

Abstract
Hematopoietic stem cells (HSCs) are a rare population of somatic stem cells that maintain blood production and are uniquely wired to adapt to diverse cellular fates during the lifetime of an organism. Recent studies have highlighted a central role for metabolic plasticity in facilitating cell fate transitions and in preserving HSC functionality and survival. This review summarizes our current understanding of the metabolic programs associated with HSC quiescence, self-renewal and lineage commitment and highlights the mechanistic underpinnings of these changing bioenergetics’ programs. It also discusses the therapeutic potential of targeting metabolic drivers in the context of blood malignancies.
Keywords: Hematopoietic stem cells, blood, quiescence, glycolysis, reactive oxygen species, fatty acid oxidation
Hematopoiesis: An Ever Changing Developmental System
The blood system is one of the most dynamic tissues in mammals, with an extremely high cellular turnover on a daily basis. Given that most mature blood cells have short lifespans, the onus of maintaining blood homeostasis rests almost entirely on the self-renewal and differentiation capabilities of a long-lived but rare population of somatic stem cells, termed hematopoietic stem cells (HSCs). HSCs can maintain themselves and generate all types of mature blood cells through production of a series of increasingly committed progenitor cells [1]. It is now recognized that the biological properties of HSCs are not constant throughout life, but vary temporally to meet the shifting demands of an evolving organism [2]. During embryonic development, fetal HSCs emerge from various hemangenic endothelium and are programmed to be highly proliferative. This cycling state allows for rapid production of mature blood cells to meet the immediate requirements of a developing embryo, while simultaneously building a reservoir of expanding fetal HSCs to colonize the emerging sites of adult hematopoiesis. In adults, HSCs are mainly found in specialized niches in the bone marrow (BM) cavity and are programmed for enforced quiescence. This dormancy state allows adult HSCs to stay poised for quick and massive production of blood cells in emergency situations, while simultaneously limiting their proliferation to homeostatic production of blood cells. Hence, at steady state, adult HSCs divide only rarely to maintain low production of committed progenitor cells and renew the HSC pool. In mice, while the majority of adult HSCs divide on average once every 30 days [3], a small subset of deeply quiescent HSCs has been found to cycle on average once every 145–193 days [4,5]. However, HSCs can rapidly respond to cues produced by the environment following stress or damage and exit from quiescence to replenish the injured blood system. Finally, with age, HSCs increase in numbers but show severe functional decline [6]. This reduced functionality of old HSCs leads to impaired blood production with characteristic anemia and immunosenescence features, and increased age-dependent incidence of varied blood disorders including BM failure, myeloproliferative neoplasms (MPN) and leukemia [7]. Therefore, one of the most striking features of HSCs is their ability to adapt to the changing needs of the blood system. In addition, each of the different cellular states in HSCs (i.e., quiescence, proliferation and differentiation) imposes a unique set of bioenergetics demands [8]. This review focuses on describing the unique bioenergetics properties of HSCs and the signaling networks that act as metabolic sensors in HSC regulation. It also highlights the major metabolic regulators that have emerged as potential therapeutic targets in blood malignancies.
HSC Biology: A Dynamic Metabolic Landscape
Cellular metabolism involves a tug-of-war between energy-producing catabolic processes and energy-consuming anabolic processes [9]. Transitioning between different cellular states requires HSCs to depend on a flexible balance among these energy processes. Therefore, one of the defining features of HSCs is their metabolic plasticity, which underlies the successful transitions of HSCs from dormancy to activity.
Quiescence - The Anaerobic Bias
Quiescence provides an effective barrier against the onset of blood disorders by preserving the integrity and function of HSCs via limitation of cellular damage from mitochondria respiration and cytotoxic agents, and by preventing HSC exhaustion due to uncontrolled cell cycle entry and excessive proliferation [10, 11]. Accumulating evidence indicates that while HSCs carry higher levels of mitochondria relative to other primitive blood cell types, they are less active, as determined by a lower mitochondrial membrane potential and ATP levels, and greater reliance on anaerobic glycolysis relative to oxidative phosphorylation and the TCA cycle [12–14]. In fact, isolation techniques based on low mitochondrial activity and high endogenous NADH fluorescence can enrich for engrafting HSCs [12], and a recent metabolomic study demonstrating elevated levels of glycolytic intermediates and byproducts such as fructose 1, 6-bisphosphate and pyruvate and relative absence of TCA cycle-related metabolites in quiescent HSCs [14], further supports the idea of a glycolytic bias in HSCs. Anaerobic glycolysis, where pyruvate (the glycolytic end product) is directed towards lactate production, is not an efficient energy-producing process since it generates only 2 ATPs per molecule of glucose as opposed to oxidative phosphorylation, where pyruvate enters the mitochondrial TCA cycle and generates 36 ATPs per glucose molecule [9]. However, it likely suffices for a quiescent population that has relatively low energy demands and a greater need for preventing production of reactive oxygen species (ROS) as byproducts of active mitochondria [15]. Understanding whether enforcement of anaerobic glycolysis in HSCs is the product of niche regulation or the result of cell intrinsic wiring is currently the topic of active research in the field. A series of publications have inspired a model where anaerobic glycolysis is driven by the residence of HSCs in low oxygen pockets in the BM, and adaptation to these ‘hypoxic niches’ remodels the metabolic profile of postnatal HSCs and induces quiescence [12;16–18]. This idea is supported by several lines of evidence including the mathematical modeling of