

Abstract
Human cystathionine β-synthase (hCBS) is a key enzyme of sulfur amino acid metabolism, controlling the commitment of homocysteine to the transsulfuration pathway and antioxidant defense. Mutations in hCBS cause inherited homocystinuria (HCU), a rare inborn error of metabolism characterized by accumulation of toxic homocysteine in blood and urine. hCBS is a complex multidomain and oligomeric protein whose activity and stability is independently regulated by the binding of S-adenosyl-methionine (SAM) to two different type of sites at its C-terminal regulatory domain. Here we study the role of surface electrostatics on the complex regulation and stability of hCBS using biophysical and biochemical procedures. We show that the kinetic stability of the catalytic and regulatory domains is significantly affected by the modulation of surface electrostatics through noticeable structural and energetic changes along their denaturation pathways. We also show that surface electrostatics strongly affect SAM binding properties to those sites responsible for either enzyme activation or kinetic stabilization. Our results provide new insight into the regulation of hCBS activity and stability in vivo with implications for understanding HCU as a conformational disease. We also lend experimental support to the role of electrostatic interactions in the recently proposed binding modes of SAM leading to hCBS activation and kinetic stabilization.
Keywords: conformational disease, homocysteine metabolism, protein kinetic stability, ligand binding, allostery, surface electrostatics
1. Introduction
Human cystathionine beta-synthase (hCBS) is a key regulator of sulfur amino acid metabolism, catalyzing the β-replacement of the hydroxyl group of L-serine by the thiolate group of homocysteine using pyridoxal 5′-phosphate (PLP) as a cofactor. Its activity is essential to control the flux of sulfur from methionine to cysteine, redox regulation, methyl-transfer reactions and H2S production [1, 2]. hCBS displays a complex architecture, forming functional homotetramers, and each monomer containing three structural domains: i) a short heme-binding N-terminal domain (residues 1-70), which is thought to modulate CBS activity and proper folding; ii) a catalytic domain (CD; residues 71-413) which contains the catalytically active PLP molecule bound to Lys119 through a Schiff base; iii) a C-terminal regulatory domain (RD), which contains two types of SAM binding sites involved in SAM mediated enzyme activation and kinetic stabilization of hCBS [1, 3, 4]. Over 160 mutations in the CBS gene have been found to cause classical homocystinuria (HCU) ([5] and http://medschool.ucdenver.edu/krauslab). Missense mutations often lead to protein instability and impaired catalytic activity and regulation [1, 4, 6-9]. Some of the mutations responsible for HCU were modeled based on the structure of the truncated CBS [10, 11], in some cases providing a structural rationale for loss-of-function [12]. More recently, we have determined the crystal structure of an optimized dimeric full-length hCBS construct lacking 10 residues in the regulatory domains [3], that provides a structural framework to explain SAM mediated regulation of hCBS function and stability. However, comprehensive structural-energetic studies on major forces contributing to hCBS stability as well the impact of disease-causing mutations on the thermodynamic and kinetic stability of hCBS are scarce [4, 6, 7].
In a recent calorimetric study, we have shown that hCBS catalytic and regulatory domains denature independently, and display largely different stabilities at physiological pH (with half-lives of about 1 day and three weeks at 37°C; [4]). Binding of SAM to a set of high affinity sites in the RDs strongly enhances their kinetic stability with no effect on the stability of the CDs [4]. HCU-causing mutations affect the kinetic stability of the RDs even if they occur within the CD, suggesting some communication between domains [4]. However, disease-causing mutations may affect SAM mediated activation of hCBS without distorting SAM thermodynamic binding properties to a large extent, suggesting that transmission of the SAM mediated allosteric signals is subtle from thermodynamic [4] and structural [3] viewpoints. SAM binding to the high affinity sites might be also of importance to understand ligand-mediated regulation of CBS steady-state levels in vivo and to prevent misfolding of HCU-causing mutants [4, 13].
The role of electrostatic interactions in a wide set of protein properties such as catalytic performance, regulation and stability is well recognized [14-19]. Particularly, surface charge-charge interactions have been shown to be important for protein thermodynamic and kinetic stabilities since they are more frequent in the native state [15, 18-23]. Interestingly, evolution seems to have tuned electrostatic interactions in proteins to fit the biologically relevant conformational stability and folding barriers to protein function [15, 24]. Particularly, surface charge-charge interactions contribute to the adaptation of proteins to high temperature or high saline environments [15, 25]. One of the most relevant features of surface charge-charge interactions is the Debye-Hückel screening exerted by salt ions that accumulate around surface charges thus strongly weakening electrostatic forces between ionizable residues of the protein (either attractive or repulsive interactions). Accordingly, protein thermodynamic and kinetic stabilities [21] as well as protein folding barriers [26] can be efficiently modulated through electrostatic screening of surface charges. Salt effects on protein stability vary according to their concentration (i.e. the ionic strength) and in some cases, on the nature of the ions. At low ionic strength (typically below 0.2 M of monovalent salt), salt ions may interact with surface charges of opposite sign causing changes in protein stability that exponentially depend on the ionic strength, even though other mechanisms such as ion specific binding may also be involved. At high ionic strength, salt ions may affect protein stability by perturbing the hydration shell of the protein (which is ion dependent, according to the Hofmeister series) and this effect scales linearly with salt concentration [21, 26, 27].
We have recently characterized the effect of SAM on the stability of hCBS (ionic strength ~10 mM; [4]). Here, we explore the role of electrostatic interactions in the function, regulation and stability of hCBS by studying these properties in the presence of different salts. We show that biologically relevant properties of hCBS, such as its kinetic stability, enzyme activity and SAM-mediated allosteric modulation are strongly affected by screening of surface electrostatic interactions. We thus provide insight into the function and regulation of hCBS in health and disease, and further structural/thermodynamic information underlying SAM mediated activation and stabilization of hCBS.
