

Abstract
Relatively few MHC class I epitopes have been identified from M. tuberculosis, but during the late stage of infection CD8+ T-cell responses to these epitopes are often primed at an extraordinary high frequency. Although clearly available for recognition during infection, their role in resistance to mycobacterial infections still remain unclear. As an alternative to DNA and viral vaccination platforms, we have exploited a novel CD8+ T-cell-inducing adjuvant, CAF05 (DDA/TDB/Poly I:C), to prime high frequency CD8 responses to the immunodominant H2-Kb-restricted IMYNYPAM epitope contained in the vaccine antigen TB10.4/Rv0288/EsxH. We report that the amino acid C-terminal to this minimal epitope plays a decisive role in proteasomal cleavage and epitope priming. The primary structure of TB10.4 is suboptimal for proteasomal processing of the epitope and amino acid substitutions in the flanking region markedly increased epitope-specific CD8+ T-cell responses. One of the optimized sequences was contained in the closely related TB10.3/Rv3019c/EsxR antigen and when recombinantly expressed and administered in the CAF05 adjuvant, this antigen promoted very high CD8+ T-cell responses. This abundant T-cell response was functionally active but provided no protection against challenge, suggesting that CD8+ T-cells play a limited role in protection against M. tuberculosis in the mouse model.
Keywords: CD8, Cross-priming, ESX, protection, TB10.3 / TB10.4
Introduction
In the mouse model, infection with M. tuberculosis (M.tb.) elicits CD8+ T-cell responses characterized by large expansions of T cells recognizing a limited number of antigens. One of the primary CD8+ T-cell targets can be found within the secreted ESX-H antigen TB10.4 (Rv0288), which contains the strong H2-Kb-restricted epitope IMYNYPAM (TB10.44-11) [1]. During chronic stages of infection, responses to this major infection-driven CD8 epitope can constitute a very large fraction of the entire pool of pulmonary CD8+ T cells [1, 2] (Fig. 1A & B). Despite being a highly immunodominant target, the relevance of this CD8 epitope for protection is unclear, and the requirements for its induction by vaccination and the protection afforded has not been rigorously studied.
Figure 1. Despite being a major infection-driven CD8+ T-cell epitope, rTB10.4+CAF05 vaccination does not stimulate responses to TB10.44-11.

(A, B) Mice (CB6F1) were aerosol challenged with M. tb. Erdman and euthanized six weeks later to evaluate responses in perfused lungs. (A) Cells isolated from perfused lung tissue were subjected to intracellular flow cytometry after stimulation (1+5 (with BFA) hours) with either medium, rTB10.4, TB10.4 P11-18 or the 9-meric peptide TB10.43-11. Cells were gated as follows: singlets > lymphocytes > CD4+/CD8+. Bars show the frequency of IFN-γ-producing antigen-specific CD4+ (white) and CD8+ (black) T cells from pooled cells (from three individual mice) from one of two experiments with similar results. (B) Non-stimulated cells from perfused lung tissue were subjected to MHC class I dextramer staining using PE-conjugated H2-Kb-IMYNYPAM. Cells were gated as follows: singlets > lymphocytes > CD19− > CD8+. TB10.44-11 (IMYNYPAM) specific cells are depicted against CD8. Histogram is representative of two independent experiments.
(C, D) CB6F1 mice were immunized three times with 5 μg rTB10.4 + CAF05 (i.p.). Ten days after the last immunization, mice were euthanized. Experiments were also repeated two times further with TB10.4-containing fusion proteins, with similar results. (C) Spleen cells were subjected to intracellular flow cytometry after 1+5 hour stimulation with either medium, rTB10.4, TB10.4 P11-18 or the 9-meric peptide TB10.43-11. Gating as in Fig. 1A. Bars show the frequency of IFN-γ producing antigen-specific CD4+ (white) and CD8+ (black) T cells from spleen cells pooled from three individual mice from one of two experiments with similar results. (D) Non-stimulated splenocytes were MHC class I dextramer-stained using PE-conjugated H2-Kb-IMYNYPAM. Gating as in Fig. 1B. Histogram is representative of two independent experiments.
In humans, the role of CD8+ T cells in immunity against tuberculosis (TB) is not fully elucidated, but they do appear to be of significance as illustrated by the fact that CD8+ T cells from humans not only recognize infected macrophages [3, 4] but also directly kill mycobacteria by granulysin-dependent mechanisms [5, 6]. Additionally, CD8+ T cells seem to be important for immunity to mycobacteria in both non-human primates [7] and cattle [8, 9]. Consequently, targeting the CD8+ T-cell compartment is actively being pursued in some vaccine candidates in clinical development [10–12]. Despite the fact that mice lack a granulysin orthologue, a number of murine studies have likewise provided compelling evidence that CD8+ T cells are required for optimal protection against M. tb. infection [13]. The majority of these murine studies have been dominated by adoptive transfer models, in which Ag-specific CD8+ T cells are transferred into irradiated or Rag-mice prior to challenge and the protection afforded subsequently evaluated, often relatively early during the course of infection (2–4 weeks) [14–18]. Early adoptive transfer studies by Orme [19] also revealed some CD8+ T-cell related protection at week 4 and again at week 10–12 post infection with M.tb. However, the levels of protection afforded was relatively modest compared with that of adoptively transferred CD4+ T cells, which is also in step with the fact that CD4+ T cells dominate in the lungs of infected mice and guinea pigs [20, 21]. A depletion study by van Pinxteren et al. [22] reported that CD8+ T cells primarily play a role in preventing reactivation in a murine latency model of TB and other studies have also argued against a significant role for CD8+ T cells for the control of early, primary M. tb. infection in mice [23–25]. Thus, during primary infection, CD8+ T cells predominantly seem to play a role during late stage infection [24]. Adding to the complexity, it has also been reported that infection-driven CD8+ T-cell responses primarily reflect infectious load and are associated with lack of control with the infection [1, 2, 26]. Consequently, considerable uncertainty exists concerning even basic questions relating to the implication of CD8+ T cells during TB, such as when – and to what extent – during the infection CD8+ T cells are important and whether vaccine-promoted CD8+ T cells can mediate protection.
In the current study we have utilized a strong CD8-inducing adjuvant to promote CD8+ T cell-responses to the TB10.4-encoded H2-Kb epitope IMYNYPAM (TB10.44-11) and studied the requirements for vaccine priming. We find that C-terminal residues flanking the minimal epitope are highly decisive for vaccine priming and show that a single amino acid substitution at position TB10.412 dramatically influences the CD8+ T-cell output. In contrast, the ESX-analogue TB10.3 (ESX-R), which also expresses the IMYNYPAM epitope (TB10.34-11), is predicted to be cleaved by the proteasome and vaccination with recombinant TB10.3 using the recently developed CD8-promoting adjuvant CAF05 (DDA/TDB/Poly I:C) leads to priming of strong CD8+ T-cell responses. Although these CD8+ T cells were functionally active, highly cytotoxic and of a very high frequency they provided no protection against a TB challenge in the mouse model.