the BM cavity, which suggests that HSCs reside in areas of low oxygen supply [19]; the enhanced incorporation of pimonidazole (Pimo), which forms adducts with cellular proteins under low oxygen states, in HSCs [12,16,20]; the stable expression of the alpha subunit of hypoxia-inducible transcription factor 1 (HIF-1α), which undergoes proteasomal degradation when oxygen levels exceed 5%, in HSCs [12,20]; and, the in vivo loss of HSCs following injection of tirapazamine, which is a toxin selective for hypoxic cells [16]. However, a comprehensive mapping of the spatial distribution of HSCs in femoral BM cavities using laser scanning cytometry demonstrates that the hypoxic profile of HSCs, based on Pimo incorporation and HIF-1α expression levels, is not related to the localization of HSCs in regions of minimal oxygen supply [21]. This study raises the possibility that HSCs can stabilize HIF-1α through oxygen-independent mechanisms [22,23], which highlights the need for caution when directly correlating these assays with oxygen defects. It also indicates that the glycolytic profile of HSCs is not merely a product of their BM microenvironment, but relies heavily on cell intrinsic mechanisms. In turn, this idea is supported by the fact that HSCs strongly express the CRIPTO protein, an extracellular factor essential for early vertebrate development, which induces expression of many glycolytic enzymes [24]; and, that HIF-1α actively promotes anaerobic glycolysis in HSCs by preventing pyruvate from entering the TCA cycle via up-regulation of several pyruvate dehydrogenase kinases (PDKs) including PDK2 and PDK4 [14]. In addition, HIF-1α levels are regulated by the homeobox transcription factor MEIS1 and deletion of either Hif-1α or Meis1 results in loss of quiescence and HSC dysfunction [12,25]. Altogether, these studies point towards the active cooperation of an array of cell-extrinsic/niche-related and cell-intrinsic mediators to cement a strong glycolytic bias in HSCs. However, it remains to be established whether HSCs solely rely on anaerobic glycolysis for maintenance of quiescence, and how this anaerobic bias is relieved when HSCs shift to a more energy-consuming active state. Recent findings summarized below have started to provide insights into these outstanding questions.
Commitment - Burning Fat to Determine Fate
As HSCs exit quiescence and re-enter the cell cycle, the choice between asymmetric and symmetric divisions constitutes one of the first important decision points governing their fate. Asymmetric division generates two cells with different fates, which allows maintenance of the HSC pool and generation of differentiating progeny during homeostatic blood production. In contrast, symmetric division generates two cells with equivalent fates and can either expand the HSC pool or increase the numbers of differentiating cells in conditions of emergency hematopoiesis. A shifted balance between asymmetric and symmetric HSC divisions is also often associated with diseases conditions including BM failures and leukemic transformation [26]. Strikingly, fatty acid oxidation (FAO) has emerged as a critical determinant of these fate decisions in HSCs. FAO consists of a series of biochemical reactions that result in the progressive shortening of fatty acids and the production of acetyl CoA, which can enter the TCA cycle and generate, via β-oxidation, both NADH and FADH2, and twice as much ATP as carbohydrate metabolism [27]. Members of the peroxisome proliferator-activated receptor (PPAR) family of nuclear receptors are important regulators of FAO [28] and PPARδ deletion or direct pharmacological inhibition of FAO results in HSC loss and concomitant accumulation of committed progenitor cells [29]. This progenitor expansion is due to a decrease in the number of asymmetric divisions with a concomitant increase in differentiation-inducing symmetric divisions, thereby impairing HSC self-renewal activity and promoting HSC exhaustion. The promyelocytic leukemia (PML) tumor suppressor protein, which is also implicated in HSC maintenance [30], is one of the key regulators of the FAO pathway in HSCs [29]. However, the mechanism by which the PML/PPARδ/FAO metabolic axis normally promotes HSC self-renewal and opposes HSC commitment still remains largely unknown. Based on the observation that both PML and FAO can sustain ATP levels in breast epithelial cells that have lost contact with the extracellular matrix [31], it isproposed that the PML/PPARδ/FAO metabolic axis might support asymmetric divisions by supplying enough ATP molecules to HSCs when they lose contact with the BM niche during cell division [29]. However, this possibility remains to be directly tested. Interestingly, PML has also been implicated in the onset of p53-dependent aging through induction of premature senescence [32]. While cellular senescence is not a readily accepted feature of aged HSCs [33,34], a p53-dependent proliferation decline is actually observed in the expanded but functionally deficient pool of old HSCs [35], hence re-enforcing the idea of a functional link between PML, FAO and HSC cell cycle regulation that could go awry with age. However, it remains to be established how PML-mediated FAO regulations control HSC cell cycle activity in both young and old HSCs. Collectively, these results indicate that the FAO pathway influences HSC commitment by allowing asymmetric cell divisions, which might be a more energy demanding fate decision than symmetric cell divisions. In this context, it is interesting to note that FAO blockade results in differentiating symmetric divisions rather than self-renewing symmetric divisions [29], and it is tempting to speculate that a block in FAO might impair anaerobic glycolysis and force HSCs to ‘switch’ to a more energy-efficient metabolic program, such as the TCA cycle, that is known to drive differentiation (as discussed below). While this remains to be tested, it will certainly be interesting to determine which alternate metabolic programs kick in when FAO is blocked. Regardless of the mechanism, it is also tempting to speculate that it might serve as a safeguard against neoplastic transformation, as symmetric self-renewing divisions resulting in large pools of long lived HSCs afford a greater risk for transformation than differentiating divisions leading to an expansion of short-lived progenitor cells [36].
