.96E-08 | 1.10 [1.06-1.14] | - | 0.0024 | 1.11 [1.04-1.20] | yes |
|
| ||||||||||||
| Top variants (p<10-5) for large artery stroke in the METASTROKE sample | ||||||||||||
| rs1333047 | 9p21.3 | CDKN2BAS | T | 1.44E-53 | 1.24 [1.20-1.27] | yes | 0.0063 | 1.05 [1.01-1.08] | yes | 1.64E-06 | 1·20 [1·11-1·29] | - |
note that the CARDIoGRAM sample represents a subsample of the CARDIoGRAMplusC4D sample.
rs3184504 and rs17696736 are in high linkage disequilibrium (r2=0.72, D’=0.91)
compared to IS or LAS respectively
compared to CAD/MI; associations reaching study-wide significance (p<0.00108, p<0.008 and p<0.0045, respectively) are shown in bold;
We further analyzed all variants that showed p-values < 10-5 with IS in METASTROKE (N=6 variants) for directionality of effects in the CARDIoGRAM meta-analyses for CAD. The choice of a more liberal p-value (p<10-5) was based on the paucity of variants reaching genome-wide significance for IS. In all cases point estimates for CAD were directionally consistent for IS (p=0.0313; for directionality; exact binomial test, two-sided; Supplementary Table II). One variant was significantly associated with CAD at the 95% confidence level following Bonferroni correction for testing of multiple variants (Table 1). Finally, we analyzed variants that showed p-values < 10-5 with the LAS subtype in METASTROKE (N=11 variants) for directionality in the CAD dataset. Again, the majority (82%) showed effects going in the same direction (p=0.065; Supplementary Table II). One variant was significantly associated with CAD (Table 1). Considering LD-pruned SNPs did not change the results. None of the three loci that showed p-values <1x10-5 with cardioembolic stroke were associated with CAD (all pCAD>0.2).
Meta-Analyses of combined data from CARDIoGRAM and METASTROKE
As a further step we carried out meta-analyses for the combined data from CARDIoGRAM (for CAD) and from METASTROKE (for IS and LAS) to identify variants that are associated with the broader vascular endpoint. This meta-analysis revealed 15 loci that exceeded the threshold for genome-wide significance for the combined CAD/IS phenotype (Table 2 and Figure 2A) and 17 loci that exceeded the threshold for genome-wide significance for the combined CAD/LAS phenotype (Table 2 and Figure 2B). All of these loci have been published previously for genome-wide significant association with CAD.13 Of note however, in the combined datasets several loci showed p-values that were more than one order of magnitude lower than those in individual meta-analyses on the individual diseases. This applied to 3 loci of the CAD/IS meta-analysis and 5 loci of the CAD/LAS meta-analysis (Table 2). All loci were still significant following FDR correction (Table 2).
Table 2. Association signals for risk loci significantly associated with the combined coronary artery disease/ stroke phenotypes in meta-analyses.
shown are loci with p<5e-8. SNPs showing the lowest meta-p-values in the respective region are reported. Data were drawn from METASTROKE and CARDIoGRAM. Results are shown for both IS and LAS
| rs_number | Band | Gene in region | CAD p-value | IS p-value | Effects go in the same direction | CAD & IS combined p-value | FDR q-value |
|---|---|---|---|---|---|---|---|
| Ischemic Stroke | |||||||
|
