

Abstract
Background
Anthrax lethal toxin (LT), secreted by Bacillus anthracis, causes severe cardiac dysfunction by unknown mechanisms. LT specifically cleaves the docking domains of MAPKK (MEKs); thus, we hypothesized that LT directly impairs cardiac function through dysregulation of MAPK signaling mechanisms.
Methods and Results
In a time-course study of LT toxicity, echocardiography revealed acute diastolic heart failure accompanied by pulmonary regurgitation and left atrial dilation in adult Sprague-Dawley rats at time points corresponding to dysregulated JNK, phospholamban (PLB) and protein phosphatase 2A (PP2A) myocardial signaling. Using isolated rat ventricular myocytes, we identified the MEK7-JNK1-PP2A-PLB signaling axis to be important for regulation of intracellular calcium (Ca2+i) handling, PP2A activation and targeting of PP2A-B56α to Ca2+i handling proteins, such as PLB. Through a combination of gain-of-function and loss-of-function studies, we demonstrated that over-expression of MEK7 protects against LT-induced PP2A activation and Ca2+i dysregulation through activation of JNK1. Moreover, targeted phosphorylation of PLB-Thr17 by Akt improved sarcoplasmic reticulum Ca2+i release and reuptake during LT toxicity. Co-immunoprecipitation experiments further revealed the pivotal role of MEK7-JNK-Akt complex formation for phosphorylation of PLB-Thr17 during acute LT toxicity.
Conclusions
Our findings support a cardiogenic mechanism of LT-induced diastolic dysfunction, by which LT disrupts JNK1 signaling and results in Ca2+i dysregulation through diminished phosphorylation of PLB by Akt and increased dephosphorylation of PLB by PP2A. Integration of the MEK7-JNK1 signaling module with Akt represents an important stress-activated signalosome that may confer protection to sustain cardiac contractility and maintain normal levels of Ca2+i through PLB-T17 phosphorylation.
Keywords: Diastolic dysfunction, Anthrax lethal toxin, JNK1/2, Akt, Phospholamban
Introduction
Systemic anthrax toxicity resulting from inhalation or ingestion of B. anthracis spores causes severe cardiac dysfunction [1, 2]. The incubation period for anthrax infection is an average of 4.5 days, while death occurring from systemic anthrax infection may occur within 4-7 days post-infection [1, 2]. Because many of the symptoms are relatively nonspecific, including cough, dyspnea, pleural effusions or enlarged mediastinum, an early diagnosis of clinical anthrax infection is challenging [1, 2]. Furthermore, no effective therapies currently exist to improve the depressed ventricular performance that accompanies anthrax toxicity.
Although Bacillus anthracis produces two separate toxins, edema toxin (ET) and lethal toxin (LT), we and others have shown LT to be primarily responsible for anthrax-associated cardiac dysfunction [3-5]. LT consists of a zinc metalloprotease, lethal factor (LF), that specifically cleaves MEK members 1, 2, 3, 4, 6 and 7 at the N-terminal MAP kinase docking-site [6, 7], causing varying degrees of disruption among MEK-MAPK family members and downstream MAPK signaling [8]. To our knowledge, however, no report has described the molecular mechanism for LT toxicity linking LF-mediated MEK cleavage with functional and molecular mechanisms associated with impaired cardiac function.
Given the quintessential roles of MAPKs in mediating the processes of remodeling, survival, growth and contractility in the myocardium, we hypothesized that LT-mediated cleavage of MEKs directly causes cardiac dysfunction through dysregulation of MAPK signaling networks. In this study, we provide evidence that LT induces acute diastolic dysfunction in rats through dysregulation of JNK and Akt signaling, enhanced PP2A-B56α activity and dephosphorylation of the SERCA2a pump regulator, phospholamban (PLB). Over-expression of cardiac myocyte MEK7 protected against LT-induced PP2A activation and Ca2+i dysregulation through activation of JNK1. Furthermore, gain-of-function studies demonstrated that phosphorylation of PLB-T17 by Akt may serve as a therapeutic strategy to improve SR Ca2+i handling and diastolic function during anthrax toxicity.
Methods
Animals
Male Sprague-Dawley (SD) rats were purchased from Charles River Laboratories (Cambridge, MA) and acclimated to the Scott and White Health Care animal facilities before experimentation. The animals were allowed food and tap water ad libitum and colony room lights were regulated on a 12:12-h lightdark cycle. All animal care and use were performed in accordance with National Institutes of Health and American Association for the Accreditation of Laboratory Animal Care (AAALAC) guidelines, and approved by the Scott and White Health Care/Texas A&M Health Science Center Institutional Animal Care and Use Committee.
Toxin preparation and administration to animals
Anthrax lethal toxin (LT) components PA and LF were produced at over 95% purity with low endotoxin level, as previously described [4]. All toxin components were diluted in filter-sterilized 1X phosphate-buffered saline (PBS) (5 mL) at a concentration of 20 μg/mL PA and 10 μg/mL LF. Rats (250-350 g) were randomized into control (n=10) and LT (n=30) treatment groups for a 2, 4, 8 and 24 h time course of LT toxicity. For experiments, conscious rats were administered a 0.5 ml bolus of PBS or LT (20 μg PA + 10 μg LF) by tail vein injection, as previously described [5].