Differentiation - The Oxidative Switch
The transition to a more proliferative state associated with differentiation imposes a unique set of metabolic demands on HSCs. Proliferating cells must not only generate energy but also activate a large number of biosynthetic processes required for cell replication [9]. Conversely, differentiation requires higher energy inputs for maintaining the specialized functions of each mature cell type [15]. The adaptation to a high-energy state relies on the modulation of the mitochondrial network, which is largely mediated via cycles of biogenesis and degradation as well as dynamic fission and fusion events [37]. Although little is currently known about HSCs, differentiated adult cardiomyocytes and neuronal precursors usually display structurally mature mitochondria with a diffuse cytoplasmic organization [38, 39]. Relative to a perinuclear localization, a diffuse configuration supports a better energy exchange and nutrient supply across the various cellular compartments [15]. The structural remodeling of the mitochondrial architecture is accompanied by a propensity for increased replication of mitochondrial DNA to support mitochondrial biogenesis, production of TCA cycle enzymes and electron transport chain subunits, and down regulation of glycolysis enzymes to ‘switch’ to a more efficient mode of ATP production. In fact, the ability of HSCs to differentiate is strictly dependent on their ability to activate mitochondrial oxidative phosphorylation as shown in mice with conditional inactivation of Ptpmt-1 [40]. The PTEN-like mitochondrial phosphatase PTPMT-1 is absolutely required for oxidative phosphorylation and its deletion results in rapid hematopoietic failure due to the inability of HSCs to undergo differentiation-associated divisions both in vivo and in ex vivo culture settings. While there appears to be a clear correlation between mitochondrial metabolism, proliferation and differentiation, the precise mechanistic link is still missing, although several lines of evidence point towards a critical role for changes in cellular redox state and ROS levels in regulating the transitions between HSC self-renewal and differentiation.
Mitochondria – The Middleman of Fate Decision
Mitochondria are the primary sources of ROS, which can not only induce oxidative stress and DNA damage when overproduced, but also act as second messengers and drive changes in cell fate at specific concentrations. In fact, the relative intracellular ROS levels can be used to enrich for ‘primitive HSCs’, with ROS-low HSCs demonstrating quiescence and increased self-renewal potential and ROS-high HSCs displaying significant exhaustion [41]. A study in Drosophila indicates that ROS can directly trigger differentiation in a population of hematopoietic progenitors similar to mammalian myeloid progenitors [42]. The differentiation effect is significantly retarded by the expression of antioxidant scavenger proteins, thereby suggesting a role for developmentally regulated ROS levels in priming progenitor cells for differentiation. The regulatory effects of ROS on the self-renewal and differentiation characteristics of HSCs and other adult somatic stem cells can be largely attributed to direct modulation of gene expression by altering the activity of either redox-sensitive transcription factors such as the p38 mitogen associate protein kinases (MAPKs), the forkhead (FoxO) family members, p53, and the nuclear factor-kappa B (NF-κB) [43–47], or major epigenetic regulators such as histone deacteylases (HDACs) and polycomb group proteins [48].
Additional evidence linking ROS to HSC function also comes from recent studies on autophagy, which is a major cellular recycling mechanism that functions in refreshing and remodeling cells by elimination of damaged organelles such as mitochondria (mitophagy) [49], and hence limits ROS production. Hematopoietic-specific loss of Atg7, a critical autophagy protein, results in the expected increase in mitochondrial numbers and elevated ROS levels in HSCs but is also accompanied by increased proliferation and DNA damage, which is thought to underlie the onset of blood malignancies observed in these mice (discussed below) [50]. It is therefore likely that the accumulation of damaged/dysfunctional mitochondria compromises the ability of Atg7-deficient HSCs to maintain quiescence and contributes to their functional impairment. However, while these effects might indeed be a function of ROS and mitochondria, they may also be the unrelated consequences of loss of autophagy since it is a global recycling mechanism for long-lived proteins and other organelles. In fact, autophagy serves a broader stress response function in HSCs, which allows them to withstand metabolic stress that normally kills other progenitor cells [51]. This response relies on a FoxO3a dependent pro-autophagic program that poises HSCs for rapid induction of autophagy and survival in starved conditions. Hijacking of this stress-response mechanism also contributes significantly to the survival of the expanded pool of dysfunctional old HSCs [51]. Therefore, a more rigorous testing is warranted to parse out the role of mitophagy in HSC biology especially for its eventual role in modulating anaerobic glycolysis and FAO, and thereby controlling HSC quiescence and self-renewal.
The need for fine-tuning the cellular redox state of HSCs also warrants the presence of stringent mechanisms for regulating ROS levels and mitochondrial fitness. A recent study implicates a possible role for Connexin-43, a component of gap junctions, in negatively regulating ROS levels in HSCs following myeloablative stress [52]. Connexin-43 serves a detoxification role by transferring endogenous ROS to the neighboring BM stromal cells, and is also critical in mediating cell contact between HSCs and stromal cells. Therefore, it will be interesting to assess whether Connexin-43 directly regulates steady state ROS levels and thereby influences HSC fate decisions. Interestingly, members of the Bcl2 family of apoptotic factors have also been implicated in regulation of HSC differentiation via modulation of mitochondrial function and ROS production [53, 54]. In addition to acting as anti-apoptotic proteins, Mcl-1 and Bcl-xL localize to the mitochondrial matrix and facilitate basic mitochondrial functions such as fission/fusion and oxidative respiration [55–58]. In this context, it will be interesting to test whether changes in their ability to drive ROS production underlie the differential function of Bcl2 family members in HSC differentiation, especially for Mcl-1, which has been shown to be critical for myeloid differentiation [55, 58]. It will also be important to test whether Bcl2 family members affect other aspect of mitochondrial energetics such as NAD/NADH ratio, iron production and calcium homeostasis, which could also play a role in regulating HSC fate decisions.
