

Abstract
Over the past decade, the early C. elegans embryo has proven to be a useful animal model to study a variety of membrane trafficking events, at least in part due to its large size, optical transparency, and ease of manipulation. Importantly, the stereotypic nature of membrane remodeling that occurs during early embryogenesis has enabled quantitative measurement of endocytic flux. In the absence of exogenous stimulation, resumption of the cell cycle triggered by fertilization is coupled to a dramatic redistribution of plasma membrane content. Numerous proteins are rapidly internalized via clathrin-mediated endocytosis, and the fate of these cargoes can be followed precisely using live imaging in utero. Key to these studies is the maintenance of animal health and their immobilization, which can become technically challenging during extended imaging sessions. Here we highlight recent advances in live imaging techniques that have facilitated the interrogation of endocytic transport in live animals. We focus on the use of transgenic C. elegans strains that stably express fluorescently-tagged proteins, including components of the endosomal system and cargo molecules that traverse this network of membranes. Our findings demonstrate the utility of the C. elegans embryo in defining regulatory mechanisms that control the numerous steps of endocytic trafficking.
1. Introduction
Endocytosis is broadly defined as the process by which cargo molecules at the cell surface are packaged into vesicular transport carriers for movement into a network of endosomal membranes. It plays an essential role in intercellular signaling, the uptake of nutrients, and membrane recycling, and defects in endocytosis are associated with numerous disease states, including oncogenesis, immune dysfunction, and neurodegeneration. One major pathway of endocytic transport is initiated by clathrin, a coat protein that polymerizes on specialized subdomains of the plasma membrane to generate vesicular carriers that can vary widely in size (as small as ~ 35 nm in diameter in yeast cells to ~ 200 nm in animals cells actively taking up viruses) [1]. Although clathrin does not associate directly with lipid bilayers, it engages several adaptor proteins, which exhibit both membrane- and cargo-binding capabilities. The fate of many cargoes following internalization depends on sorting signals that are present on their cytosol-facing domains, which are recognized by a diverse set of adaptor molecules. Ubiquitin-modification of transmembrane proteins has been shown to promote further packaging into lumenal vesicles that bud into the interior of endosomal compartments, a process that is governed by the endosomal sorting complex required for transport (ESCRT) machinery [2]. In other cases, sorting signals found in the primary amino acid sequence of cargoes can enable transport to other subcellular locations, which includes recycling to the cell surface in the case of the transferrin receptor, or transport to the Golgi in the case of the Wnt-ligand chaperone Wntless (MIG-14 in C. elegans) [3].
Although numerous approaches have been taken to study endocytosis, light microscopy-based imaging affords an excellent opportunity to interrogate the transport process from start to finish. Visualization of a wide variety of cargoes has been enhanced by the development of bright, genetically-encoded fluorescent molecules, which can be fused directly to substrates. Alternatively, components of the endocytic machinery itself can be labeled in a similar manner to study the formation and consumption of different endocytic intermediates. Using this approach, new regulatory mechanisms that control internalization and downstream sorting events have been identified, although there remains abundant controversy regarding the precise role of various components [4]. Total internal reflection fluorescence microscopy (TIR-FM) has become a standard technique to study the initial formation and budding of clathrin-coated vesicles at the plasma membrane. However, multi-plane acquisition, typically using confocal, multi-photon, or widefield imaging, is necessary to track individual particles once they leave the evanescent field. The type of imaging used is often determined by the model organism under investigation. While yeast and human tissue culture cells can be maintained close to an imaging interface that is compatible with the use of TIR-FM optics, studies of tissues that lie deep within an organism such as the intestine or uterus require distinct methodologies.
The combination of available genetic tools, stereotypic development, and optical translucency has placed C. elegans at the forefront of the endocytosis field. Our efforts have mainly been focused on the developing 1-cell stage zygote, in large part due to the substantial reorganization of plasma membrane content that is triggered only minutes after fertilization. In a brief time window, the early C. elegans embryo initiates a wave of endocytosis and multivesicular endosome biogenesis, providing an endogenously encoded ‘pulse-chase’ to study ubiquitin-dependent cargo degradation [5]. Additionally, the ability to genetically engineer animals that express a variety of fluorescently-tagged proteins has been instrumental in measuring the kinetics of endocytic flow, both in control and mutant scenarios. These studies have highlighted the critical importance of clathrin-mediated endocytoisis during early embryonic development, and also demonstrated the surprisingly extensive level of redundancy that exists in the function of clathrin adaptors proteins [6–8]. Here we present detailed protocols and approaches to study endocytosis in the C. elegans zygote, which can be performed in immobilized, transgenic animals using confocal microscopy-based imaging.
2. Challenges and solutions to long-term imaging in a whole animal
To study endocytosis during early zygotic development, transgenic animals expressing fluorescently-tagged proteins within the germline are typically necessary. However, unlike somatic tissues, the C. elegans germline is particularly prone to the loss of transgene expression, an effect commonly referred to a silencing [9]. Recent evidence indicates that foreign DNA sequences, which include genes that encode fluorescent tags, can be discriminated and silenced in the germline via an RNA-induced epigenetic silencing pathway [10]. In some cases, silencing can be circumvented by generating strains that ectopically express transgenes at low copy number. To do so, three approaches have had proven success. First, a mixture of genomic DNA and DNA encoding the transgene of interest can be injected into animals to create a complex extrachromosomal array [11]. Although this method is relatively simple, arrays typically do not exhibit Mendelian inheritance patterns and can often silence even when expressed at low levels. Subsequent studies demonstrated that the chromosomal integration of transgenes facilitates stable, long-term expression in the germline. To accomplish this, microparticle bombardment is often used to randomly insert transgene DNA [12]. One or more selectable markers are typically employed to identify putative transgenic animals. We have successfully used this approach to generate dozens of animals that express fluorescently-tagged proteins, including numerous components of the endocytic machinery and its cargoes. Drawbacks to this approach include the random nature of insertion, which can sometimes interfere with the function of an endogenous locus, and the lack of control over insertion frequency that can cause the inadvertent overexpression of transgenes. A third method known as Mos1-mediated Single Copy Insertion (MosSCI) directs the single-copy insertion of a transgene into a specific (benign) chromosomal locus [13]. Multiple insertion strains are now available, enabling the integration of transgenes into different chromosomes. However, variability exists in these insertion lines, and in some cases, they have been found to be incompatible for germline expression.