2. Materials and Methods
2.1 Protein samples
Human CBS (hCBS) was expressed and purified as previously described [9] and buffered exchanged to 20 mM Hepes pH 7.4 using PD-10 columns (GE Healthcare). Protein samples were concentrated to ~200 μM in subunit, flash frozen in liquid nitrogen and stored at −80°C. Protein concentration was measured by UV-absorption using ε280nm of 1.038·105 M−1·cm−1 [4]. To perform experiments in the presence of varying concentrations of salt, solutions of NaCl, CsCl and NaBr were prepared at 2 M in 20 mM Hepes pH 7.4.
2.2 Spectroscopic analyses
UV-visible absorption spectra were acquired using an Agilent 8453 diode array spectrophotometer, using 20 μM protein subunit in a 3 mm path length cuvette. Circular dichroism (CD) spectra were obtained using a Jasco J-715 spectropolarimeter, using 1 mm (Far-UV) and 5 mm (Near-UV/visible) path length cuvettes and 5 μM (Far-UV) and 20 μM (Near-UV/visible) protein concentration in subunit. Dynamic light scattering (DLS) measurements were carried out in a DynaPro MSX instrument (Wyatt) using a 1.5 mm path length cuvette and 10 μM protein in subunit. The hydrodynamic radius and polydispersity of the samples in DLS measurements were obtained from the autocorrelation function using the software supplied by the manufacturer and assuming a spherical shape for the protein. All experiments were performed at 25°C. Samples were prepared in 20 mM Hepes pH 7.4 and 0-1 M NaCl.
2.3 Differential scanning calorimetry
Differential scanning calorimetry (DSC) analyses were performed in a capillary VP-DSC microcalorimeter (GE Healthcare) with a cell volume of 0.135 ml. Scan were routinely performed in a 4-100°C range at 2-4°C scan rate. Samples were prepared in 20 mM Hepes pH 7.4 and 0-1 M NaCl, containing 5 μM protein concentration (per CBS subunit), 50 μM PLP and 0-100 μM SAM. For some experiments, urea was added to a final concentration up to 1.5 M and its concentration was determined by refractive index measurements.
DSC profiles were analyzed using a model including two independent thermal transitions, each of them described by a two-state irreversible model with first-order kinetics [4]:
| (Scheme 1) |
where N holds for native state and F stands for the final state which cannot fold back to N, and k is a first-order rate constant. These fittings provide, for each thermal transition, the values of the half-denaturation temperature (Tm), the denaturation enthalpy (ΔH) and the activation energy (Ea)[4]. The rate constants (k) are determined from Arrhenius plots built using the experimental DSC profiles and equation 1:
| (Equation 1) |
Where Cp(exc) and <H> stand for the excess heat capacity and the excess enthalpy at each temperature, and τ and ΔH for the scan rate and the calorimetric enthalpy, respectively. These Arrhenius plots are used to extrapolate the rate constants to 37°C.
The changes in activation free energy (ΔΔG), enthalpy (ΔΔH) and entropy (–TΔΔS) are calculated based on the transition state theory as described [28, 29] using equations 2-4:
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
To determine the contributions to activation energies (≈enthalpies) from unfolding and solvation barriers, we determined the kinetic m values (mǂ) from the urea concentration dependence of the Tm and Ea values according to [29], using the following expression:
| (Equation 5) |
Tm and Ea values in the absence of urea were used at each scan rate, and the values of dTm/d[urea] and dln(Ea/RTm2)/d[urea] values obtained from their dependence on urea concentrations. mǂ values are the means±s.d. from three experimental scan rates.
The contributions from unfolding (ΔHUNFǂ) and solvation barriers (ΔH*) to activation enthalpies were determined using equations 6-7 [28-30]:
| (Equation 6) |
| (Equation 7) |
The contributions of ΔHUNFǂ and ΔH* has been determined using meq, either calculated using the total change in accessible surface area (ΔASA) upon unfolding expected for the regulatory and catalytic domains based on their number of residues or the crystal structure of hCBS [3] and using the correlations of [31] and [32] and the experimental values of ΔH. In either case, denaturation of four regulatory or catalytic domains have been considered since hCBS tetramer does not dissociate prior to the rate-limiting step of irreversible denaturation [4]. The structural changes associated to the transition between the native and transition states are calculated from the average values for ΔHUNFǂ, and ΔH* are estimated from the relation between these two parameters and ΔASAtotal described by [31] and [29], respectively.
2.4 Isothermal titration calorimetry (ITC)
ITC experiments were performed in a VP-ITC200 microcalorimeter in 20 mM Hepes pH 7.4 with or without 200 mM NaCl. In the absence of salt, hCBS (8 μM in protein tetramer) was titrated by performing 50 stepwise injections (0.75 μL) of SAM (0.7 mM) spaced 150 s. In the presence of salt, hCBS (20 μM in protein tetramer) was titrated by performing 19 stepwise injections (2 μL) of SAM (1.06 mM) spaced 250 s. Analysis of the binding isotherms was performed using the software provided by the manufacturer as recently described [4].
2.5. Activity measurements
The CBS activity in the classical reaction was determined by a previously described radioisotope assay using L-[U-14C]serine as the labeled substrate [33]. CBS enzyme (420 ng) was assayed in a 100 μl reaction mixture for 30 min at 37°C. The reaction mixture contained 20 mM Hepes pH 7.4, 0-0.2 M NaCl, NaBr or CsCl, 0.2 mM PLP, 10 mM L-serine, 0.3 μCi L-[U-14C]serine and 0.5 mg/ml BSA. The reaction was initiated by the addition of homocysteine to a final concentration of 10 mM following a 5 min incubation at 37°C. One unit of CBS specific activity was defined as the amount of CBS that catalyzes the formation of 1 μmol of cystathionine in 1 h at 37°C under standard assay conditions.