Taken together, these recent studies point towards a rheostat model where low levels of ROS in resting HSCs could suppress commitment to differentiation, while an increase in ROS levels induced by activation of mitochondrial oxidative phosphorylation could engage key differentiation programs. Studies assessing the interplay between ROS generation and function in purified quiescent, proliferating and differentiating HSCs will be required to further test these intriguing but exciting possibilities.
HSC Bioenergetics – Emerging Paradigms
Recent studies have helped establish that metabolic programs are critical drivers of HSC fate decisions rather than passive consequences of changes in HSC cellular states. Based on these findings, we can start constructing a model of HSC bioenergetics with a ‘ground state’ geared towards prevention of ROS production and maintenance of low ATP levels through anaerobic glycolysis, which allows to maintain HSC quiescence; and an ‘active state’ geared towards production of high ATP and ROS levels through the FAO and TCA cycle, which allows for commitment to differentiation by asymmetric cell division (Figure 1). Impairment or absence of these energy generating programs preferentially drives symmetric divisions, thereby triggering either HSC or progenitor expansion. These differential energy programs thus provide a dynamic platform for HSCs to alternate between these varying fates and meet the shifting needs of the blood system. Moreover, new insights have been gained into the signals that facilitate the transitions between these metabolic programs, and fine-tuning of mitochondrial energetics and ROS levels is emerging as one of the important drivers for differentiation.
Figure 1.
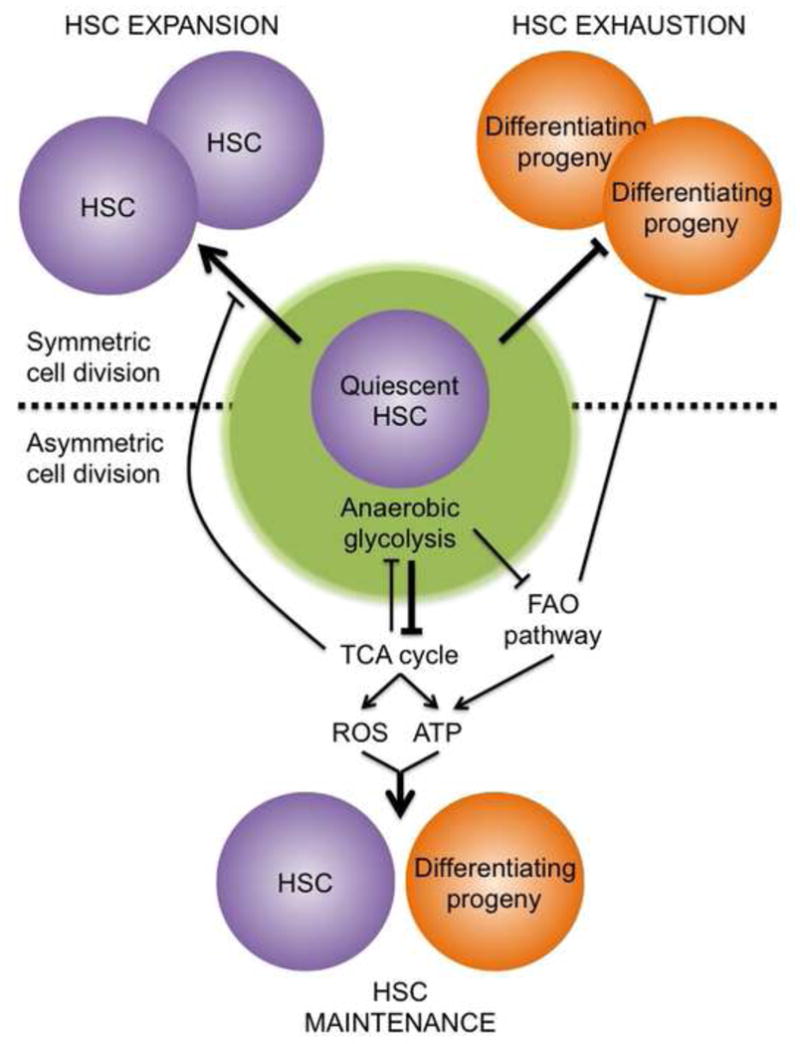
Metabolic drivers as determinants of HSC cell fate.
The ‘ground state’ of adult HSCs is a ROS-low and ATP-low quiescent state, which is enforced by anaerobic glycolysis. Activation of energy generating metabolic pathways such as the TCA cycle and FAO pathway produce high ATP and ROS levels, which drive an ‘active state’ where HSCs are pushed towards commitment and differentiation through asymmetric cell divisions. Blockade of energy generating metabolic programs triggers symmetric cell divisions, which either expand HSCs or their progeny. A shifting balance between these different metabolic programs likely contributes to HSC function in normal, regenerative and disease conditions.
Bioenergetics Sensors: The “First Responders” to Metabolic Changes
A high degree of crosstalk exists between cellular metabolism and signaling networks. Information about nutrient availability is first relayed through metabolic sensors and then channeled via a complex network of signaling pathways to ultimately impact cell fate decisions. Consequently, it is not surprising that the link between signaling mechanisms and metabolic sensors heavily influences HSC biology.