Just as important as the method chosen to generate transgenic animals is the choice of regulatory sequences used in transgene constructs. While a number of promotor and 3′UTR sequences have been defined as sufficient for germline expression, the PIE-1 regulatory sequences have been used effectively to support germline protein synthesis for more than a decade [14]. However, to best mimic the expression pattern of a particular gene of interest, it is ideal to use its native 3′UTR. Additionally, use of genomic DNA, as opposed to cDNAs that lack introns, is preferred to maintain germline expression. Fluorescent proteins (GFP and mCherry) that harbor introns and/or have been codon optimized for C. elegans expression are available and also appear to lessen the likelihood of germline silencing.
Yet another alternative to generating transgenic animals that express fluorescently-tagged proteins is the use of Cas9-mediated homologous recombination. Several labs have contemporaneously developed this technology, which enables direct editing of the C. elegans genome by taking advantage of the clustered, regularly interspersed, short palindromic repeats (CRISPR) RNA-guided Cas9 nuclease [15]. One of the major benefits of this method is the ability to modify the endogenous locus of a target gene. Through self-fertilization, one can rapidly isolate homozygous lines and determine whether fusion proteins made are functional. Over the next several years, the use of CRISPR/Cas9 will likely become standard in the field and further establish C. elegans as a leading, genetically-tractable model organism in the study of endocytosis.
Following isolation of transgenic strains, timelapse imaging of endocytosis during early zygotic development requires the immobilization of animals. Several methods have been employed to restrict the movement of worms. First, treatment with anesthetics (e.g., levamisole and tricane) can rapidly minimize the ability of animals to move during long-term imaging [16]. Drug treatment can be carried out in solution, and immobilized animals are transferred onto an agarose pad for imaging. Orientation of animals on the pad is critical to minimize light scattering, which can decrease contrast and result in poor signal-to-noise. Ideally, the germline of the animal should be placed as close as possible to the coverslip. Since the transfer of animals to an agarose pad can be difficult to control, several animals should be anesthetized simultaneously to increase the chances that an individual is oriented properly following mounting. It should be noted that the utility of anesthetics is somewhat limited, as animals typically fail to continue oocyte production following paralysis. Nonetheless, at least one or a few fertilization events can be tracked using this methodology. An alternative to drug treatment is the use of polystyrene beads. When applied to a concentrated pad of agarose, the friction created between the worm, pad and coverglass results in immobilization [17]. Again, orientation can be difficult to control, and thus the deposition of multiple animals into small pools of beads is often necessary. In most cases, worms can be easily recovered after imaging by adding an excess volume of media to the pad and then pipetting animals to a separately prepared plate. We have found this approach both simple and cost effective for long-term imaging of the oocyte-to-embryo transition. However, a potential disadvantage of approaches using agarose is the propensity of pads to dry during the imaging session, which can lead to variability in the focal plane over time or animal movement when polystyrene beads are used. Sealing coverslips with petroleum jelly and paraffin limits this effect, but can reduce oxygenation during imaging.
Instead of mounting animals onto agarose, another alternative is the use of microfluidic devices, which are typically made using the air-permeable elastomer poly (dimethylsiloxane), also known as PDMS [18]. We have successfully used this approach, inserting individual animals into single microchannels, which exhibit a diminishing diameter over their lengths. When placed under flow, animals become trapped within the channels and fail to move significantly over the course of several hours. Recovery of animals is as simple as reversing the flow within the channels. PDMS devices can be reused, as long as they are kept hydrated, although seals at the inlet and outlet holes can begin to leak over time.
3. Methods and Materials
3.1. C. elegans handling and generation of transgenic strains
All C. elegans strains were maintained at 20°C on Nematode Growth Medium (NGM) agar plates that were seeded with the E. coli strain OP50. Methods for the culturing and handling of animals were performed according to standard protocols [19]. All C. elegans strains used were derived from the Bristol strain N2. Transgenic strains expressing GFP- or mCherry-tagged fusion proteins were created using microparticle bombardment and have been described previously [5, 6, 15, 20–22]. The following N2-derivative strains were used: unc-119(ed3) III; pwIs792[ppie-1::MIG-14::GFP; unc-119 (+)], unc-119(ed3) III; hzIs100[ppie-1::GFP::CAV-1; unc-119 (+)], unc-119(ed3) III; ItIs105[ppie-1::GFP::MVB-12; unc-119 (+)], unc-119(ed3) III; hzIs172[ppie-1::GFP::HGRS-1; unc-119 (+/−)], unc-119(ed3) III; hzIs154[ppie-1::GFP::RAB-7; unc-119 (+)], unc-119(ed3) III; itIs63[ppie-1::mCherry::RAB-5; unc-119 (+)], unc-119(ed3) III; hzIs143[ppie-1::GFP::CLIC-1; unc-119 (+)], and hzIs319[ppie-1::GFP::APA-2; unc-119 (+)], aps-2(tm2912) X; hzIs319[ppie-1::GFP::APA-2; unc-119 (+)].