Results
Primary structure of ESX-H(TB10.4/Rv0288) is important for proteasomal cleavage and epitope priming
We have recently developed the CD8-inducing adjuvant CAF05 (DDA/TDB/poly I:C). This adjuvant is known to induce prominent CD8+ T-cell responses to a range of different antigens [27–29]. We consequently wanted to exploit CAF05 for selectively inducing CD8+ T-cell responses to mycobacterial antigens. In contrast to the exceedingly high CD8+ T-cell responses seen during natural infection by both intracellular staining for cytokines and by tetramer staining (Fig. 1A & B), we were repeatedly unable to raise any noticeable CD8 response to the minimal TB10.44-11 epitope (IMYNYPAM) or the 9-mer version TB10.43-11 when immunizing with rTB10.4 in the context of CAF05 (Fig. 1C & D), despite the fact that strong CD8+ T-cell responses to SIINFEKL could be obtained using ovalbumin with CAF05 (Supporting information Fig. 1). The same pattern was observed using TB10.4 containing fusion proteins (not shown). This indicated to us that there are special requirements for CD8+ T-cell induction to the minimal IMYNYPAM epitope contained within TB10.4, which prompted us to look at proteasomal cleavage of the antigen. In silico prediction algorithms using a proteasomal processing model (RANKPEP) [30] suggested that the TB10.44-11 epitope could not be cleaved from TB10.4 (Table 1). We therefore produced an 18-mer long synthetic TB10.4 P11-18 peptide containing the CD8 epitope alongside a number of peptide variants all containing the minimal epitope (Table 1). As the P1′ amino acid just C-terminal to CD8 epitopes has been reported to be of major importance for defining proteasomal cleavage by the immunoproteasome [31–33], we produced 5 variants of the TB10.4 P11-18 peptide – only differing at the P1′ position (TB10.412). Four of these variants were predicted to be cleaved (TB10.4 P11-18 P1M, P1E, P1R, P1H), whereas the fifth (P1C), containing a cysteine at P1′, was not. Immunization with these peptides in CAF05 showed a dramatic effect of substituting just one single amino acid C-terminal to the minimal epitope (Fig. 2), and significant CD8+ T-cell responses ranging from 2–20% were found to peptides predicted to be cleaved, whereas no response were found to P1C (Fig. 2A & B). Using MHC I dextramers only a small, but detectable response could be promoted by immunization with the parent TB10.4 P11-18 peptide, whereas in particular substitution of the leucine (L) group with arginine (R) or histidine (H) resulted in peak frequencies (around 20%) of IMYNYPAM-specific CD8+ T cells (Fig. 2A & B). This pattern was likewise reflected in the frequency of IFN-γ secreting CD8+ T cells at day 10 after the third immunization in spleen cells, where the peptide containing the arginine substitution (P1R) primed significantly higher responses than the predicted non-cleavable peptides, including the parent TB10.4 P11-18 peptide (Fig. 2B, p < 0.01).
Table 1.
| Parent TB10.4 P1 peptide: MSQIMYNYPAMLGHAGDM | ||
|---|---|---|
| TB10.4 P11-18 | Peptide sequence | Proteasomal clevage prediction |
| P1 | MSQIMYNYPAMLGHAGDM | NO |
| P1M | MSQIMYNYPAMMGHAGDM | YES |
| P1E | MSQIMYNYPAMEGHAGDM | YES |
| P1R | MSQIMYNYPAMRGHAGDM | YES |
| P1H | MSQIMYNYPAMHGHAGDM | YES |
| P1C | MSQIMYNYPAMCGHAGDM | NO |
Figure 2. One amino acid substitution C-terminal to the minimal TB10.44-11 epitope (P1′ position) in TB10.4 P11-18-modified peptides significantly influences CD8+ T cell output following vaccination.

Groups of three C57BL/6 mice were immunized with either the parent TB10.4 P11-18 peptide or one of five modified peptides having one amino acid substitution at the P1′ position (TB10.412) relative to P1 (P1M, P1E, P1R, P1H & P1C – see Table 1). Mice were immunized with 5 μg peptide + CAF05 (i.p.). The experiment was also repeated one time further only using the parent TB10.4 P11-18 peptide against the modified P1R peptide.
(A) Non-stimulated cells from pooled PBMCs were isolated one week post-second immunization and subjected to MHC class I dextramer staining using PE-conjugated H2-Kb-IMYNYPAM. Cells were gated as follows: singlets > lymphocytes > CD8+ > H2-Kb-IMYNYPAM+ CD62L−. Flow cytometry was performed and the plot shows the frequency of IMYNYPAM+CD8+CD62L− out of CD8+ T cells. Data are shown as frequencies of pooled PBMCs from three individual mice and representative of two independent experiments.
(B) Splenocytes from individual mice were isolated ten days post-third immunization and the frequency of IFN-γ producing CD8+ T cells was determined by intracellular flow cytometry. Cells were stimulated with the 9-meric peptide TB10.43-11 and the frequency of IFN-γ+ CD8+ T cells is depicted following background subtraction. Gating as in 1A. * p < 0.05, ** p< 0.01 (One-Way ANOVA with Bonferroni post-test).
Immunization with ESX-R (TB10.3), but not ESX-H (TB10.4), induces CD8+ T-cell responses to IMYNYPAM
After having realized the importance of the amino acids flanking the C-terminal part of the minimal epitope, we next looked for homologues of the TB10.4 (ESX-H) derived IMYNYPAM sequence in the TB genome. Two antigens –TB10.3 (ESX-R; Rv3019c) and TB12.9 (ESX-Q; Rv3017c) – were found to contain homologues to the CD8 epitope, but the epitope sequence was only completely conserved in the TB10.3 antigen (Fig. 3A). In TB10.3, the minimal epitope was found to be flanked by a methionine group at the P1′ position, and therefore proteasomal cleavage and presentation of the minimal epitope would be expected. In order to test this, two 20-mer peptides from TB10.4 and TB10.3 were subsequently produced (only differing at position 12 and 13; Fig. 3A) and used for immunization in CAF05. After two immunizations, P11-20 from TB10.3 was clearly superior in priming CD8+ T-cell responses relative to the peptide from TB10.4 resulting in more than a six-fold higher frequency of H2-Kb-IMYNYPAM-specific cells (Fig. 3B). This was likewise reflected in the frequency of IFN-γ secreting CD8+ T cells, where only TB10.3 P11-20 was found capable of priming CD8 responses (Fig. 3C). Interestingly, whereas the precise sequence of the flanking region had great impact on the ability of the antigen to prime CD8+ T-cell responses post immunization (p < 0.001), both the TB10.4 and TB10.3 version of the epitope triggered similar levels of ex vivo recall response (Fig. 3C).
Figure 3. Vaccination with TB10.3 + CAF05 primes high CD8+ T cell responses to the IMYNYPAM epitope, in contrast to TB10.4.