| |||||||
| rs1333049 | 9p21.3 | CDKN2BAS | 2.96E-56 | 0.005 | yes | 1.09E-56 | 5.00E-51 |
| rs11065987 | 12q24.12 | chr12q24/SH2B3 | 5.13E-09 | 1.62E-07 | yes | 4.05E-14 | 9.60E-10 |
| rs10455872 | 6q25.3 | SLC22A3/LPAL2/LPA | 3.15E-13 | 0.322 | yes | 1.72E-12 | 3.75E-08 |
| rs1122608 | 19p13.2 | LDLR/SMARCA4 | 3.32E-11 | 0.002 | yes | 2.59E-12 | 5.52E-08 |
| rs4714955 | 6p24.1 | PHACTR1 | 6.30E-12 | 0.498 | no | 4.24E-11 | 8.30E-07 |
| rs11556924 | 7q32.2 | ZC3HC1 | 2.55E-10 | 0.217 | yes | 9.37E-10 | 1.59E-05 |
| rs964184 | 11q23.3 | ZNF259 | 1.50E-10 | 0.871 | yes | 1.17E-09 | 1.95E-05 |
| rs579459 | 9q34.2 | ABO | 2.14E-07 | 0.0003 | yes | 1.81E-09 | 2.95E-05 |
| rs2219939 | 15q25.1 | ADAMTS7 | 2.65E-09 | 0.042 | no | 2.49E-09 | 3.97E-05 |
| rs7582720 | 2q33.1 | WDR12 | 3.76E-09 | 0.052 | no | 4.18E-09 | 6.30E-05 |
| rs599839 | 1p13.3 | SORT1 | 1.41E-09 | 0.938 | yes | 1.07E-08 | 0.00014 |
| rs12190287 | 6q23.2 | TCF21 | 2.32E-09 | 0.823 | no | 1.69E-08 | 0.00022 |
| rs12449964 | 17p11.2 | RAI1-PEMT-RASD1 | 1.64E-07 | 0.005 | yes | 2.23E-08 | 0.00028 |
| rs17114036 | 1p32.2 | PPAP2B | 9.78E-09 | 0.162 | yes | 2.66E-08 | 0.00032 |
| rs9351814 | 6q13 | C6orf155 | 1.45E-07 | 0.015 | yes | 4.93E-08 | 0.00055 |
|
| |||||||
| Large Artery Stroke | |||||||
|
| |||||||
| rs1333049 | 9p21.3 | CDKN2BAS | 2.96E-56 | 3.85E-06 | yes | 1.20E-59 | 5.92E-54 |
| rs10455872 | 6q25.3 | SLC22A3/LPAL2/LPA | 3.15E-13 | 0.009 | yes | 9.25E-14 | 2.18E-09 |
| rs2107595 | 7p21.1 | HDAC9 | 0.042 | 2.39E-12 | yes | 2.60E-12 | 5.72E-08 |
| rs1122608 | 19p13.2 | LDLR/SMARCA4 | 3.32E-11 | 0.017 | yes | 1.56E-11 | 3.31E-07 |
| rs4714955 | 6p24.1 | PHACTR1 | 6.30E-12 | 0.173 | no | 2.11E-11 | 4.42E-07 |
| rs12936587 | 17p11.2 | RAI1-PEMT-RASD1 | 1.98E-07 | 2.69E-05 | yes | 1.93E-10 | 3.56E-06 |
| rs11065987 | 12q24.12 | chr12q24/SH2B3 | 5.13E-09 | 0.002 | yes | 3.20E-10 | 5.77E-06 |
| rs11556924 | 7q32.2 | ZC3HC1 | 2.55E-10 | 0.167 | yes | 7.74E-10 | 1.33E-05 |
| rs599839 | 1p13.3 | SORT1 | 1.41E-09 | 0.023 | yes | 8.17E-10 | 1.40E-05 |
| rs964184 | 11q23.3 | ZNF259 | 1.50E-10 | 0.468 | no | 9.14E-10 | 1.55E-05 |
| rs12190287 | 6q23.2 | TCF21 | 2.32E-09 | 0.814 | yes | 1.69E-08 | 0.00023 |
| rs12413409 | 10q24.32 | CYP17A1-CNNM2-NT5C2 | 1.24E-06 | 0.0005 | yes | 1.77E-08 | 0.00023 |
| rs6841581 | 4q32.21 | EDNRA | 8.45E-07 | 0.0007 | yes | 1.78E-08 | 0.00024 |
| rs17114036 | 1p32.2 | PPAP2B | 9.78E-09 | 0.133 | yes | 2.29E-08 | 0.00029 |
| rs899997 | 15q25.1 | ADAMTS7 | 4.75E-09 | 0.391 | no | 2.41E-08 | 0.00030 |
| rs7582720 | 2q33.1 | WDR12 | 3.76E-09 | 0.682 | no | 2.55E-08 | 0.00032 |
| rs579459 | 9q34.2 | ABO | 2.14E-07 | 0.005 | yes | 2.96E-08 | 0.00036 |
In bold: p-value for the combined phenotype is more than one order of magnitude lower than in individual meta-analyses on the individual phenotypes. Note that variants at individual loci may differ from those reported in Table 1
Figure 2.
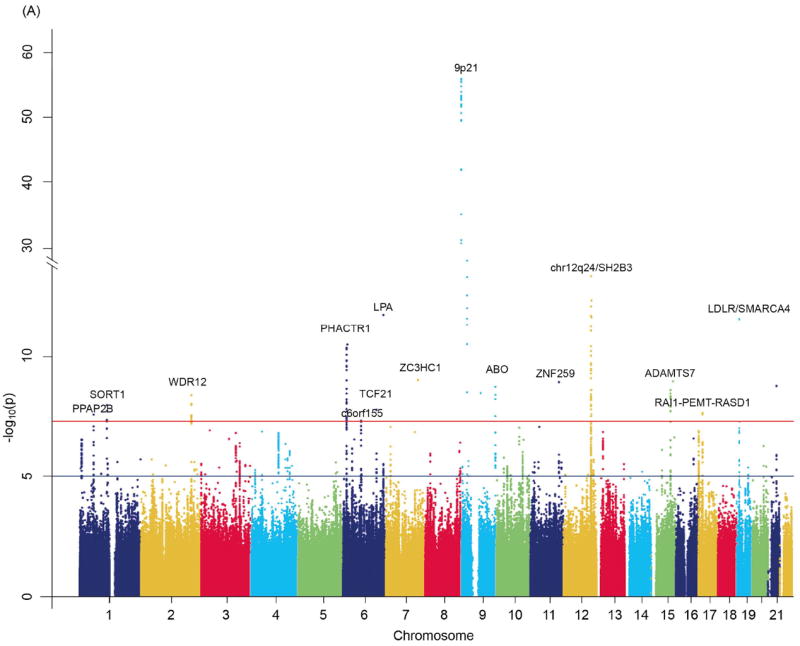
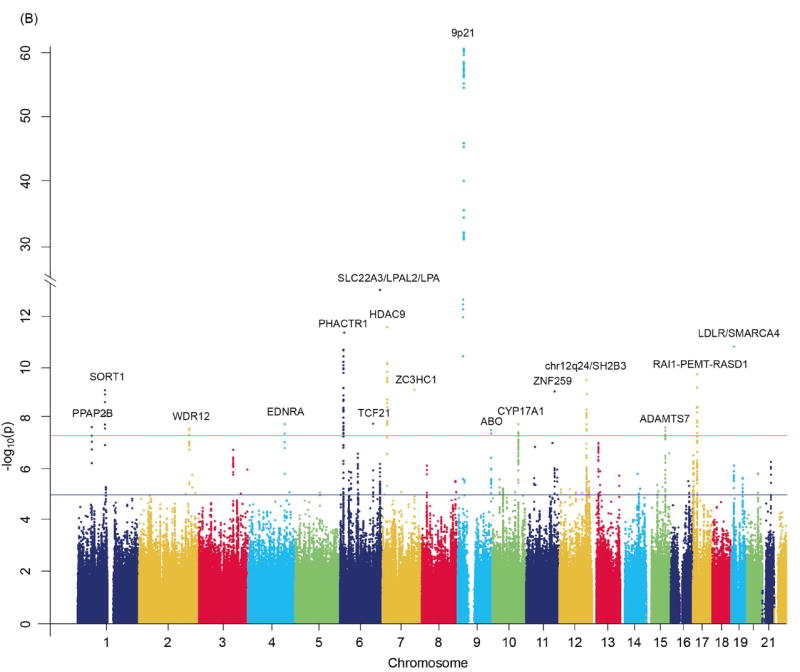
Manhattan plots of –log10(p) against genomic position: Results are shown for (A) the combined endpoint of all IS or CAD and (B) the combined endpoint of LAS or CAD. Genome-wide meta-analysis association results by genomic position at autosomal SNPs. Data were drawn from METASTROKE and CARDIoGRAM.