3.2. C. elegans mounting and immobilization
Animals were immobilized in a 4 μl suspension of polystyrene beads (Polysciences, 2.5% by volume, 0.1 μm diameter) that was placed onto the center of a 10% agarose pad and gently compressed with 18×18 mm coverglass. To make a 10% agarose mixture, 1 mL of fresh M9 buffer was added to 0.1 g of agarose (Low-EEO) and transferred to a heat block set at 95°C for 15–20 minutes. To generate a pad, three glass slides were placed onto a flat surface, and the outer two were firmly adhered using two strips of masking tape. Molten agarose (~ 200 μl) was transferred using a metal spatula onto the middle slide, and a fourth glass slide was then quickly placed perpendicular to the original three, pressing down firmly to compress the agarose into a thin, even layer. The pad was allowed to cool for ~ 20 seconds before removing the perpendicular slide. A razor blade can be used to trim excess agarose from the edge of the slide, as necessary. Once animals are placed into the polystyrene bead mixture and a coverglass is applied, the surface tension generated will hold the animals in place for long-term imaging.
To obtain embryos for ex utero analysis, animals are moved into a solution of egg salts (described in the text) and are cut using a razor blade at or near the spermatheca, which lies adjacent to the uterus. By cutting at this location, early embryos spill out into solution and can be transferred by mouth pipette onto a 1% agarose pad. To maintain the osmotic integrity of the embryo, transfer should not occur until after the embryo permeability barrier is formed. Imaging under a stereoscope should be performed to determine the timing of meiosis II (during extrusion of the second polar body). Only at this point can the embryo withstand the pressure between the agarose pad and the coverslip.
3.3. Live timelapse imaging
Images were acquired on a swept-field confocal microscope (Nikon Ti-E), using a Nikon 60x, 1.4 numerical aperture Planapo oil objective lens. Acquisition parameters were controlled by Nikon Elements software, and image analysis was conducted using Metamorph software. Images were taken every 60 seconds (single plane) using a Roper CoolSnap HQ2 CCD (charge-coupled device) camera, in the case of whole animals. Manual focus adjustments were made as necessary during the intervals between image acquisitions. For embryo filming, images were acquired every second, and the focal plane was maintained using a Nikon Perfect Focus system.
4. Results and discussion
4.1. Methods to study clathrin-mediated endocytosis
One of the best characterized pathways for cargo internalization from the cell surface is mediated by clathrin triskelions, which are composed of three heavy chains and three light chains that co-assemble to form a polyhedral lattice that surrounds an endocytic vesicle [1]. Sequence analysis indicates that the C. elegans genome encodes one heavy chain (CHC-1) and at least one light chain (CLIC-1). Surprisingly, deletion mutations in CLIC-1 do not affect viability, suggesting the potential existence of another light chain that exhibits poor sequence conservation. An alternative interpretation is that a redundant paralog of CLIC-1 does not exist, and that light chains only function in cells in a regulatory capacity. Consistent with this idea, previous work has shown that lights chains are largely dispensable for clathrin-mediated endocytosis [23], but they do function in clathrin-dependent trafficking between the trans-Golgi network and endosomes, where they regulate the actin cytoskeleton [24]. Irrespective of its precise mechanism of action, a GFP fusion to CLIC-1 provides a useful probe for clathrin-mediated endocytosis. Using microparticle bombardment, we previously generated a transgenic animal that stably expresses GFP:CLIC-1 within the germline and early embryo [6]. In oocytes, the fusion protein is largely restricted to the cell surface, although some internal compartments are also labeled, which likely highlight clathrin accumulation on Golgi ministacks and endosomes (Figure 1 and Movie S1). However, upon oocyte fertilization and movement of the zygote into the uterus (ovulation), a majority of clathrin is rapidly internalized and redistributed to internal membrane compartments and the cytoplasm (Figure 1 and Movie S1). We refer to this process as the first wave of clathrin-mediated endocytosis. Within 10 minutes, clathrin levels at the plasma membrane cannot be obviously detected above cytoplasmic fluorescence (Figure 1 and Movie S1). However, within an additional 10 minutes, a new accumulation of clathrin-coated pits at the plasma membranes is observed, which is quickly (within ~ 3 minutes) followed by another round of internalization, which we refer to as the second wave of clathrin-mediated endocytosis (Figure 1 and Movie S1). At a timepoint 30–35 minutes after ovulation, the majority of membrane-bound clathrin is observed on internal compartments, with only a weak signal remaining on subdomains of the plasma membrane (Movie S1). A similar distribution remains until after the first embryonic cell division, when again, a pool of clathrin appears to accumulate on the cell surface, particularly enriched at the interface between the daughter cells (Figure 1 and Movie S1). This conspicuous enrichment is likely a due to the presence of two, juxtaposed membrane surfaces that cannot be resolved at the level of the light microscope, as opposed to enrichment specifically at the junction between two adjacent cells. These data indicate that the re-initiation of cell cycle progression (triggered by fertilization) is coupled to multiple waves of clathrin-mediated endocytic transport.
Figure 1. Clathrin distribution during early zygotic development.

Individual images of GFP:CLIC-1 demonstrate its distribution at various times relative to zygote ovulation. Arrows highlight clathrin accumulation at the plasma membrane prior to ovulation (−3 min timepoint) and coincident with meiosis II (+20 min timepoint) or its absence at the plasma membrane (+10 min timepoint). Insets are 3x zoomed views of the areas highlighted by arrows. Scale bar, 20 μm. Inset scale bars, 5 μm.