(A) Three antigens – TB10.4 (ESXH; Rv0288), TB10.3 (ESXR; Rv3019c) and TB12.9 (ESXQ; Rv3017c) were found to contain homologues to the IMYNYPAM CD8+ T cell epitope, with the epitope being completely conserved in the two first antigens. Amino acid sequences for TB10.4 and TB10.3 have been aligned with substitutions marked in black. The 8-meric minimal H2-Kb-restricted epitope (TB10.3/44-11; IMYNYPAM) is marked by a black box, whereas the I-Ab-restricted CD4+ T cell epitope (core sequence TB10.45-13/ TB10.35-13) has been marked with a light grey box. Note the two substitutions in TB10.3 at the C-terminal part of the CD4+ epitope core sequence (TB10.312-13). Two 20-mer peptides from TB10.4 and TB10.3, highlighted by underscoring, were produced and used for immunization with CAF05.
(B, C) Groups of 3 C57BL/6 mice were immunized twice at two weeks intervals with 5 μg of either TB10.4 P11-20 + CAF05 or TB10.3 P11-20 + CAF05 by the i.p. route. Nine days after the last immunization, CD8+ T cell responses to TB10.3/44-11 were evaluated. Experiments with the 20-mer peptides (Fig. 3A, B & C) were repeated five times with similar results.
(B) MHC class I dextramer staining of non-stimulated splenocytes using PE- conjugated H2-Kb-IMYNYPAM. Representative plot of TB10.44-11 specific cells depicted against CD8. Gating as in Fig. 1B.
(C) Splenocytes were analyzed from individual mice (n = 3) by intracellular flow cytometry to establish the frequency of IFN-γ-producing CD8+ T cells. Cells were stimulated (1+5h) either with TB10.3 P11-20, TB10.4 P11-20 or the 9-meric peptide TB10.3/43-11. Background was subtracted. Cells were gated as in Fig. 1A and data are shown as mean + SEM representative of 5 independent experiments. * p < 0.05, ** p< 0.01, (Two-Way ANOVA with Bonferroni post-test).
(D) C57BL/6 mice (n = 3/group) were immunized three times using either 5 μg rTB10.3 + CAF05 or 5 μg rTB10.4 + CAF05 i.p. . Twelve days after the last immunization, mice were euthanized and CD8+ T cell responses to TB10.3/44-11 in the spleen were evaluated in individual mice by intracellular flow cytometry. Cells were stimulated (1+5h) either with rTB10.3, rTB10.4 or the 9-meric peptide TB10.3/43-11. Background was subtracted. Cells were gated as in Fig. 1A and data are shown as mean + SEM. ** p< 0.01, (Two-Way ANOVA with Bonferroni post-test). The experiment with recombinant TB10.3/TB10.4 proteins was repeated once with similar results.
We continued by producing recombinant TB10.3 in order to compare head-to-head with rTB10.4 for CD8+ T-cell induction with CAF05. After three immunizations with CAF05, significantly higher CD8+ T-cell responses could be obtained with rTB10.3 compared with rTB10.4 (p < 0.0001), which, in agreement with our earlier data (Fig. 1C & D), induced neglectable CD8 responses (Fig. 3D). A vaccination strategy simply using the 9-mer epitope TB10.3/4 P13-11 as vaccine antigen was not fruitful and was found equally inferior at priming CD8+ T-cell responses (Supporting Information Fig. 2). Similar to our observations with the peptides, once primed, both recombinant proteins could trigger comparable ex vivo recall responses (Supporting information Fig. 2). These data therefore suggests that the high levels of infection-driven responses to the CD8-epitope IMYNYPAM could arise as a consequence of priming by TB10.3 (Rv3019c; ESX-R) rather than by TB10.4 (Rv0288; ESX-H).
ESX-H CD8+ T cells are functionally active and highly cytotoxic but confer no protection to M.tb
Phenotypic characterization of the CD8+ T cells induced by rTB10.3/CAF05 revealed that the H2-Kb-IMYNYPAM-specific cells were CD62Llow, CD44high and CD127low/high – with more than half of the dextramer-specific cells expressing the IL-7 receptor (CD127) (Fig. 4A & B). This shows that immunization with rTB10.3/CAF05 induces highly activated CD8+ T cells and that a large proportion of these are endowed with a capacity to generate long-lived memory CD8+ T cells.
Figure 4. Vaccine-promoted TB10.3/43-11-specific CD8+ T cells are in activated state, express memory precursor marker and exhibit high in vivo cytotoxicity.

(A, B) Mice (C57BL/6; n = 3) were immunized three times two weeks apart with 5 μg rTB10.3 + CAF05 i.p.
(A) Unstimulated spleen cells (pooled) were stained using PE-conjugated MHC class I dextramer H2-Kb-IMYNYPAM and surface markers against (i) CD8, (ii), CD62L, (iii) CD44 and (iv) CD127 (IL-7 receptor). Cells were gated as follows: singlets > lymphocytes > CD4+ vs CD8+. Histograms are representative of two independent experiments with similar results.
(B) Pooled PBMCs were isolated after the last immunization and subjected to MHC class I dextramer staining using PE-conjugated MHC class I dextramer H2-Kb-IMYNYPAM. IMYNYPAM+ cells were gated and histogram overlays relative to IMYNYPAM+ CD8+ T cells are shown, from one of two representative experiments.
(C–E) Groups of mice (C57BL/6; n = 8/group) were immunized twice using 5 μg TB10.3 P11-20 + either CAF01 (DDA/TDB – 250/50 μg/dose), Poly I:C in PBS (50 μg/dose), or CAF05 (DDA/TDB/Poly I:C – 250/50/50 μg/dose), or left non- immunized.
(C) Nine days post-immunization, individual splenocytes (n = 3) were MHC class I dextramer-stained using PE-conjugated H2-Kb-IMYNYPAM and gated as in Fig. 2A. The frequency of IMYNYPAM+CD8+CD62L− cells from CD8+ T cells is shown. Each symbol represents an individual mouse and means ± SEM are shown. *** p < 0.001 (One-way ANOVA, Tukey’s post-test).
(D) In vivo CTL assay. Ten days after immunization, splenocytes from naïve feeder mice were isolated and either pulsed with the 9-mer CD8 epitope TB10.3/4 P13-11 and stained with a high concentration of CFSE or left unpulsed and stained with a tenfold lower CFSE concentration. These CFSEhigh and CFSElow cells were mixed at a 1:1 ratio and adoptively transferred i.v. into remaining mice from each immunization group (n = 5). One day after transfer, the cytotoxicity was determined by comparing the ratio of pulsed (CFSEhigh) to unpulsed (CFSElow) cells. Cells were gated as follows: singlets > SSC vs CFSE > CFSE+. Histogram plots (# of cells vs CFSE) from blood (RBC lysed; pooled) of each immunization group are shown.
(E) In vivo CTL activity was determined in spleens and expressed as specific lysis. Each symbol represents an individual mouse (n = 5/group, experiment was repeated two times with similar results). Means ± SEM shown. *** p < 0.001, (One-way ANOVA, Tukey’s post-test).