The PI3K/mTOR Axis – The Master Integrator
The mammalian target of rapamycin (mTOR) is a master sensor that integrates cues from nutrients and growth factors to regulate multiple processes such as translation, autophagy, and mitochondrial biogenesis [59]. mTOR exists in two distinct complexes, mTORC1 and mTORC2, which are defined by their respective scaffolding proteins RAPTOR and RICTOR, and its activity is positively regulated by the phosphatidylinositol 3-kinase (PI3K) signaling cascade. Activation of PI3K through tyrosine kinase receptors and other mechanisms results in the downstream activation of AKT, which in turn phosphorylates and inactivates the Tuberous Sclerosis Complex (TSC), a negative regulator of mTOR. On the other hand, the tumor suppressor phosphatase and tensin homolog (PTEN) negatively regulates PI3K [60] and therefore suppresses mTOR activity. Various genetic models have implied a key role for the PI3K/mTOR axis in regulating adult hematopoiesis either by loss-of-function alleles of Pten or Tsc1 or gain-of function mutations in Akt, which all result in increased HSC proliferation and depletion [61–65]. Conversely, Akt1/2 deletion promotes increased HSC quiescence [66]. A clear indication that the effect of these pathways in HSCs is mediated through metabolic perturbations comes from the Tsc1 and Akt1/2 deficiency models [63,66]. Akt1/2-deficient HSCs demonstrate increased quiescence and a differentiation defect which is rescued by pharmacologically increasing ROS levels. Conversely, mTOR hyper activation by Tsc1 deletion results in a rapid exit from quiescence and cell cycle entry, which is accompanied by an increase in mitochondrial biogenesis and ROS levels. These observations suggest a model where mTOR activation could promote mitochondrial activity and ROS generation, thereby supporting a proliferative and differentiation program, and lending support to the ROS rheostat model proposed above. Moreover, as a negative regulator of autophagy, mTOR activation could exert these effects via autophagy suppression, which in turn could also result in deregulated mitochondrial clearance and ROS generation. Furthermore, this might explain why RICTOR is dispensable for quiescent HSCs but is required for proliferation [67]. Additionally, mTOR inhibition by rapamycin partially rescues the maintenance defect in Pml-deficient HSCs [29], and has been previously shown to increase FAO in other cell types [68] via activation of members of the PPAR family [69]. Therefore, it will be interesting to examine the status of the FAO pathway in HSCs from Tsc1mutants with hyperactive mTOR since decreased FAO and increased symmetric divisions might contributes to HSC depletion in these mice.
LKB1/AMPK – The ATP Bankers
The liver kinase B (LKB1) and its downstream target, AMP-activated protein kinase (AMPK), are switched on during periods of low cellular ATP and respond by turning on catabolic processes like FAO, aerobic glycolysis and autophagy to restore energy levels [70]. Similarly, anabolic processes are turned off, at least in part, by inactivation of mTORC1. Studies examining the role of LKB1 in HSC maintenance consistently reveal a loss in quiescence accompanied by functional exhaustion of HSCs, which is demonstrated to be largely independent of AMPK and mTORC1 activity [71–73]. Moreover, unlike for Pten deficiency, the HSC depletion in Lkb1 null mice is not rescued by scavenging ROS, but is accompanied by a decrease in mitochondrial numbers and potential, and reduced ATP levels. These results indicate a ROS-independent route to mitochondrial dysfunction that might result from decreased expression of PGC-1α and β, which are master transcriptional regulators of mitochondrial biogenesis [71]. Since PGC-1 α/β are the principal co-activators for PPAR family members, it is tempting to speculate that Lkb1 deficiency might negatively impact HSC quiescence and activity by reducing FAO through its actions on the PML/PPARδ/FAO metabolic axis. Interestingly, the decrease in mitochondrial fitness but not HSC depletion, is phenocopied in Amkp deficient mice [73], and both catalytic subunits of AMPK (Prkaa1 and 2) are highly expressed in HSCs [51], leading to the possibility that AMPK might play a cooperative role with LKB1 in regulating mitochondrial fitness in concert with other kinases downstream of LKB1. In addition, since the LKB1/AMPK axis is a potent activator of autophagy, through mTOR-dependent and independent pathways [74, 75], it is possible that the altered metabolic features (particularly the changes in mitochondrial mass and decreased ATP levels) seen in the absence of LKB1 and AMPK could also be due to decreased levels of autophagy. However, it remains to be determined whether a defect in autophagy contributes to the HSC phenotypes in Lkb1 and Amkp deficient mice.
HSC Metabolic Sensors – Emerging Paradigms
Recent studies have helped establish that metabolic sensors translate information on cellular energy levels into HSC biological responses essentially via two primary signaling modules – ROS production and FAO modulation (Figure 2). The LKB1/AMPK axis primarily senses low energy levels and responds mainly via FAO activation to regulate HSC quiescence and self-renewal. On the other hand, the PI3K/mTOR signaling network, which is downstream of receptor tyrosine kinase activation, impacts HSC biology largely through modulation of ROS levels and mainly drive proliferation and commitment. However, both sets of metabolic sensors have a considerable degree of cross-regulations and impinge on each other to influence the overall biology of HSCs.
Figure 2.