3. Results
3.1 Effect of salt on the conformation and function of hCBS
To determine the effect of salt on the conformation of hCBS, we have used a battery of spectroscopy techniques. First, we have evaluated the oligomeric status at different salt concentrations by dynamic light scattering (DLS). In the absence of salt, hCBS behaves as a highly monodisperse protein with a hydrodynamic radius of ~5.2 nm (Figure 1A) consistent with a tetrameric structure based on the correlation between hydrodynamic radii and protein size (in number of residues) for globular proteins [34]. Addition of salt has no significant effect on either the hydrodynamic radius or the sample polydispersity, indicating that hCBS remains mainly as a tetramer in the presence of salt. Addition of salt has also negligible effect on the secondary structure content as assessed by Far-UV CD spectroscopy (Figure 1B). The microenvironment of bound chromophores (PLP and heme) has been monitored by UV-visible absorption and circular dichroism spectroscopies (Figure 1C and 1D), also revealing negligible effects on the tertiary structure of hCBS. These studies support that the presence of salt has no noticeable effects on the overall hCBS structure.
Figure 1. Spectroscopic characterization of the hCBS native state in the absence and presence of NaCl.

A) Polydispersity and hydrodynamic radius of hCBS in the presence of different salt concentrations obtained from DLS experiments. B) Far-UV circular dichroism spectra. Inset: molar ellipticity at 222 nm at different NaCl concentrations. C) Near-UV/visible absorption spectra. Spectra in the absence or presence of salt are undistinguishable. Inset: absorption intensities at 429 and 365 nm as a function of NaCl concentration. D) Near-UV/visible circular dichroism spectra. In panels B-D, the lines follows the legend of panel B. Errors in panel A are mean ± s.d. from three different measurements.
We have also characterized the effect of salt on the activity and regulation of hCBS (Figure 2). In the presence of 200 mM of different salts, the specific activity is reduced by 30-40% (Figure 2A). The presence of salt does not prevent hCBS from SAM mediated activation (Figure 2A), but raises the concentration of SAM required for half-activation 4-fold (Figure 2B).
Figure 2. Effect of salt on the activity and regulation of hCBS.

A) Effect of 200 mM NaCl on the specific activity of hCBS in the absence/presence of 300 μM SAM. The extent of SAM mediated activation of hCBS is indicated as a x-fold. B) SAM concentration dependence of hCBS activity in the absence and presence of 200 mM NaCl. SAM concentration required for half-activation are 3.7±0.2 μM (no salt) and 14.4±0.9μM (200 mM NaCl). Note the logarithmic scale of the x-axis. Experiments performed in 20 mM Hepes pH 7.4. Activity measurements were performed in 20 mM Hepes pH 7.4, 10 mM serine and 10 mM homocysteine. Error bars in panel B are S.D. from triplicate experiments.
3.2 Surface charge screening strongly affects the kinetic stability of the regulatory domains of hCBS
We have thus tested whether the presence of a physiological relevant ionic strength (200 mM NaCl) may affect the stability and denaturation mechanism of hCBS using differential scanning calorimetry (DSC). Representative DSC traces are displayed in Figure 3A, showing that the presence of 200 mM NaCl increases the Tm of the RDs and slightly reduces the Tm of the CDs. Denaturation of hCBS in the absence or presence of salt is described well by two independent two-state irreversible denaturation reactions with first-order kinetics (Figure 3B-D, Table 1). Remarkably, in the presence of 200 mM NaCl the kinetic stability of the RDs is increased ~300-fold (half life~100 days) while the stability of CDs slightly decreases (~5-fold, half-life ~8 days) (Table 1).
Figure 3. Thermal stability and denaturation mechanism of hCBS in the absence and presence of 200 mM NaCl.

A) DSC profiles at 3°C/min. B) Scan rate dependence of Tm values of RDs (lower panel) and CDs (lower panel). C) Consistency test for the two-state kinetic model based on the Tm dependence on scan rates [44]. D) Arrhenius plots for the denaturation of RDs and CDs in the absence/presence of 200 mM salt. The dotted vertical line indicates 37°C. In panel B, errors are smaller than data points. For the rest, data are from a single experiment at different scan rates.
Table 1.
Thermal stability parameters for hCBS in the absence or presence of 200 mM NaCl analyzed by a two independent and irreversible two-state denaturation model.
| [NaCl] (mM) |
Regulatory domains | Catalytic domains | ||||
|---|---|---|---|---|---|---|
| Tm (°C)a |
Ea (kcal·mol−1)b |
ΔH (kcal·mol−1)c |
Tm (°C)a |
Ea (kcal·mol−1)b |
ΔH (kcal·mol−1)c |
|
| 0 | 52.5±0.1 | 86±6 | 61±4 | 71.8±0.1 | 64±8 | 167±27 |
| 200 | 56.1±0.1 | 129±15 | 72±9 | 70.7±0.1 | 59±4 | 269±64 |
at 3K/min, three independent experiments.
from the three different consistency tests previously applied [4].
from at least six different experiments at different scan rates and protein concentrations
We have then studied the salt concentration dependence of the kinetic stability of RDs and CDs (Figure 4A). The RDs are strongly stabilized even at relatively low salt concentrations, while the CDs are destabilized but to a lower extent (see changes in Tm in Figure 4B). The activation energies for the denaturation of RDs and CDs are also strongly dependent on the salt concentration (Figure 4C), with a large increase for RDs (which contributes to their large kinetic stabilization) and a small decrease for CDs, suggesting an important role played by surface electrostatics on the energetics of either the native or the TS (transition state) for denaturation of both domains. The increase in ΔH for CDs in the presence of salt (Figure 4D) could be explained by a more extensively unfolded final state.
Figure 4. Dependence of thermal denaturation of hCBS on salt concentration.

A) Thermal denaturation profiles at the indicated NaCl concentrations (in mol/L); B-E) Salt concentration dependence of thermal denaturation parameters (Tm, Ea, ΔH and ln k) on salt concentrations for RDs (open circles) and CDs (closed circles). Experiments performed at 4°C/min. All data are from single DSC experiments. In panels B to D, fitting errors are smaller than the symbols.