We next focused on loci that showed the strongest independent association with IS or LAS in addition to their genome-wide significance in the combined data set. For IS these were chr12q24/SH2B3 (pIS =1.62×10-7) and ABO (pIS=2.65×10-4)(Table 2). For LAS, these were HDAC9 (pLAS=2.39×10-12), 9p21 (pLAS=3.85×10-6), RAI1-PEMT-RASD1 (pLAS=2.69×10-5), CYP17A1-CNNM2-NT5C2 (pLAS=4.92×10-4), and EDNRA (pLAS=7.29×10-4)(Table 2). In all cases p-values for individual variants within the respective genetic regions (defined as a consistent drop of p-values <0.05) significantly correlated between CAD and stroke phenotypes suggesting that the association signals originate from the same genetic variants (chr12q24/SH2B3: Spearman’s rhoIS/CAD=0.68; p=3.8×10-87; ABO: rhoIS/CAD =0.82, p=2.2×10-08; HDAC9: rhoLAS/CAD =0.83, p=4.8×10-12; 9p21: rhoLAS/CAD =0.85; p=2.9E-35; RAI1-PEMT-RASD1: rhoLAS/CAD =0.78; p=5.6×10-17; CYP17A1/CNNM2/NT5C2: rhoLAS/CAD =0.46, p=7.5×10-15; EDNRA: rhoLAS/CAD =0.85; p=6.5×10-13)(Supplementary Figure II).
A closer look at loci that were significant in the combined meta-analyses revealed that some loci showed a strong association with both phenotypes reaching a similar level of significance for the IS and CAD phenotypes (Figure 3 and Supplementary Figure II), whereas for other loci the association was largely confined to a single phenotype (Figure 4 and Supplementary Figure II).
Figure 3.
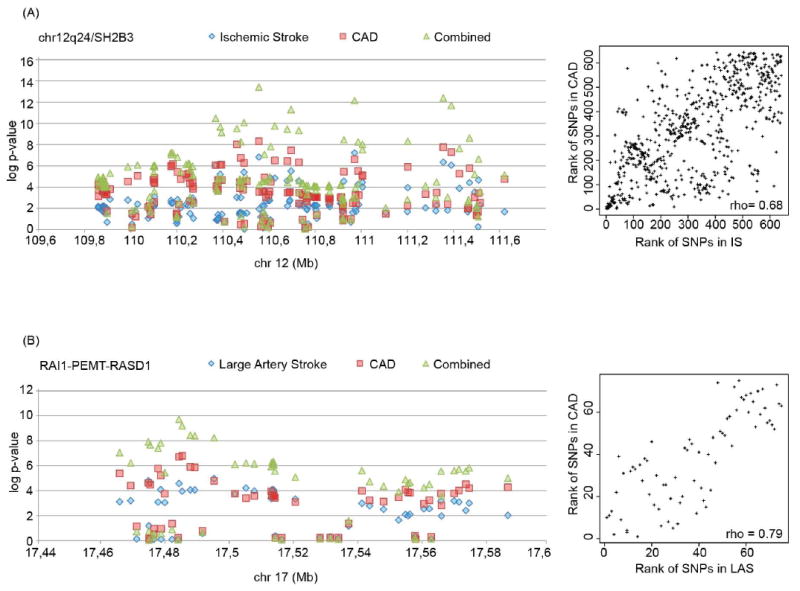
Regional association plots (left) and corresponding Spearman correlation plots (right) of p-values for individual variants of (A) the chr12q24/SH2B3 locus for IS and CAD and (B) the RAI1-PEMT-RASD1 locus for LAS and CAD. For clarity, only a subset of variants is displayed (see Supplementary Figure II for all variants). Data were drawn from METASTROKE and CARDIoGRAM.
Figure 4.
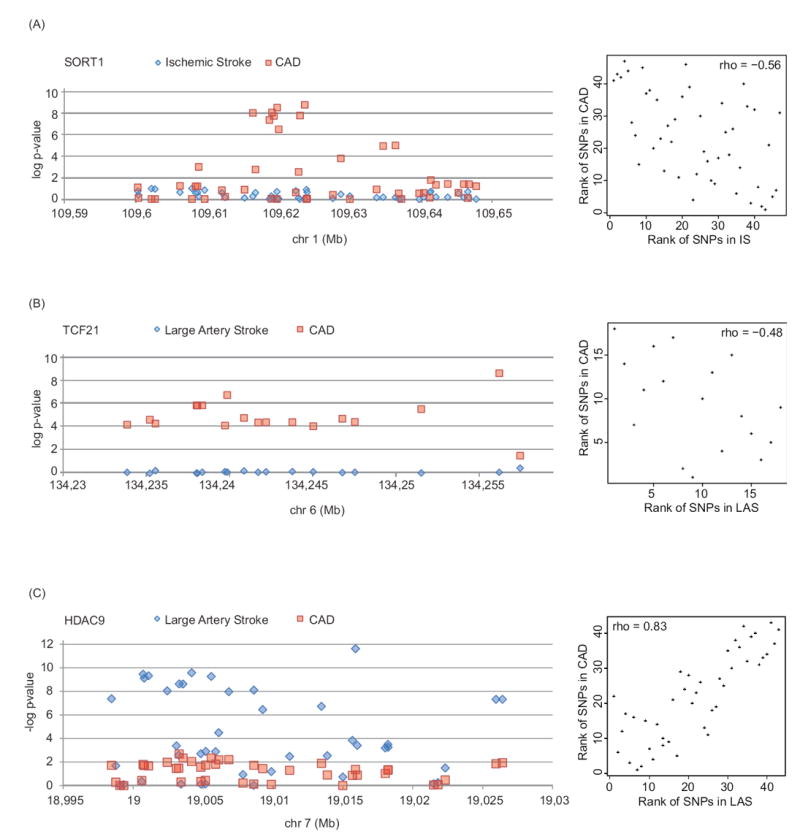
Regional association plots (left) and corresponding Spearman correlation plots (right) of (A) the SORT1 locus for IS and CAD, (B) the TCF21 locus for LAS and CAD, and (C) the HDAC9 locus for LAS and CAD. For clarity, only a subset of variants is displayed (see Supplementary Figure II for all variants). Data were drawn from METASTROKE and CARDIoGRAM.