Following fertilization, zygotes remain osmotically sensitive until after formation of a permeability barrier, which forms between the eggshell and the embryo plasma membrane [25]. This event occurs coincidently with meiosis II, just prior to initiation of the second wave of clathrin-mediated endocytic transport (~ 23–27 min after ovulation). Thus, it is feasible to obtain higher resolution imaging of this phase of endocytosis by dissecting zygotes into a specialized media (egg salts, consisting of 82.6 mM NaCl, 28 mM KCl, 2.4 mM MgCl2, 2.4 mM CaCl2, 3.5 mM HEPES pH 7.4), and transferred them onto a 1% agarose pad at the appropriate time. Using a transgenic strain expressing a GFP fusion to the alpha subunit of the AP-2 complex, one of several clathrin adaptors important for internalization of cargoes, we used confocal imaging to study assembly and internalization of individual clathrin-coated structures. For these studies, imaging was conducted at the embryo cortex, just underneath the eggshell. A near-infrared 870 nm LED and CCD line sensor were engaged to prevent focus drift during the timecourse. Our findings reveal that internalization events occur throughout the embryo cortex, but become more asymmetric (toward the anterior) as the embryo becomes polarized (Movie S2). To further confirm the specificity of the GFP fusion, we generated a mutant strain expressing it, but also harboring a null mutation in the AP-2 sigma subunit, which is required for the alpha subunit to engage the AP-2 beta and mu hemicomplex [26]. In this genetic background, the GFP:alpha fusion fails to target to clathrin-coated pits (Figure 2). Together, these data highlight the ability to analyze clathrin-mediated endocytosis using the early C. elegans embryo, both in utero and in dissected, fertilized embryos.
Figure 2. The AP-2 sigma subunit is required for localization of the alpha subunit.
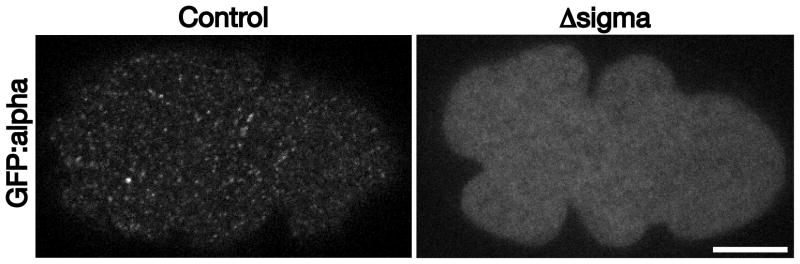
One-cell stage embryos expressing a GFP fusion to the alpha subunit of AP-2 were imaged using swept-field confocal optics at the embryo cortex. Control embryos exhibit numerous sites of clathrin-mediated endocytosis, while mutant embryos that lack expression of the AP-2 sigma subunit fail to recruit the alpha subunit to sites of endocytosis. Scale bar, 10 μm.
4.2. Methods to study endosomal sorting downstream of endocytosis
Subsequent to internalization, vesicles shed their coats and typically fuse with early endosomal compartments, which are labeled with the small GTPase RAB-5. Germline expression of fluorescently-labeled Rab5 indicates that it lies on membranes that are juxtaposed to the cell surface in oocytes, exhibiting relatively little movement (Figure 3 and Movie S3). However, following fertilization and ovulation, early endosomes undergo a dramatic redistribution, scattering throughout the zygote (Movie S3). At nearly the same time as clathrin is recruited back to the cell surface (~ 20 minutes after ovulation), RAB-5 begins to accumulate again near the plasma membrane (Figure 3). The size of RAB-5-decorated endosomes, as well as RAB-5 intensity on endosomes, during this time period is elevated relative to that found in embryos at a later stages of development, suggesting an increased rate of membrane addition. These data are consistent with the idea that an augmented rate of clathrin-mediated endocytosis results in increased RAB-5-dependent membrane fusion at early endosomes. Nevertheless, endosome size undergoes rapid reduction following the transient wave of endocytosis, and the RAB-5-labeled compartments become enriched at the embryo anterior in response to polarity establishment (Figure 3 and Movie S3) [20].
Figure 3. RAB-5 dynamics during early zygotic development.

Individual images of mCherry:RAB-5 illustrate its distribution at various times relative to zygote ovulation. Arrows highlight RAB-5 accumulation on early endosomes near the plasma membrane. Note the increased RAB-5 intensity on enlarged early endosomes during the second wave of clathrin-mediated endocytosis (+23 min timepoint), as compared to endosomes labeled with RAB-5 in later stage embryos. Insets are 3x zoomed views of the areas highlighted by arrows. Scale bar, 20 μm. Inset scale bars, 5 μm.
The decrease in early endosome size and RAB-5 intensity on endosomal compartments likely corresponds with cargo export to other subcellular locations. Cargoes leave the early endosome via several routes: recycling to the plasma membrane, transport to the Golgi, and a process known as endosome maturation, in which Rab5 levels progressively diminish, and another Rab type GTPase accumulates [27]. In the case of late endosome formation, RAB-5 is exchanged for RAB-7. Using animals stably expressing both mCherry:RAB-5 and GFP:RAB-7, we found that late endosomes appear in close apposition to RAB-5-labeled early endosomes in oocytes, mostly restricted to the cell periphery (Figure 4 and Movie S4). The intensity of RAB-7 on internal membranes diminishes rapidly after ovulation and rises again several minutes after the second wave of clathrin-mediated endocytosis (~ 35 minutes after ovulation) (Figure 4 and Movie S4). The timing of increased RAB-7 accumulation on late endosomes corresponds to the stage during which RAB-5 diminishes on early endosomes. These findings are entirely consistent with the idea of endosomal maturation, and highlight the C. elegans developing zygote as an ideal model system to study regulatory mechanisms that govern this process.
Figure 4. RAB-7 dynamics relative to RAB-5 during early zygotic development.