The dependence of kinetic stabilities for RDs and CDs on salt concentration is shown in Figure 4E. The presence of salt has opposite effects on the kinetic stabilities of RDs and CDs. Below 0.1 M salt, CDs are more kinetically stable than RD, while the opposite is observed above 0.1 M. The activation free energies for denaturation (which are proportional to ln k in Figure 4E) change exponentially on salt concentration for both domains, as it would be expected if electrostatic screening is modulating the kinetic stability of RDs and CDs. Indeed, DSC experiments performed with CsCl and NaBr (in the Hofmeister series, Cs+ is more kosmotropic than Na+ while Br− is more chaotropic than Cl−) provide similar results to those with NaCl (Figure 5), supporting that electrostatic screening effects play a major role in the modulation of RDs and CDs kinetic stabilities by salt (especially at low salt concentrations). Overall, these results lend support to a modulation of the kinetic stability of RDs and CDs by electrostatic screening of surface charges in the native state and/or the TS of the hCBS domains.
Figure 5. Effect of different monovalent salt ions on thermal denaturation of RDs and CDs.

Salt concentration dependence of the Tm (A) and Ea (B) values for denaturation of RDs (closed symbols) and CDs (open symbols) in the presence of NaCl (triangles), CsCl (squares) or NaBr (circles). In all cases, fitting errors are smaller than the symbols.
3.3 Surface charge screening causes large structural changes in the TS for denaturation of RDs
The remarkable effect of salt on the kinetic stability of RD and CD, prompted us to further investigate their structural and energetic basis. Since the kinetic stability is related to the height of the activation free energy barrier, changes in this barrier in the presence of salt (Figure 4E) may be caused either by changes in the free energy level of the native state or the denaturation TS. These changes in free energy level could imply structural changes either in the presence of salt (which are not experimentally supported for the native state; Figure 1) or by surface charge screening.
To get further insight into the changes in the free energy barriers for denaturation in the presence of salt, we have performed detailed structure-energetic analyses in the presence of low non-denaturing urea concentrations (see Figure S1 and S2 for corresponding control experiments). In the presence of low urea concentrations, denaturation rates for both domains are speeded up (i.e. increased k values). The changes in denaturation free energies on urea concentration (the kinetic m values, mǂ) are determined from the effect of urea on Tm and Ea values of each domain (Figure 6). mǂ values have also a structural interpretation, because they are proportional to the change in solvent accessible surface area (ΔASA) between the folded domain and the transition state of the rate-limiting state (TS, which is placed at the top of the denaturation free energy barrier). The equilibrium m values can be simply approximated from the size and/or the native structure of the domain (Figure S3), and the ratio mǂ /m represents the fractional solvent exposure of the transition state (Figure 7B), thus allowing detection of large structural changes in the TS by salt.
Figure 6. mǂ values are determined from the slopes of panels A and B using Equation 5 [29].
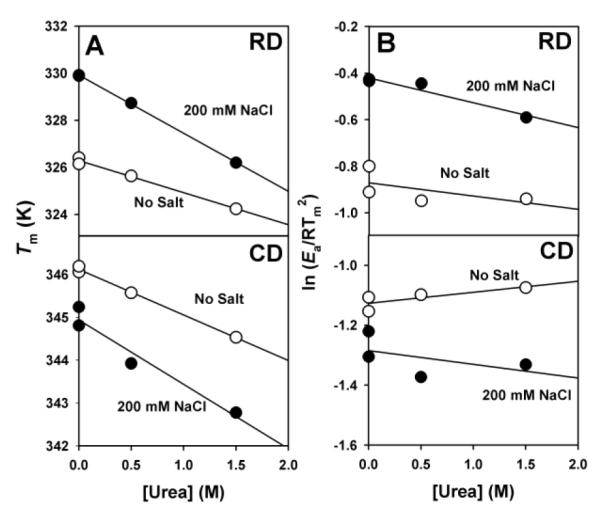
Data are from experiments at a single scan rate (4°C·min−1) and yield the following mǂ values (in kcal·mol−1·M−1): RD (no salt).- 0.41; RD (200 mM NaCl).- 1.12; CD (no salt).- 0.21; CD (200 mM NaCl).- 0.32. The contribution from panel B to mǂ values is residual (lower than 10% of the total mǂ value).
Figure 7. Structural and energetic effects of 200 mM NaCl on the kinetic stability of RDs and CDs.

A) Changes in activation energetic parameters by salt (200 mM – 0 mM NaCl). Errors are those propagated from denaturation rate constants determined at different scan rates (activation ΔΔG), from experimentally determined Ea values at different scan rates (activation ΔΔH) and from those propagated using activation ΔΔG and ΔΔH (activation -TΔΔS). B) Kinetic (mǂ) and equilibrium (meq) m values for the RDs and CDs in the absence and presence of 200 mM NaCl. mǂ values are reported as mean ± s.d. from three different scan rates determined using equation 5; C) energetic and D) structural effects from unfolding and solvation components in the absence and presence of salt. Errors in panels C and D are propagated from experimental errors in mǂ values.
First, we have studied the enthalpic and entropic contributions that lead to changes in the kinetic stability of RDs and CDs in the presence of salt (Figure 7A). The activation free energies are related to changes in activation entropies (-TΔSǂ) and enthalpies (ΔHǂ≈Ea) using the classical thermodynamic relationship (but in activation terms: ΔGǂ=ΔHǂ-TΔSǂ). The stabilization of RDs by 200 mM salt is caused by changes in these two opposing forces that almost cancel out each other: the enthalpic contribution becomes more stabilizing while the entropic contribution is destabilizing. These changes are much smaller for the destabilization of CDs (Figure 7A). The large effects observed for the enthalpic/entropic contributions to the free energy barrier of denaturation of the RDs are difficult to explain solely by effects on the structure of native RDs, even though changes in conformational dynamics cannot be excluded [35]. Thus, we invoke large structural changes on the TS of the RDs.