We next addressed to what extent CAF05-promoted CD8+ T cells with specificity to IMYNYPAM were functionally active and displayed cytotoxicity. To this end, mice were immunized twice with TB10.3 P1 either using CAF01 (DDA/TDB), free Poly IC (in PBS), CAF05 (DDA/TDB/pIC) or left unimmunized. Nine days post-immunization, only mice immunized with TB10.3 P1 in CAF05 had mounted strong responses to the epitope reaching an average frequency of approx. 8% of the CD8+ T cells in the spleen (Fig. 4C; ANOVA, Tukey post-test, p < 0.001). We continued by investigating the cytotoxic activity of the vaccine-induced CD8+ T cells in an in vivo CTL assay. One day after target cell transfer, the degree of cytotoxicity was determined and expressed as specific lysis (Fig. 4D & E). We observed cytotoxic activity in mice immunized with TB10.3 P1 in CAF05 reaching on average 90% specific lysis, which was highly significant from all other treatment groups (ANOVA, Tukey post-test p < 0.001 – Fig. 4E).
As a number of reports using different approaches find that CD8+ T cells are required for optimal protection against virulent M. tb., we continued by performing an aerosol challenge experiment in order to assess whether vaccine-induced CD8+ T cells specific for TB10.3/44-11 were protective. Groups of CB6F1 mice were immunized with TB10.3 P1 in CAF05 three times with two weeks interval by the i.p. route and both local (lung) and systemic (PBMCs and spleen) immune responses evaluated two weeks after last immunization. Mice were subsequently challenged and protective efficacy evaluated 7 weeks after challenge. A BCG group was included as a control. To ensure that the vaccine-promoted CD8+ T cells reached the lung in sufficient numbers to recognize and respond early to the bacteria, we further included a group with pulmonary delivery of the vaccine. Significant levels of IFN-γ producing CD8 T+ cells specific for TB10.33-11 could be detected after i.p. administration (~1.5%; p < 0.01), whereas CD4 responses to the I-Ab-restricted epitope were neglectable in the CB6F1 mice (Fig. 5A). Measured by MHC I dextramers, both i.p. and pulmonal delivery of the vaccines led to discernable systemic CD8+ T-cell responses, leading to frequencies of approximately 5% and 0.7% out of the CD8+CD62L− T cells in PBMCs and 3.3% and 0.93% in the spleen, respectively (Fig. 5B). Strong lung CD8+ T-cell responses were detected after i.p. delivery (approx 6%), but reached as high as 20% H2-Kb-IMYNYPAM +ve CD8+CD62L− T cells after pulmonary vaccination (Fig. 5B). Despite high CD8+ T-cell responses both systemically and in the lung, none of the vaccination groups were protected against challenge with M. tb. Erdman when evaluated 7 weeks after infection (p > 0.05). Only BCG immunized mice showed significant levels of protection equaling a Log10 reduction in CFUs of approximately 1.7 (ANOVA, Tukey post-test p < 0.001, Fig. 5C).
Figure 5. Vaccine-promoted CD8+ T cell responses to TB10.3/43-11 do not confer protection to aerosol challenge.
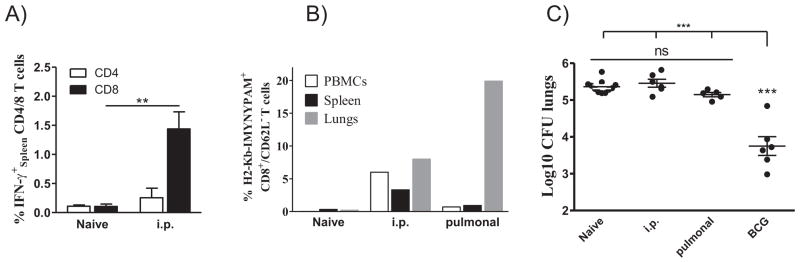
(A) CB6F1 mice (n = 4) were immunized three times with 10 μg TB10.3 P11-20 + CAF05 (i.p.) or left unimmunized. Three weeks after the last immunization, splenocytes from individual mice were analyzed by intracellular flow cytometry and the frequency of IFN-γ-producing CD4+ and CD8+ T cells determined. Cells were stimulated with 2 μg/mL of the TB10.3 P11-20 peptide and mean frequencies ± SEM following background subtraction are presented from one experiment representative of 3 independent experiments. **p < 0.01, (Student’s T-test).
(B) Groups of CB6F1 mice were immunized three times with 10 μg TB10.3 P11-20 + CAF05 either by the i.p. route or through pulmonary vaccine delivery using a Penn-Century Microsprayer aerosolizer. Non-immunized mice were included as controls. Three weeks post-immunization, pooled cells from lungs, spleen and PBMCs were isolated and non-stimulated cells subjected to ex vivo staining using PE-conjugated MHC class I dextramer H2-Kb-IMYNYPAM and surface markers. Bars show the frequency of IMYNYPAM+CD8+CD62L− cells out of CD8+ T cells in the different compartments.
(C) Seven weeks post-immunization, mice from naïve (n = 11), BCG (n = 6), pulmonal (n = 5) and i.p. (n = 6) groups were subjected to aerosol challenge (~100 CFU/mouse). Mice were euthanized at week 7 post-infection and CFU levels were determined by serial dilutions of individual lung homogenates onto Middlebrook 7H11 agar plates. Each symbol represents the Log10 CFU level of one mouse and means ± SEM are shown. *** p < 0.001 (ANOVA with Tukey’s post-test). Experiment was repeated once with similar results.