Individual images of GFP:RAB-7 illustrate its distribution at various times during to the oocyte-to-embryo transition. Arrows highlight the distinct but juxtaposed localizations of RAB-5 and RAB-7 near the cell periphery during different stages of oocyte or zygote development. Note the elevated levels of RAB-5 on endosomal membranes relative to RAB-7 levels during the second wave of endocytosis (+23 min timepoint), and the reciprocal relationship 10 minutes later. Insets are 3x zoomed views of the areas highlighted by arrows. Scale bar, 20 μm. Inset scale bars, 5 μm.
Early-to-late endosome maturation enables cargoes to be efficiently trafficked into the endolysosomal system, ultimately for degradation in most cases, when late endosomes fuse with lysosomes. The endosomal sorting complex required for transport (ESCRT) machinery has been shown to be essential for the biogenesis of multivesicular endosomes, a specialized subset of late endosomal compartments that harbor internal, cargo-laden lumenal vesicles. Upon lysosome fusion, these intralumenal vesicles are degraded. ESCRT-dependent protein sorting is a major route by which ubiquitin-modified transmembrane proteins are turned over, and thus represents a key system to maintain cellular homeostasis. The ESCRT machinery is composed of five protein complexes, and we have developed probes to study both the ESCRT-0 and ESCRT-I complexes, which function early in multivesicular endosome formation [2].
We first examined animals stably expressing a GFP fusion to HGRS-1, a subunit of the ESCRT-0 complex, which binds directly to ubiquitin-modified cargoes. Additionally, HGRS-1 harbors a FYVE domain that associates avidly with the lipid phosphatidylinositol 3-phosphate, which is enriched on early endosomes. In oocytes, we found that HGRS-1 distribution was largely cytosolic until ~ 5 minutes prior to ovulation, when it became redistributed to endosomes that were juxtaposed directly to the plasma membrane (Figure 5 and Movie S5). These data highlight the existence of an additional regulatory signal that governs ESCRT-0 localization, which has yet to be defined. Similar to RAB-5, the distribution of ESCRT-0-labeled compartments is scattered until ~ 20–25 minutes after ovulation. At this timepoint, ESCRT-0 accumulates on enlarged membrane compartments that are at or near the plasma membrane (Figure 5 and Movie S5). Both subunits of ESCRT-0 have been found to associate directly with clathrin adaptor proteins, indicating that the complex engages cargoes at clathrin-coated pits and likely directs their transport into the endolysosomal system [6]. The intensity of HGRS-1 on endosomes remains elevated for a longer period of time as compared to RAB-5 (until ~ 45 minutes after ovulation) (Movie S5). Thus, increased ESCRT-0 distribution on endosomes spans the period during which endosomal maturation occurs, consistent with the idea that the ESCRT machinery participates in the formation of specialized late endosomes [2].
Figure 5. ESCRT-0 dynamics during early zygotic development.

Images of GFP:HGRS-1 show its distribution at various times relative to zygote ovulation. Long arrows highlight the accumulation of HGRS-1 on membranes at or near the cell surface in oocytes just prior to ovulation (compare −3 min timepoint and +23 min timepoint). Short arrows highlight HGRS-1 accumulation on membranes at or near the cell surface in embryos. Note the increased HGRS-1 intensity on enlarged early endosomes during the second wave of clathrin-mediated endocytosis (+23 min timepoint), as compared to endosomes labeled with HGRS-1 in the adjacent embryo. Inset is a 3x zoomed view of the area highlighted by arrowheads. Scale bar, 20 μm. Inset scale bar, 5 μm.
To study ESCRT-I dynamics, we previously generated a GFP fusion to its MVB-12 subunit. In contrast to ESCRT-0, the ESCRT-I complex only transiently assembles on endosomal membranes and exhibits a largely cytoplasmic distribution in oocytes and embryos (Figure 6 and Movie S6). During the height of ESCRT-0 accumulation on endosomal compartments at or near the cell surface (during the second wave of clathrin-mediated endocytosis, ~23–25 minutes after ovulation), MVB-12 can also be distinguished, albeit weakly, on these membranes (Figure 6 and Movie S6). However, the increased localization of ESCRT-I to endosomes is very transient (Figure 6 and Movie S6), which is consistent with its rapid exchange between endosomal membranes and the cytoplasm. Based on these studies, GFP fusions to ESCRT-I subunits are unlikely to be as useful as other markers to study endosome maturation in embryos, since so few subcellular compartments can be distinguished above cytosolic fluorescence. However, aberrant accumulation of MVB-12 on endosomes can provide a clear indication of a perturbation to multivesicular endosome dynamics [5]. Thus, the use of animals expressing the GFP::MVB-12 fusion could provide the basis of a visual genetic screen to identify new regulators of ESCRT function.
Figure 6. ESCRT-I dynamics during early zygotic development.

Individual images of GFP:MVB-12 illustrate its distribution at various times relative to zygote ovulation. Long arrows highlight GFP:MVB-12 accumulation on early endosomes near the plasma membrane during the second wave of clathrin-mediated endocytosis (+23 min timepoint). Short arrowheads highlight the localization of ESCRT-I to midbodies. Inset is a 3x zoomed view of the area highlighted by an arrow. Scale bar, 20 μm. Inset scale bar, 5 μm.
Unlike its brief accumulation on endosomes, ESCRT-I is found to stably associate with midbodies that form during cytokinesis [28]. These structures, which are essential to the completion of cell division, are internalized into daughter cells and ultimately degraded. Depletion of ESCRT-I impairs midbody internalization, suggesting a key role for the ESCRT machinery in midbody degradation [28]. However, the mechanism by which ESCRT-I binds to midbodies in C. elegans is unclear. In mammals, the Tsg101 subunit of ESCRT-I associates directly with the midbody component Cep55 [29]. However, no Cep55 homologues appear to exist in worms. Thus, C. elegans should prove a useful model to define alternative mechanisms by which the ESCRT machinery is recruited and functions during cytokinesis.