The calculated mǂ values for RDs and CDs are shown in Figure 7B. Salt has little effect on the mǂ values of CDs, with fractional m∕mǂ values of ~0.015 (no salt) to ~0.02 (200 mM Salt)(Figure 7B), thus showing that the TS for CDs denaturation is native-like in terms of solvent exposure. However, in the presence of 200 mM salt the m∕mǂ values of RDs increases from ~0.06 to ~0.17 (Figure 7B), implying a more unstructured TS in the presence of salt for the RDs. Thus, the free energy barrier for denaturation of RDs in the presence of salt is raised because the number of interactions that must be broken and solvated is larger, thus kinetically stabilizing the domain. Note that, we are considering the CBS tetramer, and a 10% increase in solvent exposure for four regulatory domains may involve large structural differences, in the order of several thousands of Å2. Indeed, analyses of the contributions from unfolding (unfolded and solvated regions in the TS) and solvation (unfolded but not yet solvated regions in the TS)(Figure 7C) barriers clearly show significant structural changes in the TS in the presence of salt for the RDs (Figure 7D).
3.4 Salt effect of SAM binding and stabilization of RDs
SAM binds hCBS at two different types of sites in the absence of salt: a low-affinity site (four per CBS tetramer; Kd~600 nM) that causes CBS activation, and high-affinity site (two per CBS tetramer; Kd~10 nM), that kinetically stabilizes the RDs (Figure 9A and C, Table 2 and [4]). Titrations performed in the presence of 200 mM NaCl reveal a completely different binding pattern (Figure 8B and 8D). Due to the small calorimetric signals, we have performed titrations in the presence of 200 mM NaCl using higher injection volumes, which increase the signal-to-noise ratio but provide less detail in the binding isotherm (Figure 8D). Using this procedure, we systematically observed deviations (over four independent experiments) from a single type of independent sites model at low ligand:protein ratios, supporting the existence of two independent type of sites in the presence of 200 mM NaCl, and this behavior is independent on the buffer used (Figure S4). However, the low number of experimental data points does not provide convergent fittings when all parameters are allowed to float. However, the binding isotherm in the presence of salt is described well by this model when a few of the parameters are fixed (actually fixing those for the high-affinity sites allow convergence of those for the low affinity sites). Thus, in the presence of salt, it seems that the two types of sites are preserved, but salt changes the SAM binding properties (Table 2). The low binding enthalpies in the presence of salt easily explain the difficult detection and characterization of the two types of sites, and the somewhat lower affinity of the high affinity sites could also contribute to this. The apparent Kd values for the low-affinity sites is somewhat higher in the presence of salt (≈2.5-fold; Table 2), in good agreement with the increased in the SAM concentration required for half-activation (≈4-fold; Figure 2B).
Figure 9. Effect of SAM on hCBS thermal denaturation in the absence or presence of 200 mM NaCl.

Thermal denaturation profiles in the absence (A) and presence (B) of salt. The concentrations of SAM are indicated alongside the DSC profiles. C) SAM concentration dependence of the Tm of the RD in the absence (closed circles) or presence of 200 mM NaCl (open circles). Fitting errors are smaller than the symbols. Lines are meant to guide the eye and have no theoretical meaning. D) Plots used to calculate ν (the number of SAM molecules released per CBS tetramer prior to the rate-limiting step of denaturation) values of 1.9±0.1 (no salt; closed circles) and 2.4±0.2 (200 mM NaCl; open circles) using the procedure previously described [4, 45].
Table 2.
Thermodynamic parameters for SAM binding to hCBS in the absence and presence of 0.2 M NaCl. N is expressed as number of SAM molecules bound per tetramer, Kd in nM, and ΔH and −TΔS in kcal·mol−1. The values for the two-types of sites model with 0.2 M are rough estimates obtained by fixing some of the parameters during different rounds of fitting.
|
[NaCl]
(M) |
Site 1 | Site 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| N | Kd | ΔH | −TΔS | N | Kd | ΔH | −TΔS | |
| 0 | 2.10 | 13 | −12.4 | 1.6 | 4.14 | 610 | −6.0 | −2.4 |
| 0.2 | −1.5 | 100 | −2.4 | −7.2 | ~3.5 | 1500 | −3.3 | −4.6 |
Figure 8. Titrations of hCBS with SAM in the absence or presence of 200 mM NaCl by ITC.

A) and B) show raw calorimetric data, while C and D) display binding isotherms, with the corresponding fittings to two (solid line) or one (dashed line) independent type of sites. Experiments were performed using 8.5 μM (no salt) and 20 μM enzyme (200 mM NaCl) calculated per hCBS tetramer and 0.7 mM (no salt) or 1.0 mM SAM (200 mM NaCl) .
Interestingly, the Kd values for the high-affinity sites derived from ITC (Table 2) would imply a lower concentration dependence of SAM mediated stabilization of the RDs. Hence, we have investigated whether SAM mediated kinetic stabilization of the RDs might be also modulated by surface charge screening. SAM binding stabilizes the RDs also in the presence of salt (Figure 9A-B) but its dependence on SAM concentration is slightly lower than in the absence of salt (in terms of Tm up-shift; Figure 9C), consistent with our ITC results. Nevertheless, SAM mediated stabilization of RDs is still consistent with the release of two SAM molecules prior to the denaturation TS (Figure 9D).
4. Discussion
Regulation of the stability and activity of hCBS in vitro and in vivo depends on the binding of its allosteric regulator SAM to the RDs, thus controlling the commitment of methionine to the transsulfuration pathway [4, 13]. In this work we show that the kinetic stability of the RDs and CDs and SAM mediated enzyme activation and stabilization of RDs are strongly modulated by the presence of physiological concentrations of salt, thus allowing for further understanding of the dynamics of hCBS activity in vivo. Salt mediated effects are observed even at low salt concentration and they are largely independent on the nature of salt ions, thus suggesting that these effects are mostly mediated by the ability of salt to screen surface charge-charge interactions. Binding of SAM to two structurally different types of sites at the C-terminal RDs leads to enzyme activation and kinetic stabilization of the regulatory domains [4]. Screening of surface charges does not abolish the function of these two types of sites (Figure 2 and 9), but SAM binding is weaker and display different thermodynamic signatures in the presence of 200 mM NaCl (Table 2). This lower binding affinity explains the higher SAM concentrations required for activation and kinetic stabilization (Figure 2 and 9), and most likely, originates from the screening of favorable interactions between solvent exposed ionizable groups in hCBS and SAM in the protein:ligand complex. A structural rationale for this interpretation is provided by the recently solved crystal structure of a full-length hCBS [3]. In the absence of SAM, two plausible and structurally different types of SAM binding sites (named sites S1 and S2) are found, with potentially favorable electrostatic interactions between SAM and the binding pockets in hCBS: in S1 (which may correspond to the sites responsible for enzyme activation), the carboxylate group of SAM may interact with positively charge side chain of H507, whereas in S2 (which may be those sites leading to kinetic stabilization), favorable interactions could be established between D538 with the ribose of SAM, and an acidic cluster (D444 and E201) with the positively charged sulphonium group in SAM (see Figure 5 in [3]). More generally, charged residues in nucleotide binding sites of CBS domains are known to provide a proper conformation of the binding site and to interact favorably with the nucleotide [36, 37]. These electrostatic (as well as pH effects, see [4]) may help to explain the wide range of binding affinities reported for SAM binding at the regulatory sites of hCBS (this work, [4, 8, 38].