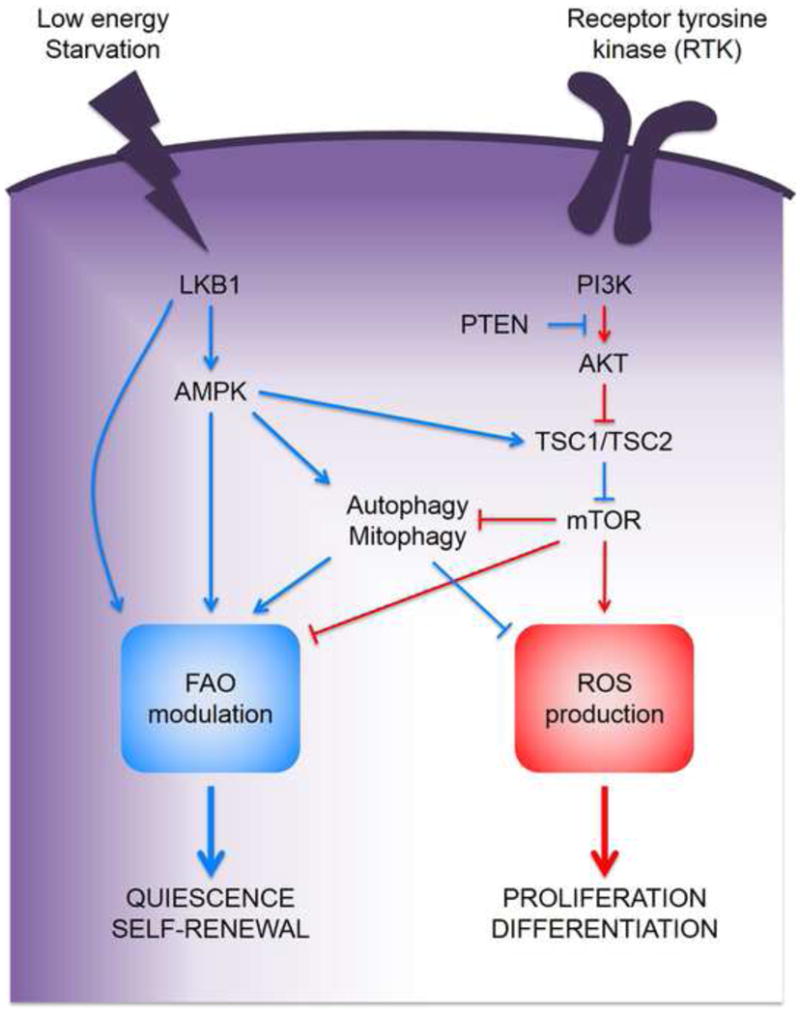
Cross-antagonistic and self-re-enforcing metabolic signaling in HSCs.
Upon activation by receptor tyrosine kinases (RTKs), the PI3K-mTOR axis drive HSC proliferation and differentiation by increasing ROS production and repressing the FAO signaling module. In contrast, low energy levels activate the LKB1/AMKP pathway that promote HSC quiescence and self-renewal via FAO modulation and inhibition of the ROS signaling module.
Metabolic Reprograming: An Escape Route for Leukemic Stem Cells
An altered balance between quiescence, self-renewal and differentiation underlies the onset of most blood malignancies. Myeloid disorders such as chronic or acute myeloid leukemia (CML and AML) are clonal and develop from transformed HSCs with leukemia-initiating stem cell (LSC) properties and unrestricted self-renewal and expansion potential [76]. LSCs are thought to be the drivers of leukemia initiation and therapeutic resistance, and therefore the focus of intense research to try to eradicate them [77]. Recent studies on HSC metabolism have provided new insights into the potential link between metabolic dysfunction in HSCs and the onset of blood malignancies, and opened new avenues for anti-LSC therapies.
The longstanding concept of the ‘Warburg effect’ implies an increased dependence of cancer cells on anaerobic glycolysis, even in the presence of oxygen. While this may hold true for most tumor types, LSCs in AML have an increased dependence on mitochondrial respiration for their survival [78]. In fact, ‘quiescent’ LSCs fail to ramp up or utilize the glycolytic machinery when mitochondrial energetics is blocked. This is in stark contrast with normal HSCs and offers a valuable therapeutic opportunity for specifically targeting the ‘rogue’ stem cells in leukemia. The PML/PPARδ/FAO pathway offers an exciting opportunity in this regard. Targeting PML with compounds such as arsenic trioxide pushes LSCs out of quiescence and induces cell cycle entry [30], which suggests that FAO inhibitors could be used to promote LSC exhaustion in combination with chemotherapeutics targeting proliferating cells [29]. In fact, while mitochondrial function is essentially intact in leukemic cells, ATP synthesis is uncoupled from oxygen consumption, which leads to decreased pyruvate entry into the TCA cycle and increased oxidation of alternate carbon sources such as fatty acids [79]. This increased dependence on FAO has already been exploited to sensitize leukemic cells to apoptotic inducers [79]. However, these studies were performed on bulk populations and it remains to be seen if similar dependencies exist in LSCs. Furthermore, given the importance of the FAO pathway for normal HSC differentiation, it will also be important to establish how LSCs differ from HSCs in their responseto FAO pathway modulation. Mitochondrial function and ROS levels are also being exploited as attractive modulators of LSC function and survival. Low ROS levels and energy metabolism rates now characterize the majority of functionally defined LSCs in AML [78], which also display high expression levels of pro-survival Bcl2 genes as already found in normal HSCs [80]. However, these ROS-low LSCs fail to utilize glycolysis when mitochondrial oxidative respiration is inhibited, thereby making their survival uniquely dependent on intact mitochondrial function [78]. Interestingly, this study has identified a novel cytotoxic role for Bcl2 inhibition, which relies on blocking oxidative phosphorylation and selectively inhibiting energy generation in the ROS-low LSC population thereby triggering cell death. This sheds light on the existence of important crosstalk between commonly studied regulators of cell survival and the unique metabolic machinery in LSCs that could provide selective therapeutic targets. Going forward, the dependence of LSCs on oxidative phosphorylation could be exploited by