Discussion
The H2-Kb-restricted IMYNYPAM epitope from the TB10.4 vaccine antigen (EsxH/Rv0288) probably represents the strongest murine CD8+ T-cell epitope known from M. tb. [1, 34]. During the natural infection, exceedingly high levels of TB10.44-11-specific CD8+ T-cell responses are found [1, 2] and the magnitude of the response seems to correlate with bacterial virulence and in part on the presence of a functional secretion apparatus [1]. This and other studies have thus been used as a support for the potential importance of cytosolic access of M. tb. for proper MHC I loading [1, 18, 35, 36]. Despite reported examples of proteasome-independent processing of mycobacterial CTL antigens [37, 38], most M. tb. antigens may therefore require proteasomal degradation, TAP transport, MHC I loading and ER-Golgi egress [35]. During subunit vaccination with recombinant protein or long peptides in CAF05, CD8+ T-cell induction would be accomplished strictly through cross-priming. Although this represent a different cellular mechanism than antigen presentation during the natural infection, cross-presentation of the CTL epitope still to a large extent depend on processing by the multicatalytic proteasome complex complemented by cytosolic peptidases such as tripeptidyl peptidase II and thimet oligopeptidase [39]. Hence, both proteasome specificity and epitope trimming would be expected to be important processes leading to the presentation of the TB10.44-11 CTL epitope after vaccination [32, 39–41]. Given the strong immunodominance of the TB10.44-11 epitope, we were initially surprised by the lack of vaccine primed responses to this CD8 epitope. Based on the Netchop-20S 3.0 database, the average cleavage score of peptides having a methionine group at the P1 position have been reported to be relatively high, which would indeed suggest a peptide available for class I presentation [40]. However, although the TB10.44-11 epitope do carry a methionine group at P1 (IMYNYPAM), the amino acids flanking the C-terminal proteasome cleavage site (P1′ position) have also been reported to profoundly influence epitope processing [31, 33, 42]. By exchanging single amino acids at the P1′ position (TB10.412), we discovered that the leucine group in TB10.412 prevents efficient proteasomal processing in contrast to substitutions with methionine (M), glutamic acid (E), arginine (R) or histidine (H) at the same position, which all both increased the predicted cleavage score of TB10.44-11 and the CD8+ T-cell output. In this context, it was recently reported that a C-terminal extension of an immunodominant CD8 epitope from the intracellular parasite Toxoplasma gondii with one single leucine (L) group (P1′ position) likewise severely disrupted its presentation, whereas a lysine (K) extension only had a mild impact [42]. Importantly, our predictions on the importance of antigen processing for CTL priming did not only adhere to the synthetic versions of the TB10.4 P1 peptide, but was shown to have a natural analogue in TB10.3, which carries a methionine group at the P1′ position relative to the CTL epitope and accordingly promoted very high CD8+ T-cell responses after CAF05-immunization. Thus, our observations go beyond theoretical considerations on synthetic vaccine antigens, but also relate to naturally processed antigens. Therefore, our finding suggests that the major CD8+ T-cell response promoted during infection in the mouse model instead of being driven by the vaccine antigen TB10.4 is derived from its close relative TB10.3. This might also have human relevance and implications, as the predicted cleavage score of TB10.3 (0.76) is notably higher than TB10.4 (0.59), based on calculations from the human proteasome cleavage prediction server Netchop 3.1-20S 3.0 [40]. TB10.4 is currently a vaccine candidate in clinical trials, but if the primary aim is to induce CD8+ T-cell responses by subunit vaccination, TB10.3 would probably represent a better antigen candidate. Although being a poorer proteasomal substrate, TB10.4 is however expressed at 100 times higher levels than TB10.3 both during in vitro cultivation and early during infection [43], and the processed epitope output from TB10.4 might therefore still be adequate for priming. Interestingly, we observed that once primed, CD8+ T-cell responses to IMYNYPAM could be recalled ex vivo with the apparent same efficiency using TB10.4 or TB10.3. As activation threshold for priming is expected to be higher than for recall, even suboptimal processing of the CTL epitope from TB10.4 would seem adequate for expansion of already primed responses and this probably play an important role for the very large expansion of TB10.3/44-11 specific cells seen during natural infection. Therefore, we also anticipate that specific CD8+ T cells would recognize the CTL epitope on the surface of infected cells regardless of being derived from either TB10.3 or TB10.4.
At a glance, our data might seem at odds with the ease by which adenoviral (Ad5 or Ad35) delivery of TB10.4 or TB10.4-containing constructs prime strong CD8+ T-cell responses to the TB10.44-11 epitope [44, 45]. As cross-priming in our system prevail, it seems likely that access to MHC I loading would be less efficient in comparison with viral vectors, where the antigens are directly synthesized by the host cell translation machinery. Accordingly, any parameter which diminishes or abrogates efficient processing (such as L12 in TB10.4) may become the determining factor for MHC I presentation and hence CTL output. In addition, it has been reported that C-terminal flanking amino acids might differentially impact direct presentation vs cross-presentation of the same CTL epitope [46]. TB10.4 might therefore be a good substrate for direct priming of CTL responses to IMYNYPAM, but poor at priming responses through cross-presentation. By employing a subunit vaccination strategy for induction of CD8+ T cells, problems with vector immunity could be circumvented and combined CD4+ and CD8+ responses potentially be achieved without the need for mixed modality vaccinations.
Using TB10.3 as an antigen in CAF05, we were able to induce very strong functionally active cytotoxic CD8+ T cell responses to the IMYNYPAM epitope. Yet, upon challenge, mice were not protected against infection. This is in contrast to adoptive transfer models, where CD8+ T cells have been shown to protect against M. tb. infection [15, 18], but in agreement with other studies showing no or limited protective effect of vaccine promoted CD8+ T cells [34, 47] and with data showing that CD8+ T cells do not add significantly to the protection afforded by CD4+ T cells [44, 48]. In a recent study, Woodworth et al. used a rVV (Vaccinia Virus) approach to prime CD8+ T-cell responses to the H2-Kd-restricted TB10.420-28 epitope [34], which were greatly expanded by the infection but in agreement with our data, this response had no impact on bacterial loads. These observations of a lack of protection by T cell epitopes that are clearly available during infection suggest that either do the CD8+ T cells recognize infected cells but their effector functions (cytotoxicity and cytokine production) are insufficient for bacterial killing or alternatively that TB10.4-derived CTL epitopes are not primarily recognized on infected macrophages but on non-infected DCs that cross-present antigens. In this regard, Srivastava et al. [49] recently highlighted the importance of direct contact with infected cells by cytotoxic CD4+ T cells for control of intracellular M. tb. in vivo. CD8+ T cells would likewise act through contact-dependent cytolysis and secretion of effector cytokines, but this subset typically occupies discrete spatial niches in the periphery of the lung granulomas at a distance from the majority of the intracellular bacteria [50]. The different localization of CD4+ and CD8+ T cells have been observed both in human, cattle, guinea pig and mouse granulomas [21, 50–52], and it was originally suggested that specific lysis by CD8+ T cells of chronically infected macrophages in the periphery of the granuloma would enable uptake of extracellular bacilli by newly recruited monocytes [53]. In the light of our data we suggest that the CD8+ T cell expansion that eventually leads to their extreme immunodominance are driven by uninfected DCs that cross-present mycobacterial antigens derived from apoptotic cells and that targeting non-infected cross-presenting DCs in the periphery of the granuloma have no impact on the growth of M. tb. If this hypothesis is true, the CD8+ T-cell response in TB could be seen as a collateral consequence of the dynamic turnover taking place in an immunological active site like the granuloma during a chronic infection like TB, instead of as an effector subset relevant to target by vaccination.
Materials and methods
Animals
Female C57BL/6 and CB6F1 mice aged 6–8 weeks were purchased from Harlan Scandinavia (Allerød, Denmark). All manipulations were conducted in accordance with the regulations of the Danish Ministry of Justice and animal protection committees under permits 2004/561–868, 2009/561–1655 & 2012-15-2934-00272 and in compliance with European Community Directive 86/609. Once infected, animals were housed in cages contained within laminar flow safety enclosures (Scantainer; Scanbur) in a separate biosafety level 3 facility.