The remarkable effects of surface charge screening on the stability and regulation by SAM may provide insight into physiological and pathological aspects of hCBS. We have previously proposed a relationship between kinetic stability of the RDs (towards thermal denaturation) and hCBS protein turnover inside cells due to their similar kinetics [4]. However, the presence of physiological concentrations of salt dramatically enhance the kinetic stability of RDs (to a half-life~100 days), far exceeding protein turnover kinetics inside cells (half-life~1 day; [13]). Thus, it seems that hCBS turnover inside cells may not be coupled to the kinetic stability of RDs towards denaturation, and thus, regulated by other conformational or dynamic features of hCBS. Interestingly, SAM binding and preactivating disease-associated mutations are known to enhance protein dynamics and increase sensitivity to proteolysis [6, 7]. Therefore, protein dynamics and stability of low-populated conformational states may play a major role in both hCBS activation and protein turnover, as found for other allosteric proteins [35, 39]. SAM binding sites responsible for hCBS activation and stabilization of the RDs are still operational at physiological salt concentrations, but their affinities are somewhat lower (2 to 5-fold, Table 2 and Figure 9), thus shifting the SAM concentration range for response in terms of activation and kinetic stabilization to higher values. In the case of SAM mediated activation, the concentration range of response to SAM (~1-100 μM, with half-maximal activation at 14 μM; see Figure 2) seems to be appropriate to keep homocysteine at low levels under various metabolic conditions, since a very low affinity for these regulatory sites may rise homocysteine levels to pathological levels [40].
Our study also provides insight into the energetic and structural basis for the salt mediated changes in kinetic stability of hCBS, and to our knowledge, hCBS is the largest and most complex protein for which the contribution of unfolding and solvation barriers to protein kinetic stability has been determined so far. Surface charge screening at physiological salt concentrations (200 mM NaCl) causes a remarkable stabilization of RDs (~300-fold; corresponding to a ΔΔG≠~+3.5 kcal·mol−1), while the CDs are slightly destabilized (by ~10-fold, ΔΔG~-1.4 kcal·mol−1). These changes in the denaturation free energy barriers may arise from charge-charge screening in either the native or the denaturation TS for the denaturation. Changes in the native state stability might be estimated from a protein crystal structure using a suitable model for electrostatic interactions, but this approach cannot be used for the TS, for which the high-resolution structure is unknown (for examples, see [15, 20, 21]). Interestingly, the structural changes occurring along the denaturation pathway of RDs, but not for CDs (Figure 7), strongly support significant changes in the structure of the TS for RDs denaturation rather than in the native state (Figure 1). The denaturation TS of RDs is quite native-like without salt, with a change in the total ΔASA of ~7000 Å2 (Figure 7D), and its denaturation is dominated by a solvation barrier contribution (i.e. networks of broken interactions but not yet solvated; [29]); however, in the presence of salt, the TS becomes more unfolded in terms of solvent exposure, with change in the total ΔASA of ~21000 Å2. These additional ~14000 Å2 of total surface area exposed in the TS in the presence of salt corresponds to extra ~35 residues solvent-exposed in the TS per RD compared to the absence of salt (based on the correlations by [31]). This behavior agrees with the Hammond postulate for protein folding/unfolding: the TS becomes less native-like as the kinetic stability of the native state is increased [41]. Interestingly, those HCU-causing mutants with lower kinetic stability in their RDs (A114V, R125Q, E176K and particularly P422L; [4]) also show lower Ea values, thus suggesting a similar Hammond behavior for these destabilizing mutants. A similar relationship between kinetic stability and Ea values has been found in mutations causing human phosphoglycerate kinase 1 deficiency [28] and congenital erythropoietic porphyria [42], which might imply that this Hammond behavior is a frequent phenomenon for kinetically destabilizing mutations in loss-of-function conformational diseases. Thus, a certain degree of plasticity of the TS for denaturation might be an important factor modulating protein kinetic stability, with implications to understand mutational effects in conformational diseases as well as in the evolutionary changes and species-dependent adaptation of protein kinetic stability [28, 30, 43].
In conclusion, we show that salts are able to modulate the stability, activity and regulation of hCBS likely through screening of surface charge-charge interactions. Such screening enhances the kinetic stability of RDs through structural changes in the denaturation TS, revealing a significant structural plasticity of the TS which may also contribute to understand the destabilizing effect of disease-causing mutations in HCU [4]. Our results also support the role of electrostatic interactions in the recently proposed binding modes of SAM to those sites responsible for enzyme activation and stabilization. We also provide functional and binding thermodynamic information to be correlated with structural information obtained upon determination of high-resolution structures of hCBS in the presence of SAM.
Supplementary Material
Highlights.
- Surface electrostatics plays an important role in hCBS SAM-mediated regulation.
- Domain stabilities are differently modulated by charge-charge interactions.
- Modulation of domain stability occurs through changes in denaturation transition state.
- Activation and stabilization by SAM are differently driven by surface electrostatics.