4.3. Methods to study endocytic cargo transport
The ESCRT machinery engages ubiquitin-modified transmembrane cargoes for deposition into multivesicular endosomes and subsequent lysosomal degradation. We and others previously demonstrated that CAV-1, the C. elegans homologue of caveolin-1, undergoes clathrin-mediated endocytosis, ubiquitin modification and degradation in an ESCRT-dependent manner [6, 21]. Thus, GFP:CAV-1 is an effect tool in the study of ubiquitin-dependent sorting in the endocytic pathway. In oocytes, CAV-1 is localized mainly to the plasma membrane and cytoplasmic cortical granules (Figure 7 and Movie S7). However, during meiosis II and polar body extrusion (~ 20 minutes after ovulation), cortical granules fuse en masse with the plasma membrane (Figure 7 and Movie S7). Notably, during an identical time period, clathrin begins to assemble at the cell surface. Within ~ 3 minutes, CAV-1 begins to accumulate in peripheral, enlarged endosomes and is degraded, as indicated by a loss of GFP signal, within an additional 20–30 minutes (Figure 7 and Movie S7). Again, it is striking to note that the degradation timecourse is nearly identical to that of endosomal maturation. Additionally, RNA interference-mediated depletion of ESCRT components has been shown to dramatically slow the kinetics of CAV-1 degradation, but fails to affect its rate of internalization [5, 6]. Together, these data highlight the utility of the C. elegans early embryo in the study of cortical granule exocytosis, clathrin-mediated endocytosis, multivesicular endosome biogenesis, and ESCRT-mediated protein turnover, all in a manner that is independent of artificial stimuli.
Figure 7. CAV-1 transport through the endolysosomal system.

Images of GFP:CAV-1 illustrate its distribution at various times relative to zygote ovulation. Long arrows highlight the rapid degradation of CAV-1 following its initial clathrin-mediated endocytosis (+23 min timepoint), as indicated by the diminution in GFP fluorescence (+45 and +64 min timepoints). Inset is a 3.5x zoomed view of the area highlighted by an arrow. Scale bar, 20 μm. Inset scale bar, 5 μm.
In contrast to CAV-1, which depends on ubiquitin modification for trafficking through the endolysosomal system, the Wntless homologue MIG-14 uses the retromer complex to mediate its transport from endosomes to the Golgi [3]. In the absence of retromer function, MIG-14 is aberrantly degraded in lysosomes instead of undergoing recycling [22]. We examined the dynamics of a GFP fusion to MIG-14 during ovulation and early zygotic development to enable a comparison with the trafficking of CAV-1. In oocytes, MIG-14 is distributed on the Golgi, endosomes and the plasma membrane (Figure 8 and Movie S8). However, similar to CAV-1, the cell surface labeling of MIG-14 is rapidly lost upon fertilization and ovulation, in a manner that depends on clathrin (Figure 8 and Movie S8) [6, 22]. During the second wave of clathrin-mediated endocytosis, MIG-14 accumulates on peripheral, enlarged endosomes, again similar to CAV-1 (Figure 8 and Movie S8). However, instead of undergoing subsequent degradation, MIG-14 remains associated with internal membrane compartments (likely a combination of Golgi ministacks and endosomes) (Figure 8 and Movie S8). A significant portion of MIG-14 is then secreted back to the cell surface, as indicated by its accumulation at cell junctions in multicellular embryos (Figure 8 and Movie S8). Together, these data demonstrate the existence and tractability of distinct sorting pathways downstream of clathrin-mediated endocytosis in the C. elegans embryonic system. By using a combination of forward and reverse genetic approaches, ongoing work will undoubtedly define additional, new regulators of this essential membrane network.
Figure 8. MIG-14 trafficking during the oocyte-to-embryo transition.

Images of MIG-14:GFP highlight its changing distribution at various times relative to zygote ovulation. The long arrow highlights the accumulation of MIG-14 in peripherally distributed, enlarged endosomes that form during the second wave of clathrin-mediated endocytosis (+23 min timepoint). The short arrow highlights the secretion of MIG-14 back to the plasma membrane following the first embryonic cytokinesis (+64 min timepoint). Inset is a 3.5x zoomed view of the area highlighted by an arrow. Scale bar, 20 μm. Inset scale bar, 5 μm.
4.4. Alternative venues to study endocytosis in C. elegans
We have highlighted the utility of the C. elegans early embryo as a model system to study endocytic transport. However, several other tissues exist in the animal, which also provide useful platforms for similar lines of investigation. For example, the steady state accumulation of yolk proteins (vitellogenins) in oocytes provides a useful indicator of endocytic flux. Yolk proteins, complexed with a variety of lipids, are initially synthesized in the adult intestine and secreted into the body cavity, from where they are internalized in a receptor-mediated process into oocytes following cellularization within the germline. The yolk proteins are then stored in vesicles for later use as a nutrient source by larvae [30]. Creation of transgenic animals expressing a GFP-tagged vitellogenin (VIT-2::GFP) has enabled a series of genetic screens (both forward and reverse approaches) that have identified numerous, previously uncharacterized regulators of endocytic trafficking [31, 32]. Specifically, diminution of GFP fluorescence in oocytes following the inhibition of a particular factor (e.g., clathrin heavy chain, dynamin, etc.) reflects a requirement for that protein in endocytosis. Although the process of yolk uptake can be visualized in time lapse imaging experiments, many immobilization techniques inhibit continued cellularization of oocytes, resulting in a challenge to define specific changes in the kinetics of endocytic transport.