Antigens, adjuvants and immunizations
Synthetic peptides were purchased at GenScript or JPT at a purity of >85% or >90% and dissolved at a concentration of 1 mg/mL in either 10% DMSO or 10% DMF in 10 mM Tris buffer pH 7.4. Recombinant TB10.3 was produced as described in [29] using chemically synthesized DNA constructs codon-optimized for expression in E. coli. CAF05 (DDA/TDB/Poly I:C – 250 μg/50 μg/50 μg per dose) was prepared as described in Korsholm et al [29]. Mice were immunized three times with two week intervals using 5 μg recombinant protein or peptide in CAF05 in a vaccine volume of 200 μL. Mice immunized by the intraperitoneal route were treated with the analgesic Temgesic prior to injection. For data in Figure 5, mice received 10 μg TB10.3 P1 antigen and one group of mice with pulmonary vaccine delivery was included. Pulmonary vaccines were given in a volume of 50 μl (CAF05 dose 250/50/50) using a Penn-Century Microsprayer aerosolizer. Mice were fully sedated with Zoletil during this procedure.
Cell preparations and flowcytometric analysis
Blood was obtained by submandibular bleeding and the PBMCs were purified using Lympholyte (Cederlane, Burlington, NC). Splenic cell preparations were obtained by homogenization through a cell strainer and washed twice in RPMI-1640 (Gibco Invitrogen, Taastrup, Denmark). Perfused lungs were likewise homogenized through cell strainers and washed twice in RPMI-1640. MHC class I multimer stainings (Dextramer: PE-H2-Kb-IMYNYPAM, Immudex; Pentamer: PE-H2-Kb-SIINFEKL, ProImmune) were immediately performed on unstimulated cells in Nuclon V-bottom microtitre plates (96-well plates Nunc, Roskilde, Denmark) plates containing 1–2 × 106 cells/well for 10 min at rt before the cells were stained for surface markers (30 min at 4°C). All cell cultures for intracellular flowcytometry were performed in Nuclon V-bottom microtitre plates (96-well plates Nunc, Roskilde, Denmark) containing 1–2 × 106 cells/well in a volume of 200 μl RPMI-1640 supplemented with 5 × 10−5 M 2-mercaptoethanol, 1 mM glutamine, 1% pyruvate, 1% penicillin-streptomycin, 1% HEPES and 10% fetal calf serum (FCS) (all Gibco Invitrogen, Taastrup, Denmark) as earlier described [26]. Stimulants were used in concentrations of either 5 (protein antigens) or 2 (peptides) μg/mL. The following antibodies were used at a dilution of 1:600 unless otherwise stated: anti-CD4 APC-eF780 (clone GK1.5), anti-CD8-PerCp-Cy5.5 (clone 53-6.7), anti-CD44-FITC or APC (clone IM7), anti-CD127-PE-Cy7 (clone A7R34, 1:50), anti–IFN-γ–PE–Cy7 (clone XMG1.2; 1:200), anti–TNF-α–PE (MP6-XT22n; 1:200), anti–IL-2–APC (clone JES6-5h4, 1:200) (all eBiosciences) and anti-CD62L-FITC (clone MEL-14, 1:200, BD Pharmingen).
In vivo CTL assay
Ten days after last immunization, mice were injected intravenously (i.v.) with ~10 × 106splenocytes from naïve feeder mice of which one half had been previously stained with 4 μM carboxyfluorescein succinimidyl ester (CFSE, Invitrogen) (termed CFSElo) and the other with 40 μM CFSE (termed CFSEhi), which were also pulsed for 90 min with 10 μg/mL QIMYNYPAM peptide. After 1 day the mice were killed and the percentage of CFSEhi cells (antigen-pulsed) and CFSElo cells (unpulsed) out of the total CFSE+ cells determined. CTL activity was determined in spleens from individual mice and expressed as specific lysis using the formula: % Specific lysis = [1 − ((pulsed/unpulsed)immunized/(pulsed/unpulsed)naïve)] × 100.
Experimental infection
Seven weeks post immunization, mice were challenged by the aerosol route using the Glas-Col inhalation exposure system (Inhalation Exposure System, Glas-Col, Terre-Haute, IN) calibrated to deliver ~100 CFU/mice (5 ×106 CFU/mL Erdman). Mice were killed 7 weeks into the infection and CFU levels determined as earlier described [26].
Statistical analysis
For comparative analysis of immunogenicity, data were either tested by a Student’s t-test or by one-way analysis of variance (ANOVA). When significant differences were found, differences between means were either determined by Tukey’s or Bonferroni’s multiple comparison tests. In cases of multiple variable parameters, a two-way ANOVA was performed. All statistical analyses were carried out in GraphPad Prism version 5.03 (GraphPad Software Inc., La Jolla, CA).
Supplementary Material
Acknowledgments
This work was supported in part by the European Commission contract FP7-HEALTH-F3-2009-241745 under the NEWTBVAC consortium and in part through the Centre for Nano-vaccines supported by the Danish Strategic Research Council (09-067052). Additional financial support was obtained from NIH grant number AI 105422 – ‘An adjuvant that promotes TH1/TH17 and CD8 T cells in a tuberculosis vaccine’. We would like to thank Linda Christensen, Rune Fledelius Jensen, Sandra Isling, Merete Henriksen and Vivi Andersen for their excellent technical help. The animal technicians at the Statens Serum Institut are likewise gratefully acknowledged for their excellent technical assistance. Karen Korsholm is specially thanked for numerous fruitful discussions on adjuvants and CD8+ T-cell priming.
Abbreviations
- BCG
bacille Calmette-Guérin
- CAF01
Cationic Adjuvant Formulation 01
- CAF05
Cationic Adjuvant Formulation 05
- DDA
dimethyldioctadecylammonium
- ESX
mycobacterial type VII secretion system
- M.tb.