Acknowledgment
We thank Prof. Jose Manuel Sanchez-Ruiz for support. We also thank Dr. Francisco Conejero-Lara and Dr. Bertrand Morel for access to the DLS instrument and help with these experiments. This work was supported by grants from MINECO (CSD2009-00088 and BIO2012-34937), Junta de Andalucia (CTS2011-07187) and the National Institutes of Health (HL065217). ALP is supported by a Ramón y Cajal research contract from MINECO (RYC2009-04147).
Abbreviations
- RD
regulatory domain
- CD
catalytic domain
- DSC
differential scanning calorimetry
- ITC
isothermal titration calorimetry
- hCBS
human cystathionine beta-synthase
- HCU
classical homocystinuria
- TS
transition-state of the rate-limiting denaturation step
- Hepes
4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid
- SAM
S-adenosyl-methionine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Miles EW, Kraus JP. Cystathionine beta-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem. 2004;279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- [2].Singh S, Banerjee R. PLP-dependent H(2)S biogenesis. Biochim Biophys Acta. 2011;1814:1518–1527. doi: 10.1016/j.bbapap.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ereno-Orbea J, Majtan T, Oyenarte I, Kraus JP, Martinez-Cruz LA. Structural basis of regulation and oligomerization of human cystathionine beta-synthase, the central enzyme of transsulfuration. Proc Natl Acad Sci U S A. 2013;110:E3790–E3799. doi: 10.1073/pnas.1313683110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pey AL, Majtan T, Sanchez-Ruiz JM, Kraus JP. Human cystathionine beta- synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): complex regulation of CBS activity and stability by SAM. Biochem J. 2013;449:109–121. doi: 10.1042/BJ20120731. [DOI] [PubMed] [Google Scholar]
- [5].Kozich V, Sokolova J, Klatovska V, Krijt J, Janosik M, Jelinek K, Kraus JP. Cystathionine beta-synthase mutations: effect of mutation topology on folding and activity. Hum Mutat. 2010;31:809–819. doi: 10.1002/humu.21273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hnizda A, Jurga V, Rakova K, Kozich V. Cystathionine beta-synthase mutants exhibit changes in protein unfolding: conformational analysis of misfolded variants in crude cell extracts. J Inherit Metab Dis. 2012;35:469–477. doi: 10.1007/s10545-011-9407-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hnizda A, Majtan T, Liu L, Pey AL, Carpenter JF, Kodicek M, Kozich V, Kraus JP. Conformational properties of nine purified cystathionine beta-synthase mutants. Biochemistry. 2012;51:4755–4763. doi: 10.1021/bi300435e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Janosik M, Oliveriusova J, Janosikova B, Sokolova J, Kraus E, Kraus JP, Kozich V. Impaired heme binding and aggregation of mutant cystathionine beta-synthase subunits in homocystinuria. Am J Hum Genet. 2001;68:1506–1513. doi: 10.1086/320597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Majtan T, Liu L, Carpenter JF, Kraus JP. Rescue of cystathionine beta-synthase (CBS) mutants with chemical chaperones: purification and characterization of eight CBS mutant enzymes. J Biol Chem. 2010;285:15866–15873. doi: 10.1074/jbc.M110.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. Structure of human cystathionine beta-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Taoka S, Lepore BW, Kabil O, Ojha S, Ringe D, Banerjee R. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- [12].Meier M, Oliveriusova J, Kraus JP, Burkhard P. Structural insights into mutations of cystathionine beta-synthase. Biochim Biophys Acta. 2003;1647:206–213. doi: 10.1016/s1570-9639(03)00048-7. [DOI] [PubMed] [Google Scholar]
- [13].Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R. S- adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci U S A. 2006;103:6489–6494. doi: 10.1073/pnas.0509531103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bonaventura C, Arumugam M, Cashon R, Bonaventura J, Moo-Penn WF. Chloride masks effects of opposing positive charges in Hb A and Hb Hinsdale (beta 139 Asn-->Lys) that can modulate cooperativity as well as oxygen affinity. J Mol Biol. 1994;239:561–568. doi: 10.1006/jmbi.1994.1395. [DOI] [PubMed] [Google Scholar]
- [15].Sanchez-Ruiz JM, Makhatadze GI. To charge or not to charge? Trends Biotechnol. 2001;19:132–135. doi: 10.1016/s0167-7799(00)01548-1. [DOI] [PubMed] [Google Scholar]
- [16].Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- [17].Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochim Biophys Acta. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- [18].Grimsley GR, Shaw KL, Fee LR, Alston RW, Huyghues-Despointes BM, Thurlkill RL, Scholtz JM, Pace CN. Increasing protein stability by altering long- range coulombic interactions. Protein Sci. 1999;8:1843–1849. doi: 10.1110/ps.8.9.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pace CN, Grimsley GR, Scholtz JM. Protein ionizable groups: pK values and their contribution to protein stability and solubility. J Biol Chem. 2009;284:13285–13289. doi: 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Loladze VV, Ibarra-Molero B, Sanchez-Ruiz JM, Makhatadze GI. Engineering a thermostable protein via optimization of charge-charge interactions on the protein surface. Biochemistry. 1999;38:16419–16423. doi: 10.1021/bi992271w. [DOI] [PubMed] [Google Scholar]
- [21].Pey AL, Rodriguez-Larrea D, Bomke S, Dammers S, Godoy-Ruiz R, Garcia-Mira MM, Sanchez-Ruiz JM. Engineering proteins with tunable thermodynamic and kinetic stabilities. Proteins. 2008;71:165–174. doi: 10.1002/prot.21670. [DOI] [PubMed] [Google Scholar]