Another cell type that has been particularly useful in the study of endocytosis in C. elegans is the coelomocyte, a scavenger cell within the body cavity that constitutively takes up fluid and macromolecules. Transgenic animals expressing a secretory signal sequence fused to GFP (ssGFP; secreted from body-wall muscle cells) have been used to study endocytic transport in coelomocytes [33]. Similar to assays focused on yolk protein uptake by oocytes, genetic screens in coelomocytes have identified numerous endocytic components, which are required to efficiently clear ssGFP that is secreted into the body cavity. However, also akin to oocyte-based assays, coelomocytes lack an endogenous, developmental trigger that initiates endocytosis, as is observed in the one-cell stage embryo, which makes it more challenging to characterize alterations in the rate at which various cargoes are internalized and trafficked to intracellular compartments in depletion-based studies. Although coelomocytes are significantly smaller than oocytes and one-cell stage embryos, endosomal compartments within these scavenger cells are significantly larger than those found in other tissues [33]. This difference may be exploited to study the formation and turnover of endosomal microdomains that are important for cargo sorting, which has been difficult to study in other contexts without artificially altering endosome morphology.
Coelomocytes, oocytes and one-cell stage embryos all share the characteristic of being unpolarized cell types. In contrast, C. elegans intestinal cells provide an ideal setting to study polarized membrane trafficking events. The apical surface of the intestine is responsible for the uptake of nutrients from the diet and involves a high rate of clathrin-mediated endocytosis, steps of which can be dissected using forward and reverse genetic approaches. Basolateral internalization events are largely coupled to endocytic recycling, affording a tractable platform to investigate the steps of this pathway [34]. However, sorting in intestinal cells is constitutive, making it difficult to conduct similar ‘pulse-chase’ experiments as described for early zygotic development. Nevertheless, by combining assays in multiple cell types within C. elegans, ongoing studies should continue to reveal the identity of new, conserved regulators of endocytosis and endosomal trafficking, some of which will likely function in a tissue-specific manner.
In addition to the variety of cell types that can be studied in C. elegans, alternative approaches beyond the analysis of genetically encoded, fluorescently-labeled proteins exist to examine endocytic events, including the use of vital dyes, immunofluorescence, and electron microscopy. In live animal imaging studies, the injection of FM dyes (e.g., into the body cavity or germline) enables detailed characterization of fluid-phase endocytosis in several tissues [35]. In fixed animals, the distributions of endogenous endocytic factors can be readily defined using antibodies. Typically, the tissue under examination is extruded prior to fixation, to enable antibody accessibility. For example, decapitation using a scalpel is routinely used to extrude the intestine and germline, while a precise incision near the spermatheca allows early embryos to spill out of the animal. Dissections are performed on poly-lysine coated glass slides, and following extrusion (into a 4 μL solution of egg salts), a small coverslip (e.g., 10 × 10 mm square) is applied and the slide is rapidly frozen in liquid nitrogen. After removal of the coverslip, slides can be fixed in cold methanol (−20°C). The duration of fixation varies for different antibodies (typically 20–60 minutes), prior to blocking and antibody incubation. Alternatively, tissues can be fixed using formaldehyde for subsequent antibody staining. Detailed protocols have been published, which thoroughly describe each approach [36].
To obtain high resolution views of endocytic transport steps, electron microscopy-based approaches are becoming more routine. The use of high pressure freezing results in the best preservation of membrane-bound structures, and whole animals can be subjected to this procedure. Briefly, animals are deposited into a shallow (100 μm) aluminum platelet in a suspension of E. coli and frozen rapidly (e.g., using a BalTec HPM 10). Freeze substitution is carried out typically using automated instrumentation, which preserves animals in plastic blocks that can be sectioned using an ultramicrotome prior to visualization under an electron microscope [37, 38]. In contrast to live animal imaging, electron microscopy provides only a static representation of a highly dynamic process. However, the resolution gain afforded by this approach often balances out its negatives and can provide a very complementary set of data to enable further understanding of a specific membrane transport step.