Mycobacterium tuberculosis
- Poly I
C, Poly (inositic:cytidylic) acid
- TDB
trehalose dibehenate
- TB
Tuberculosis
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Billeskov R, Vingsbo-Lundberg C, Andersen P, Dietrich J. Induction of CD8 T cells against a novel epitope in TB10. 4: Correlation with mycobacterial virulence and the presence of a functional region of difference-1. Journal of Immunology. 2007;179:3973–3981. doi: 10.4049/jimmunol.179.6.3973. [DOI] [PubMed] [Google Scholar]
- 2.Hoang T, Nansen A, Roy S, Billeskov R, Aagaard C, Elvang T, Dietrich J, et al. Distinct differences in the expansion and phenotype of TB10. 4 specific CD8 and CD4 T cells after infection with Mycobacterium tuberculosis. PLoS ONE. 2009;4:e5928. doi: 10.1371/journal.pone.0005928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewinsohn DM, Zhu LQ, Madison VJ, Dillon DC, Fling SP, Reed SG, Grabstein KH, et al. Classically restricted human CD8+ T lymphocytes derived from Mycobacterium tuberculosis-infected cells: Definition of antigenic specificity. Journal of Immunology. 2001;166:439–446. doi: 10.4049/jimmunol.166.1.439. [DOI] [PubMed] [Google Scholar]
- 4.Shams H, Klucar P, Ewer SE, Lalvani A, Moonan PK, Safi H, Wizel B, et al. Characterization of a Mycobacterium tuberculosis peptide that is recognized by human CD4+ and CD8+ T cells in the context of multiple HLA alleles. Journal of Immunology. 2004;173:1966–1977. doi: 10.4049/jimmunol.173.3.1966. [DOI] [PubMed] [Google Scholar]
- 5.Ernst WA, Thoma-Uszynski S, Teitelbaum P, Ko C, Hanson DA, Clayberger C, Krensky AM, et al. Granulysin, a T cell product, kills bacteria by altering membrane permeability. Journal of Immunology. 2000;165:7102–7108. doi: 10.4049/jimmunol.165.12.7102. [DOI] [PubMed] [Google Scholar]
- 6.Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, Ganz T, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science. 1998;282:121–125. doi: 10.1126/science.282.5386.121. [DOI] [PubMed] [Google Scholar]
- 7.Chen CY, Huang D, Wang RC, Shen L, Zeng GC, Yao SY, Shen Y, et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. Plos Pathogens. 2009;5:e1000392. doi: 10.1371/journal.ppat.1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer MV, Waters WR, Thacker TC. Lesion development and immunohistochemical changes in granulomas from cattle experimentally infected with Mycobacterium bovis. Veterinary Pathology. 2007;44:863–874. doi: 10.1354/vp.44-6-863. [DOI] [PubMed] [Google Scholar]
- 9.Villarreal-Ramos B, McAulay M, Chance V, Martin M, Morgan J, Howard CJ. Investigation of the role of CD8+ T cells in bovine tuberculosis in vivo. Infection and Immunity. 2003;71:4297–4303. doi: 10.1128/IAI.71.8.4297-4303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abel B, Tameris M, Mansoor N, Gelderbloem S, Hughes J, Abrahams D, Makhethe L, et al. The novel tuberculosis vaccine, AERAS-402, induces obust and polyfunctional CD4+ and CD8+ T cells in adults. American Journal of Respiratory and Critical Care Medicine. 2010;181:1407–1417. doi: 10.1164/rccm.200910-1484OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoft DF, Blazevic A, Stanley J, Landry B, Sizemore D, Kpamegan E, Gearhart J, et al. A recombinant adenovirus expressing immunodominant TB antigens can significantly enhance BCG-induced human immunity. Vaccine. 2012;30:2098–2108. doi: 10.1016/j.vaccine.2012.01.048. [DOI] [PubMed] [Google Scholar]
- 12.Magalhaes I, Sizemore DR, Ahmed RK, Mueller S, Wehlin L, Scanga C, Weichold F, et al. rBCG induces strong antigen-specific T cell responses in rhesus macaques in a prime-boost setting with an adenovirus 35 tuberculosis vaccine vector. PLoS ONE. 2008;3:e3790. doi: 10.1371/journal.pone.0003790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodworth JSM, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells and their role in immunity. Critical Reviews in Immunology. 2006;26:317–352. doi: 10.1615/critrevimmunol.v26.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonato VLD, Lima VMF, Tascon RE, Lowrie DB, Silva CL. Identification and characterization of protective T cells in hsp65 DNA-vaccinated and Mycobacterium tuberculosis-infected mice. Infection and Immunity. 1998;66:169–175. doi: 10.1128/iai.66.1.169-175.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng CG, Britton WJ. CD4+ and CD8+ T cells mediate adoptive immunity to aerosol infection of Mycobacterium bovis bacillus Calmette-Guerin. Journal of Infectious Diseases. 2000;181:1846–1849. doi: 10.1086/315466. [DOI] [PubMed] [Google Scholar]
- 16.Green AM, DiFazio R, Flynn JL. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. Journal of Immunology. 2013;190:270–277. doi: 10.4049/jimmunol.1200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silva CL, Lowrie DB. Identification and characterization of murine cytotoxic T cells that kill Mycobacterium tuberculosis. Infection and Immunity. 2000;68:3269–3274. doi: 10.1128/iai.68.6.3269-3274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodworth JS, Wu Y, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells require perforin to kill target cells and provide protection in vivo. J Immunol. 2008;181:8595–8603. doi: 10.4049/jimmunol.181.12.8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orme IM. The kinetics of emergence and loss of mediator T lymphocytes acquired in response to infection with Mycobacterium tuberculosis. Journal of Immunology. 1987;138:293–298. [PubMed] [Google Scholar]
- 20.Henao-Tamayo MI, Ordway DJ, Irwin SM, Shang SB, Shanley C, Orme IM. Phenotypic definition of effector and memory T-lymphocyte subsets in mice chronically infected with Mycobacterium tuberculosis. Clinical and Vaccine Immunology. 2010;17:618–625. doi: 10.1128/CVI.00368-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ordway D, Palanisamy G, Henao-Tamayo M, Smith EE, Shanley C, Orme IM, Basaraba RJ. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. Journal of Immunology. 2007;179:2532–2541. doi: 10.4049/jimmunol.179.4.2532. [DOI] [PubMed] [Google Scholar]
- 22.van Pinxteren LAH, Cassidy JP, Smedegaard BHC, Agger EM, Andersen P. Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. European Journal of Immunology. 2000;30:3689–3698. doi: 10.1002/1521-4141(200012)30:12<3689::AID-IMMU3689>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Cooper AM, D’Souza CD, Frank AA, Orme IM. The course of Mycobacterium tuberculosis infection in the lungs of mice lacking expression of either perforin- or granzyme-mediated cytolytic mechanisms. Infection and Immunity. 1997;65:1317–1320. doi: 10.1128/iai.65.4.1317-1320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner J, D’Souza CD, Pearl JE, Marietta P, Noel M, Frank AA, Appelberg R, et al. CD8-and CD95/95L-dependent mechanisms of resistance in mice with chronic pulmonary tuberculosis. American Journal of Respiratory Cell and Molecular Biology. 2001;24:203–209. doi: 10.1165/ajrcmb.24.2.4370. [DOI] [PubMed] [Google Scholar]
- 25.Urdahl KB, Liggitt D, Bevan MJ. CD8+ T cells accumulate in the lungs of Mycobacterium tuberculosis-infected Kb−/−Db−/− mice, but provide minimal protection. Journal of Immunology. 2003;170:1987–1994. doi: 10.4049/jimmunol.170.4.1987. [DOI] [PubMed] [Google Scholar]
- 26.Lindenstrøm T, Knudsen NPH, Agger EM, Andersen P. Control of chronic Mycobacterium tuberculosis infection by CD4 KLRG1− IL-2-secreting central memory cells. Journal of Immunology. 2013;190:6311–6319. doi: 10.4049/jimmunol.1300248. [DOI] [PubMed] [Google Scholar]