To account for the overlap in controls between the stroke and CAD samples, we further performed conventional sample-size dependent meta-analyses 21 for two different scenarios covering the estimated number of controls that overlapped between the two samples (Supplementary Table III). The results compared well with the primary subtype-specific meta-analysis except for HDAC9 (for all IS/CAD) and SORT1 (for LAS/CAD), which reached genome-wide significance in the respective subtype-specific meta-analyses but not in the conventional sample-size dependent meta-analyses.
Discussion
This study demonstrates that common variants at a substantial number of genetic loci influence risk of both ischemic stroke and coronary artery disease. This conclusion is supported by the results of several approaches: First, selecting common variants that had reached a nominal threshold (p<0.01) of significance in previous studies and testing them for association with the respective other vascular phenotype; second, analyzing common variants that had reached a high threshold of significance in previous studies; and third, meta-analysis of the combined vascular endpoint of CAD and IS as well as CAD and LAS.
The QQ-plots suggest that multiple variants at multiple loci including variants reaching a low threshold of significance for association with IS, CAD, or both, and thus not previously reported as risk loci for arterial disease, contribute to shared genetic susceptibility to IS and CAD. This agrees with the growing evidence that common traits are affected by a large number of causative alleles with very small effects.23 As illustrated both by the QQ-plots and the analysis of variants meeting a high threshold of significance the excess of shared signals between CAD and LAS was more pronounced than the excess of signals between CAD and cardioembolic stroke. This might indicate, that some of the shared risk variants for CAD and LAS act through mechanism that are relatively specific for atherosclerotic disease.
A number of loci thus far not identified in isolated GWAS of IS or LAS showed a strong and consistent signal when considered jointly with CAD. Several lines of statistical evidence support a role for these loci in ischemic stroke risk: i) p-values for individual variants were <1×10-3, ii) the combined p-value in the joint meta-analysis with CAD was genome-wide significant and at least one order of magnitude below the p-value found for CAD alone, and iii) p-values for individual variants significantly correlated between CAD (where these loci reached genome wide significance) and IS or LAS.
Loci reaching genome-wide significance in the joint meta-analyses can be broadly classified into three categories: loci that showed a clear signal for both IS and CAD (e.g. chr12q24/SH2B3; Figure 3A), loci that showed a clear signal for both LAS and CAD (e.g. RAI1-PEMT-RASD1; Figure 3B), and loci for which the association was confined to CAD (e.g. SORT1 or TCF21; Figure 4).
The locus with the strongest association signal for IS was at chr12q24/SH2B3 and had so far not been reported for this phenotype. This locus also showed one of the strongest signals in the combined meta-analysis indicating that chr12q24/SH2B3 is a major susceptibility locus for cardiovascular disease. Variants in this region have previously been shown to be associated with various other traits including blood pressure 24, 25, blood lipids 26, platelet count 27, and type-1 diabetes.28 Several of these traits are linked to IS, CAD, or both. Odds ratios for IS and CAD were similar and p-values for individual variants for IS and CAD significantly correlated indicating that the association signals for the two phenotypes originate from the same genetic variants.
Variants at ABO, the locus with the second strongest signal for IS, have likewise been associated with a variety of traits including low-density lipoprotein (LDL-C)26, von Willebrand factor 15, and venous thromboembolism 29. Again, p-values for individual variants for IS and CAD significantly correlated and the odds ratios for IS, CAD, and LAS were all very similar with no significant heterogeneity (Supplementary Table II). Several observations suggest that the effects of this locus on vascular risk are mediated by an influence on end-stage coagulation and thrombosis 15, 29, 30, which would be consistent with shared mechanisms in CAD and the broader phenotype of IS.15
Loci significantly associated both with CAD and the more restricted phenotype of LAS included 9p21.3, the locus with the strongest signal in the combined meta-analysis, HDAC9, and several loci not previously reported to be associated with LAS. Among the most significant loci is RAI1-PEMT-RASD1 (17p11.2), which to date has not been reported as a risk locus for LAS. Once again, p-values for individual variants for LAS and CAD significantly correlated at this locus. Variants at RAI1-PEMT-RASD1 also significantly associated with IS but the OR and level of significance was lower than for LAS, suggesting that the association with IS is driven by the association with LAS. Interestingly, the RAI1-PEMT-RASD1 locus has so far not been associated with other traits or diseases known to relate to the vascular system. Another locus significantly associated with both LAS and CAD and not previously reported as being associated with LAS is EDNRA. This locus has been associated with carotid artery atherosclerosis 31 suggesting that this locus acts by promoting early atherogenesis.
Finally, several loci displayed highly significant associations with CAD, while showing no association with LAS or IS. This included TCF21 (6q23.2), PHACTR1 (6p24.1), and WDR12 (2q33.1), which are among the strongest signals for CAD.12, 13 The finding suggests partially distinct mechanisms by which common genetic variants contribute to the risk of CAD and LAS.
Our findings must be interpreted in light of the known comorbidity between IS and CAD. We did not control for comorbid vascular disease because the information was not available for most of the participants. However, the pattern of association between established CAD loci and IS differs from what would be expected based on comorbidity or referral bias in that the chr12q24/SH2B3 locus displayed a similar strength of association with IS and CAD, and RAI1-PEMT-RASD1 (17p11.2) showed a similarly strong association with LAS and CAD. Also, several of the top signals for CAD displayed no association with IS or LAS. We can largely exclude a referral bias favoring the selection of stroke patients with a diagnosis of CAD since the majority of subjects included into METASTROKE were recruited through acute stroke services or through population-based studies. There may have been some enrichment for patients with a history of stroke among subjects recruited into CARDIoGRAM/C4D. However, with the exception of HDAC9, all top signals in the combined meta-analysis showed stronger associations with CAD than with IS, which cannot be explained by comorbidity or referral bias.
Our data add to the understanding of familial aggregation of IS and CAD. A parental history of CAD is a risk factor for stroke and a family history of stroke is a risk factor for CAD and acute coronary syndromes.8 In fact, a family history of stroke was found to be as common in acute coronary syndromes as in patients with acute cerebrovascular events.8 Our finding of shared genetic influences between IS and CAD provides some explanation for the aggregation of different arterial phenotypes within families.