5. Conclusions
Here we have defined useful approaches in the study of endocytosis that take advantage of the inherent properties of the C. elegans reproductive system. Specifically, fertilization and ovulation trigger two sequential waves of clathrin-mediated endocytosis, which are tractable both genetically and optically. Key fluorescent probes to interrogate this pathway have been established, and mounting methods described enable long-term imaging. Although we have focused on the use of confocal microscopy to study endocytosis in utero, an alternative approach would be the use of light sheet microscopy [39]. The combination of high-speed acquisition and low photo-toxicity that is characteristic of light sheet imaging should enable higher frame rates than conducted here, and foster multi-plane, long-term imaging studies during early zygotic development. Nevertheless, the utility of swept-field and spinning disk confocal microscopy is clear, and impressive signal-to-noise ratios can be achieved easily, especially in the context of single plane image acquisition. Together with the advanced genetic tools available in C. elegans, growth in the use of this system to study membrane trafficking events is anticipated.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH: GM088151 to AA. We thank members of the Audhya lab for critically reading this manuscript, and we thank Barth Grant, Shohei Mitani, and Jeff Hardin for strains and reagents used to generate transgenic animals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McMahon HT, Boucrot E. Nat Rev Mol Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 2.Schuh AL, Audhya A. Rev Biochem Mol Biol. 2014 doi: 10.3109/10409238.2014.881777Crti. [DOI] [Google Scholar]
- 3.Yang PT, Lorenowicz MJ, Silhankova M, Coudreuse DY, Betist MC, Korswagen HC. Dev Cell. 2008;14:140–147. doi: 10.1016/j.devcel.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Swan LE. Bioessays. 2013;35:425–429. doi: 10.1002/bies.201200129. [DOI] [PubMed] [Google Scholar]
- 5.Audhya A, McLeod AIX, Yates JR, Oegema K. PLoS One. 2007;2:e956. doi: 10.1371/journal.pone.0000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mayers JR, Wang L, Pramanik J, Johnson A, Sarkeshik A, Wang Y, Saengsawang W, Yates JR, Audhya A. Proc Natl Acad Sci USA. 2013;110:11857–11862. doi: 10.1073/pnas.1302918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato K, Ernstrom GG, Wantanabe S, Weimer RM, Chen CH, Sato M, Siddiqui A, Jorgensen EM, Grant BD. Proc Natl Acad Sci USA. 2009;106:1139–1144. doi: 10.1073/pnas.0809541106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato M, Sato K, Liou W, Pant S, Harada A, Grant BD. EMBO J. 2008;27:1183–1196. doi: 10.1038/emboj.2008.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Wynsberghe PM, Maine EM. Adv Exp Med Biol. 2013;757:373–403. doi: 10.1007/978-1-4614-4015-4_13. [DOI] [PubMed] [Google Scholar]
- 10.Lee HC, Gu W, Shirayama M, Youngman E, Conte D, Mello CC. Cell. 2012;150:78–87. doi: 10.1016/j.cell.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly WG, Xu S, Montgomery MK, Fire A. Genetics. 1997;146:227–238. doi: 10.1093/genetics/146.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Praitis V, Casey E, Collar D, Austin J. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Nat Genetics. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merritt C, Rasoloson D, Ko D, Seydoux G. Curr Biol. 2008;18:1476–1482. doi: 10.1016/j.cub.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frokjaer-Jensen C. Genetics. 2013;195:635–642. [Google Scholar]
- 16.Green RA, Audhya A, Pozniakovsky A, Dammermann A, Pemble H, Monen J, Portier N, Hyman A, Desai A, Oegema K. Methods Cell Biol. 2008;85:179–218. doi: 10.1016/S0091-679X(08)85009-1. [DOI] [PubMed] [Google Scholar]
- 17.Kim E, Sun L, Gabel CV. C Fang-Yen PLoS One. 2013;8:e53419. doi: 10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hulme SE, Shevkoplyas SS, Apfeld J, Fontana W. GM Whitesides Lab Chip. 2007;7:1515–1523. doi: 10.1039/b707861g. [DOI] [PubMed] [Google Scholar]
- 19.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audhya A, Desai A, Oegema K. J Cell Biol. 2007;178:43–56. doi: 10.1083/jcb.200701139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato K, Sato M, Audhya A, Oegema K, Schweinsberg P, Grant BD. Mol Biol Cell. 2006;17:3085–3094. doi: 10.1091/mbc.E06-03-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi A, Sun L, Banerjee R, Tobin M, Zhang Y, Grant BD. EMBO J. 2009;28:3290–3302. doi: 10.1038/emboj.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang F, Khvorova A, Marshall W, Sorkin A. J Biol Chem. 2004;279:16657–16661. doi: 10.1074/jbc.C400046200. [DOI] [PubMed] [Google Scholar]
- 24.Poupon V, Girard M, Legendre-Guillemin V, Thomas S, Bourbonniere L, Philie J, Bright NA, McPherson PS. Proc Natl Acad Sci USA. 2008;105:169–173. doi: 10.1073/pnas.0707269105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olson SK, Greenan G, Desai A, Muller-Reichert T, Oegema K. J Cell Biol. 2012;198:731–748. doi: 10.1083/jcb.201206008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu M, Liu Q, Watanabe S, Sun L, Hollopeter G, Grant BD, Jorgensen EM. Elife. 2013;2:e00190. doi: 10.7554/eLife.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 28.Green RA, Mayers JR, Wang S, Lewellyn L, Desai A, Audhya A, Oegema K. J Cell Biol. 2013;203:505–520. doi: 10.1083/jcb.201306036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. Science. 2008;322:576–580. doi: 10.1126/science.1162042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matyash V, Geier C, Henski A, Mukherjee S, Hirsh D, Thiele C, Grant B, Maxfield FR, Kurzchalia TV. Mol Biol Cell. 2001;12:1725–1736. doi: 10.1091/mbc.12.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant B, Hirsh D. Mol Biol Cell. 1999;10:4311–4326. doi: 10.1091/mbc.10.12.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balklava Z, Pant S, Fares H, Grant BD. Nat Cell Biol. 2007;9:1066–1073. doi: 10.1038/ncb1627. [DOI] [PubMed] [Google Scholar]
- 33.Fares H, Greenwald I. Genetics. 2001;159:133–145. doi: 10.1093/genetics/159.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fares H, Grant BD. Traffic. 2002;3:11–19. doi: 10.1034/j.1600-0854.2002.30103.x. [DOI] [PubMed] [Google Scholar]
- 35.Grant B, Zhang Y, Paupard MC, Lin SX, Hall DH, Hirsh D. Nat Cell Biol. 2001;3:573–579. doi: 10.1038/35078549. [DOI] [PubMed] [Google Scholar]
- 36.Duerr JS. WormBook. 2006;19:1–61. doi: 10.1895/wormbook.1.105.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller-Reichert T, Mancuso J, Lich B, McDonald K. Methods Cell Biol. 2010;96:331–361. doi: 10.1016/S0091-679X(10)96015-9. [DOI] [PubMed] [Google Scholar]
- 38.Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR, Eimer S, Audhya A. Nat Cell Biol. 2011;13:550–558. doi: 10.1038/ncb2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomer R, Khairy K, Keller PJ. Methods Mol Biol. 2013;931:123–137. doi: 10.1007/978-1-62703-056-4_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.