- [22].Schweiker KL, Makhatadze GI. Protein stabilization by the rational design of surface charge-charge interactions. Methods Mol Biol. 2009;490:261–283. doi: 10.1007/978-1-59745-367-7_11. [DOI] [PubMed] [Google Scholar]
- [23].Zarrine-Afsar A, Zhang Z, Schweiker KL, Makhatadze GI, Davidson AR, Chan HS. Kinetic consequences of native state optimization of surface-exposed electrostatic interactions in the Fyn SH3 domain. Proteins. 2012;80:858–870. doi: 10.1002/prot.23243. [DOI] [PubMed] [Google Scholar]
- [24].Halskau O, Jr., Perez-Jimenez R, Ibarra-Molero B, Underhaug J, Munoz V, Martinez A, Sanchez-Ruiz JM. Large-scale modulation of thermodynamic protein folding barriers linked to electrostatics. Proc Natl Acad Sci U S A. 2008;105:8625–8630. doi: 10.1073/pnas.0709881105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tadeo X, Lopez-Mendez B, Trigueros T, Lain A, Castano D, Millet O. Structural basis for the aminoacid composition of proteins from halophilic archea. PLoS Biol. 2009;7:e1000257. doi: 10.1371/journal.pbio.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Desai TM, Cerminara M, Sadqi M, Munoz V. The effect of electrostatics on the marginal cooperativity of an ultrafast folding protein. J Biol Chem. 2010;285:34549–34556. doi: 10.1074/jbc.M110.154021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sedlak E, Stagg L, Wittung-Stafshede P. Effect of Hofmeister ions on protein thermal stability: roles of ion hydration and peptide groups? Arch Biochem Biophys. 2008;479:69–73. doi: 10.1016/j.abb.2008.08.013. [DOI] [PubMed] [Google Scholar]
- [28].Pey AL, Mesa-Torres N, Chiarelli LR, Valentini G. Structural and Energetic Basis of Protein Kinetic Destabilization in Human Phosphoglycerate Kinase 1 Deficiency. Biochemistry. 2013;52:1160–1170. doi: 10.1021/bi301565m. [DOI] [PubMed] [Google Scholar]
- [29].Rodriguez-Larrea D, Minning S, Borchert TV, Sanchez-Ruiz JM. Role of solvation barriers in protein kinetic stability. J Mol Biol. 2006;360:715–724. doi: 10.1016/j.jmb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- [30].Costas M, Rodriguez-Larrea D, De Maria L, Borchert TV, Gomez-Puyou A, Sanchez-Ruiz JM. Between-species variation in the kinetic stability of TIM proteins linked to solvation-barrier free energies. J Mol Biol. 2009;385:924–937. doi: 10.1016/j.jmb.2008.10.056. [DOI] [PubMed] [Google Scholar]
- [31].Robertson AD, Murphy KP. Protein Structure and the Energetics of Protein Stability. Chem Rev. 1997;97:1251–1268. doi: 10.1021/cr960383c. [DOI] [PubMed] [Google Scholar]
- [32].Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kraus JP. Cystathionine beta-synthase (human) Methods Enzymol. 1987;143:388–394. doi: 10.1016/0076-6879(87)43068-1. [DOI] [PubMed] [Google Scholar]
- [34].Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- [35].Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- [36].Gomez-Garcia I, Oyenarte I, Martinez-Cruz LA. The crystal structure of protein MJ1225 from Methanocaldococcus jannaschii shows strong conservation of key structural features seen in the eukaryal gamma-AMPK. J Mol Biol. 2010;399:53–70. doi: 10.1016/j.jmb.2010.03.045. [DOI] [PubMed] [Google Scholar]
- [37].Tuominen H, Salminen A, Oksanen E, Jamsen J, Heikkila O, Lehtio L, Magretova NN, Goldman A, Baykov AA, Lahti R. Crystal structures of the CBS and DRTGG domains of the regulatory region of Clostridiumperfringens pyrophosphatase complexed with the inhibitor, AMP, and activator, diadenosine tetraphosphate. J Mol Biol. 2010;398:400–413. doi: 10.1016/j.jmb.2010.03.019. [DOI] [PubMed] [Google Scholar]
- [38].Taoka S, Widjaja L, Banerjee R. Assignment of enzymatic functions to specific regions of the PLP-dependent heme protein cystathionine beta-synthase. Biochemistry. 1999;38:13155–13161. doi: 10.1021/bi990865t. [DOI] [PubMed] [Google Scholar]
- [39].Tzeng SR, Kalodimos CG. Allosteric inhibition through suppression of transient conformational states. Nat Chem Biol. 2013;9:462–465. doi: 10.1038/nchembio.1250. [DOI] [PubMed] [Google Scholar]
- [40].Prudova A, Martinov MV, Vitvitsky VM, Ataullakhanov FI, Banerjee R. Analysis of pathological defects in methionine metabolism using a simple mathematical model. Biochim Biophys Acta. 2005;1741:331–338. doi: 10.1016/j.bbadis.2005.04.008. [DOI] [PubMed] [Google Scholar]
- [41].Matouschek A, Fersht AR. Application of physical organic chemistry to engineered mutants of proteins: Hammond postulate behavior in the transition state of protein folding. Proc Natl Acad Sci U S A. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fortian A, Castano D, Ortega G, Lain A, Pons M, Millet O. Uroporphyrinogen III synthase mutations related to congenital erythropoietic porphyria identify a key helix for protein stability. Biochemistry. 2009;48:454–461. doi: 10.1021/bi801731q. [DOI] [PubMed] [Google Scholar]
- [43].Aguirre Y, Cabrera N, Aguirre B, Perez-Montfort R, Hernandez-Santoyo A, Reyes-Vivas H, Enriquez-Flores S, de Gomez-Puyou MT, Gomez-Puyou A, Sanchez-Ruiz JM, Costas M. Different contribution of conserved amino acids to the global properties of Triosephosphate isomerases. Proteins. 2014;82:323–335. doi: 10.1002/prot.24398. [DOI] [PubMed] [Google Scholar]
- [44].Sanchez-Ruiz JM, Lopez-Lacomba JL, Cortijo M, Mateo PL. Differential scanning calorimetry of the irreversible thermal denaturation of thermolysin. Biochemistry. 1988;27:1648–1652. doi: 10.1021/bi00405a039. [DOI] [PubMed] [Google Scholar]
- [45].Sanchez-Ruiz JM. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys J. 1992;61:921–935. doi: 10.1016/S0006-3495(92)81899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.