identifying the targetable mitochondrial enzymes that underlie the reliance of LSCs on mitochondrial oxidative pathways. ROS modulation has also emerged as one of the key drivers for the therapeutic benefit of differentiation strategies. For instance, iron deprivation therapy, which decreases leukemic blasts by promoting increased mature monocytic differentiation, relies on the generation of ROS by iron chelators to trigger differentiation in a JNK dependent manner [81]. It now remains to be determined whether other therapies influencing differentiation use a similar mechanism and whether the differentiation effect is solely mediated by ROS or requires cooperation with other, still unknown mediators. In addition, oxidative damage is thought to contribute to an invasive MPN with AML features in Atg7-deficient mice [50]. However, while the MPN is transplantable, it does not cause lethality in secondary hosts, which is proposed to reflect a need for autophagy in transformed cells after the malignancy is established. Although the diagnosis of frank leukemia vs. deregulated myeloid cell production remains to be formally established, these findings might have interesting implications for targeting the autophagy pathway, especially as a means to curtail oxidative damage or preserve mitochondrial function. Moreover, they underscore the need to assess the shifting autophagic dependence of LSCs through various stages of cancer progression. Lastly, modulating the PI3K/mTOR axis may provide a way to selectively deplete LSCs while sparing normal HSCs. Conditional deletion of Pten leads to leukemogenesis but also promotes HSC depletion in a mTOR-dependent manner, with rapamycin not only depleting LSCs but also restoring normal HSC function [61, 67, 82]. Interestingly, conditional deletion of Pten from fetal HSCs does not trigger leukemia onset until around 8 weeks of age [83], presumably due to the fact that PTEN does not regulate the PI3K/mTOR axis in fetal HSCs [67]. However, the mechanisms governing the differences between fetal and adult HSCs remain to be identified and could lead to different treatment strategies for childhood and adult leukemia. Overall, these studies emphasize the central role of the metabolic machinery in the maintenance and survival of ‘rogue’ stem cells, and exemplify how their unique metabolic requirement could be successfully targeted to selectively target or kill LSCs (Figure 3).
Figure 3.
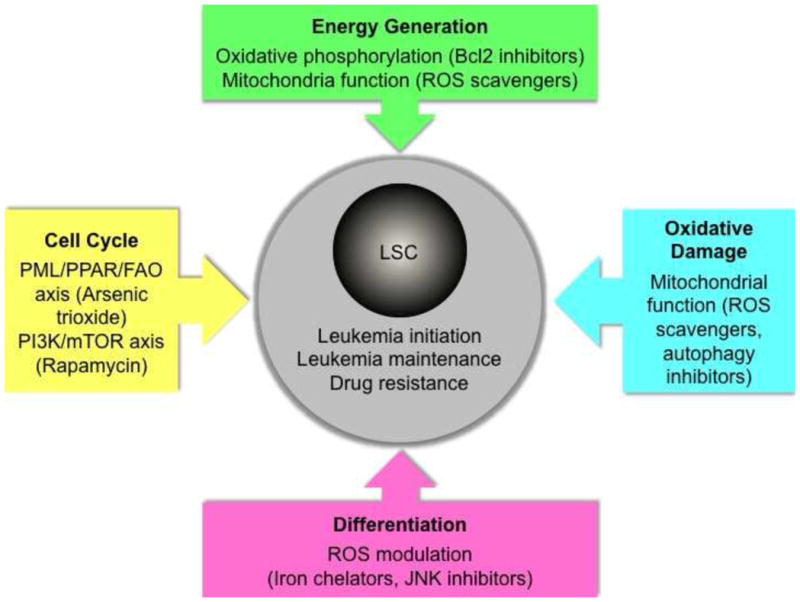
Targeting the metabolic machinery in LSCs.
LSCs are major drivers of leukemia initiation and maintenance and underlie drug resistance. Therapeutic strategies aimed at inducing cell cycle entry, disrupting energy generation, attenuating oxidative damage and promoting differentiation have been proposed as ways to selectively target or kill LSCs while sparing normal HSCs.
Concluding remarks
The rapidly emerging field of HSC metabolism has added a new dimension to our understanding of the unique adaptations that contribute to the success story of a resilient stem cell population that must constantly contend with change. Recent works have provided novel insights into the metabolic responses favored by HSCs at different stages of their life cycle. Additional studies are now required to dissect the mechanistic underpinnings of these responses as well as to understand the metabolic differences that underlie the proliferative phenotype of fetal HSCs vs. the quiescent features of adult HSCs, and to develop better therapeutic strategies for blood disorders. In fact, attacking the altered metabolic energetics of LSCs might prove to be the Achilles heel of leukemia. Finally, we still have an incomplete picture of the changes occurring with age in the HSC compartment, and future studies will help gain an understanding of the metabolic framework of old HSCs that could be used for rejuvenation purpose and anti-aging therapies.
Highlights.