- 27.Nordly P, Rose F, Christensen D, Nielsen HM, Andersen P, Agger EM, Foged C. Immunity by formulation design: Induction of high CD8+ T-cell responses by poly(I:C) incorporated into the CAF01 adjuvant via a double emulsion method. Journal of Controlled Release. 2011;150:307–317. doi: 10.1016/j.jconrel.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Hansen J, Lindenstrøm T, Lindberg-Levin J, Aagaard C, Andersen P, Agger EM. CAF05: cationic liposomes that incorporate synthetic cord factor and poly(I:C) induce CTL immunity and reduce tumor burden in mice. Cancer Immunology Immunotherapy. 2012;61:893–903. doi: 10.1007/s00262-011-1156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korsholm KS, Karlsson I, Tang ST, Brandt L, Agger EM, Aagaard C, Andersen P, et al. Broadening of the T-Cell Repertoire to HIV-1 Gag p24 by Vaccination of HLA-A2/DR Transgenic Mice with Overlapping Peptides in the CAF05 Adjuvant. PLoS ONE. 2013:8. doi: 10.1371/journal.pone.0063575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56:405–419. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 31.Beekman NJ, van Veelen PA, van Hall T, Neisig A, Sijts A, Camps M, Kloetzel PM, et al. Abrogation of CTL epitope processing by single amino acid substitution flanking the C-terminal proteasome cleavage site. Journal of Immunology. 2000;164:1898–1905. doi: 10.4049/jimmunol.164.4.1898. [DOI] [PubMed] [Google Scholar]
- 32.Mo AXY, van Lelyveld SFL, Craiu A, Rock KL. Sequences that flank subdominant and cryptic epitopes influence the proteolytic generation of MHC class I-presented peptides. Journal of Immunology. 2000;164:4003–4010. doi: 10.4049/jimmunol.164.8.4003. [DOI] [PubMed] [Google Scholar]
- 33.Shastri N, Serwold T, Gonzalez F. Presentation of endogenous peptide MHC class-I complexes is profoundly influenced by specific C-terminal flanking residues. Journal of Immunology. 1995;155:4339–4346. [PubMed] [Google Scholar]
- 34.Woodworth JS, Shin D, Volman M, Nunes-Alves C, Fortune SM, Behar SM. Mycobacterium tuberculosis directs immunofocusing of CD8+ T cell responses despite vaccination. Journal of Immunology. 2011;186:1627–1637. doi: 10.4049/jimmunol.1002911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grotzke JE, Siler AC, Lewinsohn DA, Lewinsohn DM. Secreted immunodominant Mycobacterium tuberculosis antigens are processed by the cytosolic pathway. Journal of Immunology. 2010;185:4336–4343. doi: 10.4049/jimmunol.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewinsohn DM, Grotzke JE, Heinzel AS, Zhu L, Ovendale PJ, Johnson M, Alderson MR. Secreted proteins from Mycobacterium tuberculosis gain access to the cytosolic MHC class-I antigen-processing pathway. Journal of Immunology. 2006;177:437–442. doi: 10.4049/jimmunol.177.1.437. [DOI] [PubMed] [Google Scholar]
- 37.Neyrolles O, Gould K, Gares MP, Brett S, Janssen R, O’Gaora P, Herrmann JL, et al. Lipoprotein access to MHC class I presentation during infection of murine macrophages with live mycobacteria. Journal of Immunology. 2001;166:447–457. doi: 10.4049/jimmunol.166.1.447. [DOI] [PubMed] [Google Scholar]
- 38.Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, Modlin RL, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nature Medicine. 2003;9:1039–1046. doi: 10.1038/nm906. [DOI] [PubMed] [Google Scholar]
- 39.Kessler JH, Khan S, Seifert U, Le Gall S, Chow KM, Paschen A, Bres-Vloemans SA, et al. Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes. Nature Immunology. 2011;12 :45–U67. doi: 10.1038/ni.1974. [DOI] [PubMed] [Google Scholar]
- 40.Nielsen M, Lundegaard C, Lund O, Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 41.Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nature Immunology. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- 42.Feliu V, Vasseur V, Grover HS, Chu HH, Brown MJ, Wang J, Boyle JP, et al. Location of the CD8 T cell epitope within the antigenic precursor determines immunogenicity and protection against the Toxoplasma gondii parasite. Plos Pathogens. 2013;9:e1003449. doi: 10.1371/journal.ppat.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knudsen NPH, Nørskov-Lauritsen S, Dolganov GM, Schoolnik GK, Lindenstrøm T, Andersen P, Agger EM, et al. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:1096–1101. doi: 10.1073/pnas.1314973111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elvang T, Christensen JP, Billeskov R, Hoang T, Holst P, Thomsen AR, Andersen P, et al. CD4 and CD8 T cell responses to the M. tuberculosis Ag85B-TB10. 4 promoted by adjuvanted subunit, adenovector or heterologous prime boost vaccination. PLoS ONE. 2009;4:e5139. doi: 10.1371/journal.pone.0005139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radosevic K, Wieland CW, Rodriguez A, Weverling GJ, Mintardjo R, Gillissen G, Vogels R, et al. Protective immune responses to a recombinant adenovirus type 35 tuberculosis vaccine in two mouse strains: CD4 and CD8 T-cell epitope mapping and role of gamma interferon. Infection and Immunity. 2007;75:4105–4115. doi: 10.1128/IAI.00004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma XY, Serna A, Xu RH, Sigal LJ. The amino acid sequences flanking an antigenic determinant can strongly affect MHC class I cross-presentation without altering direct presentation. Journal of Immunology. 2009;182:4601–4607. doi: 10.4049/jimmunol.0803806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennekov T, Dietrich J, Rosenkrands I, Stryhn A, Doherty TM, Andersen P. Alteration of epitope recognition pattern in Ag85B and ESAT-6 has a profound influence on vaccine-induced protection against Mycobacterium tuberculosis. European Journal of Immunology. 2006;36:3346–3355. doi: 10.1002/eji.200636128. [DOI] [PubMed] [Google Scholar]
- 48.Baldwin SL, Ching LK, Pine SO, Moutaftsi M, Lucas E, Vallur A, Orr MT, et al. Protection against tuberculosis with homologous or heterologous protein/vector vaccine approaches is not dependent on CD8+ T cells. Journal of Immunology. 2013;191:2514–2525. doi: 10.4049/jimmunol.1301161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivastava S, Ernst JD. Cutting Edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. Journal of Immunology. 2013;191:1016–1020. doi: 10.4049/jimmunol.1301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Juarrero M, Turner OC, Turner J, Marietta P, Brooks JV, Orme IM. Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis. Infection and Immunity. 2001;69:1722–1728. doi: 10.1128/IAI.69.3.1722-1728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liebana E, Marsh S, Gough J, Nunez A, Vordermeier HM, Whelan A, Spencer Y, et al. Distribution and activation of T-lymphocyte subsets in tuberculous bovine lymph-node granulomas. Veterinary Pathology. 2007;44:366–372. doi: 10.1354/vp.44-3-366. [DOI] [PubMed] [Google Scholar]
- 52.Van den oord JJ, De Wolf-peeters C, Facchetti F, Desmet VJ. Cellular composition of hypersensitivity-type granulomas: Immunohistochemical analysis of tuberculous and sarcoidal lymphadenitis. Human Pathology. 1984;15:559–565. doi: 10.1016/s0046-8177(84)80010-6. [DOI] [PubMed] [Google Scholar]
- 53.Kaufmann SHE, Flesch IEA. The role of T cell - macrophage interactions in tuberculosis. Springer Seminars in Immunopathology. 1988;10:337–358. doi: 10.1007/BF02053845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.