Translating findings from genetic association studies into clinical practice remains a challenge. Recent GWAS have revealed a large number of loci that are associated with classical vascular risk factors 24 and genetic risk scores based on multiple SNPs for blood pressure 24 or lipid levels are associated with vascular endpoints including stroke and CAD. Up to know, however, the clinical utility of such scores in predicting vascular risk is rather limited. This may change as additional information from even more markers is added. More importantly, identification of the biological pathways and mechanisms by which shared genetic influences modulate vascular risk might eventually lead to novel therapeutic strategies with a broad impact on vascular disease.
Our study has limitations. First, sample sizes for IS and CAD differed substantially. Second, there was some overlap in controls between the IS and CAD studies. We attempted to account for these limitations through the use of appropriate analytic algorithms and found that our results were remarkably stable when performing meta-analyses assuming a wide range in the proportion of overlapping controls. The statistical strength of the subtype-specific meta-analysis 19 is illustrated by the results for HDAC9, which showed a strong association in the joint subtype-specific meta-analysis despite a weak signal in CAD.
In conclusion, this is the first study examining shared genetic influences between ischemic stroke and coronary artery disease by meta-analyzing GWAS data. Our data provide insights into shared mechanisms and may in part explain why vascular events in one organ predict vascular events in the other organ.
Supplementary Material
Acknowledgments
The authors want to thank Dr. Dmitri Zaykin for generously sharing scripts used in this analysis.
Funding Sources
The Australian Stroke Genetics Collaboration (ASGC) Australian population control data was derived from the Hunter Community Study. We also thank the University of Newcastle for funding and the men and women of the Hunter region who participated in this study. This research was funded by grants from the Australian National and Medical Health Research Council (NHMRC Project Grant ID: 569257), the Australian National Heart Foundation (NHF Project Grant ID: G 04S 1623), the University of Newcastle, the Gladys M Brawn Fellowship scheme and the Vincent Fairfax Family Foundation in Australia. The Atherosclerosis Risk in Communities Study (ARIC) is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; National Institutes of Health contract HHSN268200625226C and National Heart, Lung, and Blood Institute contracts N01-HC-55015, N01-HC-55016, N01-HC-55018, N01-HC-55019, N01-HC-55020, N01-HC-55021, N01-HC-55022, and grants R01-HL087641, U01 HL096917 (Mosley) and R01-HL093029 (Fornage). Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. ARIC analyses performed as part of the METASTROKE project were supported by grant HL-093029 to M Fornage. Bio-Repository of DNA in Stroke (BRAINS) is partly funded by a Senior Fellowship from the Dept of Health (UK) to Dr Pankaj Sharma, the Henry Smith Charity and the UK-India Education Research Institutive (UKIERI) from the British Council. Cardiovascular Health Study (CHS) research was supported by National Heart, Lung, and Blood Institute contracts N01-HC- 85079, N01-HC-85080, N01-HC-85081, N01-HC-85082, N01-HC-85083, N01-HC-85084, N01-HC-85085, N01-HC-85086; N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133, N01-HC-85239, and by HHSN268201200036C and National Heart, Lung, and Blood Institute grants HL080295, HL087652, HL105756 with additional contribution from the National Institute of Neurological Disorders and Stroke. Additional support was provided through AG-023629, AG-15928, AG-20098, and AG-027058 from the National Institute on Aging. See also http://www.chs-nhlbi.org/pi.htm. DNA handling and genotyping at Cedars-Sinai Medical Center was supported in part by the National Center for Research Resources, grant UL1RR033176, and is now at the National Center for Advancing Translational Sciences, Clinical and Translational Science Institute grant UL1TR000124; in addition to the National Institute of Diabetes and Digestive and Kidney Disease grant DK063491 to the Southern California Diabetes Endocrinology Research Center. deCODE Genetics Work performed at deCODE was funded in part through a grant from the European Community’s Seventh Framework Programme (FP7/2007-2013), the ENGAGE project grant agreement HEALTH-F4-2007- 201413. Framingham Heart Study (FHS) This work was supported by the dedication of the Framingham Heart Study participants, the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract Nos. N01-HC-25195 and N02-HL-6-4278) and by grants from the National Institute of Neurological Disorders and Stroke (NS17950), the National Heart, Lung and Blood Association (HL93029) and the National Institute of Aging (AG033193). The Genetics of Early Onset Stroke (GEOS) Study, Baltimore, USA was supported by the National Institutes of Health Genes, Environment and Health Initiative (GEI) Grant U01 HG004436, as part of the GENEVA consortium under the National Institutes of Health Genes, Environment and Health Initiative , with additional support provided by the Mid-Atlantic Nutrition and Obesity Research Center (P30 DK072488); and the Office of Research and Development, Medical Research Service, and the Baltimore Geriatrics Research, Education, and Clinical Center of the Department of Veterans Affairs. Genotyping services were provided by the Johns Hopkins University Center for Inherited Disease Research (CIDR), which is fully funded through a federal contract from the National Institutes of Health to the Johns Hopkins University (contract number HHSN268200782096C). Assistance with data cleaning was provided by the GENEVA Coordinating Center (U01 HG 004446; PI Bruce S Weir). Study recruitment and assembly of datasets were supported by a Cooperative Agreement with the Division of Adult and Community Health, Centers for Disease Control and by grants from the National Institute of Neurological Disorders and Stroke and the NIH Office of Research on Women’s Health (R01 NS45012, U01 NS069208-01). Heart Protection Study (HPS) (ISRCTN48489393) was supported by the UK Medical Research Council, British Heart Foundation, Merck & Co (manufacturers of simvastatin), and Roche Vitamins Ltd (manufacturers of vitamins). Genotyping was supported by a grant to Oxford University and CNG from Merck & Co. Jemma C Hopewell acknowledges support from the British Heart Foundation Centre of Research Excellence, Oxford (RE/08/004). The Heart and Vascular Health Study (HVH) research reported in this article was funded by National Heart, Lung, and Blood Institute grants R01 HL085251 and R01 HL073410. The Ischemic Stroke Genetics Study (ISGS)/ Siblings With Ischemic Stroke Study (SWISS) The ISGS/SWISS study was supported in part by the Intramural Research Program of the National Institute on Aging, NIH project Z01 AG-000954-06. ISGS/SWISS used samples and clinical data from the NIH- National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds), human subjects protocol numbers 2003-081 and 2004-147. ISGS/SWISS used stroke-free participants from the Baltimore Longitudinal Study of Aging (BLSA) as controls. The inclusion of Baltimore Longitudinal Study of Aging samples was supported in part by the Intramural Research Program of the National Institute on Aging, NIH project Z01 AG-000015-50, human subjects protocol number 2003-078.The ISGS study was funded by NIH- National Institute of Neurological Disorders and Stroke Grant R01 NS-42733 (J. F. Meschia, P.I.). The SWISS study was funded by NIH- National Institute of Neurological Disorders and Stroke Grant R01 NS-39987 (J. F. Meschia, P.I.). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH (http://biowulf.nih.gov). The MGH Genes Affecting Stroke Risk and Outcome Study (MGH-GASROS) GASROS was supported by The National Institute of Neurological Disorders and Stroke (U01 NS069208), the American Heart Association/Bugher Foundation Centers for Stroke Prevention Research 0775010N, the National Institutes of Health and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program (R01 HL087676) and a grant from the National Center for Research Resources. The Broad Institute Center for Genotyping and Analysis is supported by grant U54 RR020278 from the National Center for Research resources. Milano - Besta Stroke Register Collection and genotyping of the Milan cases within CEDIR were supported by Annual Research Funding of the Italian Ministry of Health (Grant Numbers: RC 2007/LR6, RC 2008/LR6; RC 2009/LR8; RC 2010/LR8). FP6 LSHM-CT-2007-037273 for the PROCARDIS control samples. The Rotterdam Study was supported by the Nether-lands Organization of Scientific Research (175.010.2005.011), the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Ageing (050-060-810), the Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development, the Research Institute for Diseases in the Elderly, the Ministry of Education, Culture, and Science, the Ministry for Health, Welfare, and Sports, the European Commission, and the Municipality of Rotterdam to the Rotterdam Study. Further funding was obtained from the Netherlands Heart Foundation (Nederlandse Hartstichting) 2009B102. The Wellcome Trust Case-Control Consortium 2 (WTCCC2) The principal funding for the WTCCC2 stroke study was provided by the Wellcome Trust, as part of the Wellcome Trust Case Control Consortium 2 project (085475/B/08/Z and 085475/Z/08/Z and WT084724MA). The Stroke Association provided additional support for collection of some of the St George’s, London cases. The Oxford cases were collected as part of the Oxford Vascular Study which is funded by the Medical Research Council, Stroke Association, Dunhill Medical Trust, National Institute of Health Research (NIHR) and the NIHR Biomedical Research Centre, Oxford. The Edinburgh Stroke Study was supported by the Wellcome Trust (clinician scientist award to Dr. Sudlow), and the Binks Trust. Sample processing occurred in the Genetics Core Laboratory of the Wellcome Trust Clinical Research Facility, Western General Hospital, Edinburgh. Much of the neuroimaging occurred in the Scottish Funding Council Brain Imaging Research Centre (www.sbirc.ed.ac.uk), Division of Clinical Neurosciences, University of Edinburgh, a core area of the Wellcome Trust Clinical Research Facility and part of the SINAPSE (Scottish Imaging Network – A Platform for Scientific Excellence) collaboration (www.sinapse.ac.uk), funded by the Scottish Funding Council and the Chief Scientist Office. Collection of the Munich cases and data analysis was supported by the Vascular Dementia Research Foundation. Martin Farrall acknowledges support from the BHF Center of Research Excellence in Oxford and the Wellcome Trust core award (090532/Z/09/Z).
The ADVANCE study was supported by a grant from the Reynold’s Foundation and National Heart, Lung, and Blood Institute grant HL087647. Genetic analyses of CADomics were supported by a research grant from Boehringer Ingelheim. Recruitment and analysis of the CADomics cohort was supported by grants from Boehringer Ingelheim and PHILIPS medical Systems, by the Government of Rheinland-Pfalz in the context of the “Stiftung Rheinland-Pfalz für Innovation”, the research program “Wissenschaft Zukunft” and by the Johannes-Gutenberg University of Mainz in the context of the “Schwerpunkt Vaskuläre Prävention” and the “MAIFOR grant 2001”, by grants from the Fondation de France, the French Ministry of Research, and the Institut National de la Santé et de la Recherche Médicale. The deCODE CAD/MI Study was sponsored by NIH grant, National Heart, Lung and Blood Institute R01HL089650-02. The German MI Family Studies (GerMIFS I-III (KORA)) were supported by the Deutsche Forschungsgemeinschaft and the German Federal Ministry of Education and Research (BMBF) in the context of the German National Genome Research Network (NGFN-2 and NGFN-plus) and the EU funded integrated project Cardiogenics (LSHM-CT-2006-037593). LURIC has received funding from the EU framework 6 funded Integrated Project “Bloodomics” (LSHM-CT-2004-503485), the EU framework 7 funded Integrated Project AtheroRemo (HEALTHF2-2008-201668) and from Sanofi/Aventis, Roche, Dade Behring/Siemens, and AstraZeneca. The MIGen study was funded by the US National Institutes of Health (NIH) and National Heart, Lung, and Blood Institute’s STAMPEED genomics research program through R01 HL087676. Ron Do from the MIGen study is supported by a Canada Graduate Doctoral Scholarship from the Canadian Institutes of Health Research. Recruitment of PennCATH was supported by the Cardiovascular Institute of the University of Pennsylvania. Recruitment of the MedStar sample was supported in part by the MedStar Research Institute and the Washington Hospital Center and a research grant from GlaxoSmithKline. Genotyping of PennCATH and Medstar was performed at the Center for Applied Genomics at the Children’s Hospital of Philadelphia and supported by GlaxoSmithKline through an Alternate Drug Discovery Initiative research alliance award (M. P. R. and D. J. R.) with the University of Pennsylvania School of Medicine. The Ottawa Heart Genomic Study was supported by Canadian Institutes of Health Research #MOP--82810 (R. R.), Canada Foundation for Innovation #11966 (R.R.), Heart and Stroke Foundation of Ontario #NA6001 (R. McP.), Canadian Institutes of Health Research #MOP172605 (R. McP.), Canadian Institutes of Health Research #MOP77682 (A. F. R. S.). The WTCCC Study was funded by the Wellcome Trust. Recruitment of cases for the WTCCC Study was carried out by the