A dynamic metabolic state underlies fate transitions in HSCs
A glycolytic bias enforces quiescence and fatty acid oxidation influences commitment
Mitochondria and ROS control the balance between self- renewal and differentiation
Metabolic energetics offer new strategies to target blood malignancies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709–20. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Passegué E, et al. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202(11):1599–611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–29. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 5.Foudi A, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27(1):84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–389. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 7.Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132(4):681–96. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 8.Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development. 2013;140(12):2535–47. doi: 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115–28. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 11.Bakker ST, Passegué E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp Hematol. 2013;41(11):915–23. doi: 10.1016/j.exphem.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norddahl GL, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8(5):499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Takubo K, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folmes CD, et al. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11(5):596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parmar K, et al. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104(13):5431–6. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eliasson P, Jönsson JI. The hematopoietic stem cell niche: low in oxygen but a nice place to be. J Cell Physiol. 2010;222:17–22. doi: 10.1002/jcp.21908. [DOI] [PubMed] [Google Scholar]
- 18.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9(4):298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Chow DC, et al. Modeling pO2 Distributions in the Bone Marrow Hematopoietic Compartment. II. Modified Kroghian Models. Biophys J. 2001;81(2):685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takubo K, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 21.Nombela-Arrieta C, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15(5):533–43. doi: 10.1038/ncb2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sandau KB, et al. Regulation of the hypoxia-inducible factor 1alpha by the inflammatory mediators nitric oxide and tumor necrosis factor-alpha in contrast to desferroxamine and phenylarsine oxide. J Biol Chem. 2001;276(43):39805–11. doi: 10.1074/jbc.M107689200. [DOI] [PubMed] [Google Scholar]
- 23.Haeberle HA, et al. Oxygen-independent stabilization of hypoxia inducible factor (HIF)-1 during RSV infection. PLoS One. 2008;3(10):e3352. doi: 10.1371/journal.pone.0003352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miharada K, et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell. 2011;9(4):330–44. doi: 10.1016/j.stem.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 25.Kocabas F, et al. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood. 2012;120(25):4963–72. doi: 10.1182/blood-2012-05-432260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu M, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1(5):541–54. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227–32. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takahashi S, Tanaka T, Sakai J. New therapeutic target for metabolic syndrome: PPARdelta. Endocr J. 2007;54(3):347–57. doi: 10.1507/endocrj.kr-99. [DOI] [PubMed] [Google Scholar]
- 29.Ito K, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–8. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito K, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453(7198):1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carracedo A, et al. A metabolic prosurvival role for PML in breast cancer. J Clin Invest. 2012;122(9):3088–100. doi: 10.1172/JCI62129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vernier M, et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011;25(1):41–50. doi: 10.1101/gad.1975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8(9):703–13. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 34.Attema JL, et al. Hematopoietic stem cell ageing is uncoupled from p16 INK4A-mediated senescence. Oncogene. 2009;28:2238–2243. doi: 10.1038/onc.2009.94. [DOI] [PubMed] [Google Scholar]
- 35.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 37.Xu X, et al. Mitochondrial regulation in pluripotent stem cells. Cell Metab. 2013;18(3):325–32. doi: 10.1016/j.cmet.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Facucho-Oliveira JM, et al. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci. 2007;120(Pt 22):4025–34. doi: 10.1242/jcs.016972. [DOI] [PubMed] [Google Scholar]
- 39.Suhr ST, et al. Mitochondrial rejuvenation after induced pluripotency. PLoS One. 2010;5(11):e14095. doi: 10.1371/journal.pone.0014095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu WM, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12(1):62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461(7263):537–41. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ito K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–51. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 44.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Le Belle JE, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8(1):59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asai T, et al. The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J Cell Physiol. 2011;226(9):2215–21. doi: 10.1002/jcp.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12(86) doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doyle K, Fitzpatrick FA. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2 and 3 and antagonizes their transcriptional repressor function. J Biol Chem. 2010;285(23):17417–24. doi: 10.1074/jbc.M109.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joshi A, Kundu M. Mitophagy in hematopoietic stem cells: the case for exploration. Autophagy. 2013;9(11):1737–49. doi: 10.4161/auto.26681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mortensen M, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208(3):455–67. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warr MR, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494(7437):323–7. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taniguchi Ishikawa E, et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc Natl Acad Sci USA. 2012;109(23):9071–6. doi: 10.1073/pnas.1120358109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maryanovich M, et al. The ATM-BID pathway regulates quiescence and survival of hematopoietic stem cells. Nat Cell Biol. 2012;14(5):535–41. doi: 10.1038/ncb2468. [DOI] [PubMed] [Google Scholar]
- 54.Maryanovich M, Gross A. A ROS rheostat for cell fate regulation. Trends Cell Biol. 2013;23(3):129–34. doi: 10.1016/j.tcb.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Opferman JT, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307(5712):1101–4. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 56.Chen YB, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner mitochondrial potential. J Cell Biol. 2011;195(2):263–76. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alavian KN, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1F0 ATP synthase. Nat Cell Biol. 2011;13(10):1224–33. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perciavalle RM, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. 2012 doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(20):3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–96. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 61.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 63.Chen C, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gan B, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105(49):19384–9. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kharas MG, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115(7):1406–15. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Juntilla MM, et al. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magee JA, et al. Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell. 2012;11(3):415–28. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown NF, et al. The mammalian target of rapamycin regulates lipid metabolism in primary cultures of rat hepatocytes. Metabolism. 2007;56:1500–1507. doi: 10.1016/j.metabol.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 69.Sengupta S, et al. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468(7327):1100–4. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- 70.Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010;6(3):457–70. doi: 10.2217/fon.09.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gan B, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468(7324):701–4. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gurumurthy S, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468(7324):659–63. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468(7324):653–8. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Egan D, et al. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–4. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Passegue E, et al. Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11842–9. doi: 10.1073/pnas.2034201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355(12):1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 78.Lagadinou ED, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–41. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Samudio I, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142–56. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mohrin M, et al. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7(2):174–85. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Callens C, et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med. 2010;207(4):731–50. doi: 10.1084/jem.20091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalaitzidis D, et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11(3):429–39. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guo W, et al. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008;453(7194):529–33. doi: 10.1038/nature06933. [DOI] [PMC free article] [PubMed] [Google Scholar]