British Heart Foundation (BHF) Family Heart Study Research Group and supported by the British Heart Foundation and the UK Medical Research Council. N. J. S. and S. G. B. hold chairs funded by the British Heart Foundation. N. J. S. and A.H.G are also supported by the Leicester National Institute for Health Research Biomedical Research Unit in Cardiovascular Disease and the work described in this paper is part of the research portfolio of the Leicester National Institute for Health Research Biomedical Research Unit. The Age, Gene/Environment Susceptibility Reykjavik Study has been funded by NIH contract N01-AG-12100, the NIA Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). The Cleveland Clinic GeneBank study was supported by NIH grants P01 HL098055, P01HL076491- 06, R01DK080732, P01HL087018, and 1RO1HL103931-01. The collection of clinical and sociodemographic data in the Dortmund Health Study was supported by the German Migraine- & Headache Society (DMKG) and by unrestricted grants of equal share from Astra Zeneca, Berlin Chemie, Boots Healthcare, Glaxo-Smith-Kline, McNeil Pharma (former Woelm Pharma), MSD Sharp & Dohme and Pfizer to the University of Muenster. Blood collection was done through funds from the Institute of Epidemiology and Social Medicine, University of Muenster. The EPIC-Norfolk study is supported by the Medical Research Council UK and Cancer Research UK. The EpiDREAM study is supported by the Canadian Institutes of Health Research, Heart and Stroke Foundation of Ontario, Sanofi-Aventis, GlaxoSmithKline and King Pharmaceuticals. Funding for Andrew Lotery from the LEEDS study was provided by the T.F.C. Frost charity and the Macular Disease Society. The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University Rotterdam; the Netherlands Organization for Scientific Research; the Netherlands Organization for Health Research and Development (ZonMw); the Research Institute for Diseases in the Elderly; The Netherlands Heart Foundation; the Ministry of Education, Culture and Science; the Ministry of Health Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam. Support for genotyping was provided by the Netherlands Organization for Scientific Research (NWO) (175.010.2005.011, 911.03.012), the Netherlands Genomics Initiative (NGI)/ NWO project nr. 050-060-810 and Research Institute for Diseases in the Elderly (RIDE). Abbas Dehghan is supported by a grant from NWO (Vici, 918-76-619). The SAS study was funded by the British Heart Foundation. The Swedish Research Council, the Swedish Heart & Lung Foundation and the Stockholm County Council (ALF) supported the SHEEP study. SMILE was funded by the Netherlands Heart foundation (NHS 92345). Dr Rosendaal is a recipient of the Spinoza Award of the Netherlands Organisation for Scientific Research (NWO) which was used for part of this work. The Verona Heart Study was funded by grants from the Italian Ministry of University and Research, the Veneto Region, and the Cariverona Foundation, Verona. The KORA (Kooperative Gesundheitsforschung in der Region Augsburg) research platform was initiated and financed by the Helmholtz Zentrum München - National Research Center for Environmental Health, which is funded by the German Federal Ministry of Education, Science, Research and Technology and by the State of Bavaria. Part of this work was financed by the German National Genome Research Network (NGFN-2 and NGFNPlus) and within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ. Work described in this paper is part of the research portfolio supported by the Leicester NIHR Biomedical Research Unit in Cardiovascular Disease. This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit which is supported and funded by the National Institute of Health Research. The Coronary Artery Disease Genetics Consortium (C4D) includes GWAS data contributed by the PROCARDIS, LOLIPOP and HPS studies. PROCARDIS was supported by the European Community Sixth Framework Program (LSHM-CT-2007-037273), AstraZeneca, the British Heart Foundation, the Oxford British Heart Foundation Centre of Research Excellence, the Wellcome Trust (075491/Z/04), the Swedish Research Council, the Knut and Alice Wallenberg Foundation, the Swedish Heart-Lung Foundation, the Torsten and Ragnar Söderberg Foundation, the Strategic Cardiovascular Program of Karolinska Institutet and Stockholm County Council, the Foundation for Strategic Research and the Stockholm County Council (560283). The LOLIPOP study supported by the National Institute for Health Research (NIHR) Comprehensive Biomedical Research Centre Imperial College Healthcare NHS Trust, the National Institute for Health Research Cardiovascular Biomedical Research Unit of Royal Brompton and Harefield NHS Foundation Trust, the British Heart Foundation (SP/04/002), the Medical Research Council (G0601966, G0700931), the Wellcome Trust (084723/Z/08/Z) the NIHR (RP-PG-0407-10371), European Union FP7 (EpiMigrant, 279143) and Action on Hearing Loss (G51). We thank the participants and research staff who made the study possible.
Appendix
Collaborators/second tier of authors:
Andreas Gschwendtner MD1 ,Steve Bevan PhD44, Yu-Ching Chen PhD49, Anita L. DeStefano PhD21, Eugenio A Parati MD26, Tom Quertermous MD10, Andreas Ziegler PhD3, Eric Boerwinkle PhD15, Hilma Holm MD8, Marcus Fischer MD20, Thorsten Kessler MD48, Christina Willenborg MSc22, Reijo Laaksonen MD50, Benjamin F. Voight PhD6,29, Alexandre F.R. Stewart PhD34, Daniel J. Rader MD51,52, Alistair S. Hall MD53, Jaspal S. Kooner FRCP54
Affiliations (extended list for collaborators/second tier of authors):
49 Baltimore VA Medical Center & Dept of Medicine, University of Maryland School of Medicine
50 Science Center, Tampere University Hospital, Tampere, Finland;
51 The Cardiovascular Institute, University of Pennsylvania, Philadelphia, PA, USA
52 The Institute for Translational Medicine and Therapeutics, School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
53 Division of Cardiovascular and Neuronal Remodelling, Multidisciplinary Cardiovascular Research Centre, Leeds Institute of Genetics, Health and Therapeutics, University of Leeds, UK
54 National Heart and Lung Institute, Imperial College London
Footnotes
Authors’ contributions statement
MD, RM, HS, IRK, and MF designed the experiment. RM and IRK did the statistical analysis. RM generated the figures. MD, JR, RC, SG, GTG, BDM, TLA, CL, CJOD, MF, UT, BMP, CH, SS, JE, JBB, AP, GBB, WM, JFM, SK, MAI, RMcP, KS, CS, MPR, JRT, PS, JCH, JCC, HW, PMR, RR, AG, SB, YCC, ALD, EAP, TQ, AZ, EB, HH, MF, TK, CW, RL, BFV, AFRS, DJR, ASH and JSK were responsible for the collection, phenotyping, or preparation of the METASTROKE, CARDIoGRAM and C4D data. MD, MF and HS were responsible for communication with the three consortia. MD, RM, and HS wrote the first draft of the report. All authors reviewed and commented on the report.
Conflicts of interest
All authors affiliated with deCODE are employees of deCODE, a biotechnology company. Some deCODE employees own stock options in deCODE. The other authors declare that they have no conflicts of interest
References
- 1.Bonita R. Epidemiology of stroke. Lancet. 1992;339:342–344. doi: 10.1016/0140-6736(92)91658-u. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet. 2013;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kannel WB, Wolf PA, Verter J. Manifestations of coronary disease predisposing to stroke. The framingham study. JAMA : the journal of the American Medical Association. 1983;250:2942–2946. [PubMed] [Google Scholar]
- 4.Sacco RL, Benjamin EJ, Broderick JP, Dyken M, Easton JD, Feinberg WM, et al. Prevention and rehabilitation of stroke. Risk factors. Stroke; a journal of cerebral circulation; American heart association prevention conference. Iv; 1997. pp. 1507–1517. [DOI] [PubMed] [Google Scholar]
- 5.Iso H, Jacobs DR, Jr, Wentworth D, Neaton JD, Cohen JD. Serum cholesterol levels and six-year mortality from stroke in 350,977 men screened for the multiple risk factor intervention trial. The New England journal of medicine. 1989;320:904–910. doi: 10.1056/NEJM198904063201405. [DOI] [PubMed] [Google Scholar]
- 6.Dichgans M. Genetics of ischaemic stroke. Lancet neurology. 2007;6:149–161. doi: 10.1016/S1474-4422(07)70028-5. [DOI] [PubMed] [Google Scholar]
- 7.Roberts R, Stewart AF. The genetics of coronary artery disease. Current opinion in cardiology. 2012;27:221–227. doi: 10.1097/HCO.0b013e3283515b4b. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee A, Lim CC, Silver LE, Welch SJ, Banning AP, Rothwell PM. Familial history of stroke is associated with acute coronary syndromes in women. Circulation Cardiovascular genetics. 2011;4:9–15. doi: 10.1161/CIRCGENETICS.110.957688. [DOI] [PubMed] [Google Scholar]
- 9.Traylor M, Farrall M, Holliday EG, Sudlow C, Hopewell JC, Cheng YC, et al. Genetic risk factors for ischaemic stroke and its subtypes (the metastroke collaboration): A meta-analysis of genome-wide association studies. Lancet neurology. 2012;11:951–962. doi: 10.1016/S1474-4422(12)70234-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holliday EG, Maguire JM, Evans TJ, Koblar SA, Jannes J, Sturm JW, et al. Common variants at 6p21.1 are associated with large artery atherosclerotic stroke. Nature genetics. 2012;44:1147–1151. doi: 10.1038/ng.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikram MA, Seshadri S, Bis JC, Fornage M, DeStefano AL, Aulchenko YS, et al. Genomewide association studies of stroke. The New England journal of medicine. 2009;360:1718–1728. doi: 10.1056/NEJMoa0900094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nature genetics. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.CARDIoGRAMplusC4D Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gschwendtner A, Bevan S, Cole JW, Plourde A, Matarin M, Ross-Adams H, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Annals of neurology. 2009;65:531–539. doi: 10.1002/ana.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams FM, Carter AM, Hysi PG, Surdulescu G, Hodgkiss D, Soranzo N, et al. Ischemic stroke is associated with the abo locus: The euroclot study. Annals of neurology. 2013;73:16–31. doi: 10.1002/ana.23838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in europeans and south asians identifies five new loci for coronary artery disease. Nature genetics. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 17.Willer CJ, Li Y, Abecasis GR. Metal: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preuss M, Konig IR, Thompson JR, Erdmann J, Absher D, Assimes TL, et al. Design of the coronary artery disease genome-wide replication and meta-analysis (cardiogram) study: A genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circulation Cardiovascular genetics. 2010;3:475–483. doi: 10.1161/CIRCGENETICS.109.899443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magi R, Lindgren CM, Morris AP. Meta-analysis of sex-specific genome-wide association studies. Genetic epidemiology. 2010;34:846–853. doi: 10.1002/gepi.20540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magi R, Morris AP. Gwama: Software for genome-wide association meta-analysis. BMC bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaykin DV, Kozbur DO. P-value based analysis for shared controls design in genome-wide association studies. Genetic epidemiology. 2010;34:725–738. doi: 10.1002/gepi.20536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin DY, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. American journal of human genetics. 2009;85:862–872. doi: 10.1016/j.ajhg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common snps. Nature genetics. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wain LV, Verwoert GC, O’Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nature genetics. 2011;43:1005–1011. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qayyum R, Snively BM, Ziv E, Nalls MA, Liu Y, Tang W, et al. A meta-analysis and genome-wide association study of platelet count and mean platelet volume in african americans. PLoS genetics. 2012;8:e1002491. doi: 10.1371/journal.pgen.1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nature genetics. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tregouet DA, Heath S, Saut N, Biron-Andreani C, Schved JF, Pernod G, et al. Common susceptibility alleles are unlikely to contribute as strongly as the fv and abo loci to vte risk: Results from a gwas approach. Blood. 2009;113:5298–5303. doi: 10.1182/blood-2008-11-190389. [DOI] [PubMed] [Google Scholar]
- 30.Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, et al. Identification of adamts7 as a novel locus for coronary atherosclerosis and association of abo with myocardial infarction in the presence of coronary atherosclerosis: Two genome-wide association studies. Lancet. 2011;377:383–392. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, et al. Meta-analysis of genome-wide association studies from the charge consortium identifies common variants associated with carotid intima media thickness and plaque. Nature genetics. 2011;43:940–947. doi: 10.1038/ng.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.