Echocardiography measurements
Echocardiography was performed to determine effects of LT on cardiac function. At 12–24 h prior to toxin administration, rats were subjected to echocardiography to establish baselines and exclude any animals with abnormal cardiac function. Echocardiography was again performed immediately prior to tissue harvest for each LT treatment group and controls. We used a previously established echocardiography protocol [9] to determine systolic and diastolic function in the rats.
Tissue harvest
Control and LT-treated rats were administered 20 μL heparin (1000 U/mL) at the time of ketamine injection. Immediately upon removal, hearts were perfused with 4 °C 0.1 M glucose/PBS buffer, after which left ventricles were dissected and stored at -180 °C in liquid N2. Approximately 5 mL of blood from the inferior vena cava was obtained and stored in 2-mL tubes containing 10 μl heparin, 5 μl of 0.5 M EDTA and 5 μL 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF). Plasma was separated by centrifugation at 4°C and stored at -180 °C in liquid N2.
Plasma analysis
Plasma was analyzed using an Abaxis VetScan VS2 analyzer (Union City, CA). Approximately 100 μL plasma was injected into a Comprehensive Diagnostic Profile (Cat No: 500-0038-24) reagent rotor designed to determine concentrations of albumin, alkaline phosphatase, alkaline aminotransferase, amylase, total bilirubin, blood urea nitrogen, calcium, phosphorus, creatinine, glucose, sodium, potassium, total protein and globulin in the rat.
Preparation of left ventricular tissue lysates
Tissue was homogenized using a Tissuemiser Homogenizer (Fisher Scientific, Pittsburgh, PA). Approximately 0.02 g left ventricular tissue from each rat was homogenized in 200 μL of ice-cold PBS homogenization buffer containing 1 mmol/L dithiothreitol (DTT), 10 mM sodium bisulfate, 4 mmol/L sodium orthovanadate, 100 mmol/L sodium fluoride, 20% glycerol, 0.1 % triton-X and one tablet Complete Mini-Protease Inhibitor (Roche Applied Science) per 10 mL buffer. Insoluble material was removed by centrifugation for 15 min at 24 g and samples were boiled with loading buffer. Total protein concentration was determined using a kit (BioRad DC Protein Assay, Hercules, CA) according to the manufacturer's recommendation.
Antibodies and reagents
Phospho-p38-T180/Y182 antibody (9211), p38 antibody (9217), phospho-SAPK/JNK-T183/Y185 antibody (9251), JNK antibody (9252), phospho-ERK-T44/Y42 antibody (9106), ERK antibody (4695), phospho-MKK7-S271/T275 antibody (4171), MKK7 antibody (4172) phospho-PDK1-S241 antibody (3061), PDK1 antibody (3062), phospho-Akt-T308 antibody (9275), phospho-Akt-T473 antibody (9271), pan-Akt antibody (9272), phospho-GSK-3-beta-S9 antibody (9323), GSK-3-beta antibody (9315), PP2A/c antibody (2259), phospho-troponin-I-S23/S24 (cardiac) antibody (4004), troponin-I antibody (4002) and horseradish peroxidase-conjugated secondary antibodies (7074) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Phospho-PLB-S16 antibody (07-052) and PLB antibody (05-205) were purchased from Millipore (Billerica, MA). Phospho-PLB-T17 antibody (A010-13) was purchased from Badrilla, Ltd. (Leeds, U.K.). GAPDH antibody, phospho-PKAα antibody, PKAα-antibody, PP2A-B56a antibody and PP1 antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Bovine serum albumin (BSA, diagnostic grade K) was obtained from Celluliance (Kanakee, IL). Enhanced chemiluminescence (ECL) reagent (Western Lightning™) was obtained from PerkinElmer Life Science (Boston, MA).
Isolation of cardiac cells
Cultures of neonatal rat ventricular myocytes (NRVM) were prepared from 0-3-day-old Sprague-Dawley rat pups using enzymatic dispersion and Percoll purification methods, as previously described [10]. The NRVM were plated at a density of 4.8 × 106 cells/well in 6-well plastic dishes. Cytosine arabinoside (0.1 μmol/L final concentration) was added to prevent proliferation of nonmyocytes.
LT treatment of NRVM
Approximately 24 h after NRVM plating, NRVM were treated with LT in a 2:1 ratio of PA:LF prepared in dispersion medium. PA (activated, 63 kD segment) and LF were purchased from List Biological Laboratories (Campbell, CA) and each reconstituted in glycerol at a concentration of 0.5 mg/mL.
NRVM lysate preparation
For Western blotting and phosphatase activity studies, NRVM were harvested on ice with 1X Cell Lysis Buffer (Cell Signaling Technology, Inc.) supplemented with 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mmol/L 2-(2-aminoethyl)-benzenesulfonyl fluoride, hydrochloride and 1 mmol/L sodium orthovanadate. Insoluble material was removed by centrifugation for 15 min at 14,000 g. Total protein concentration was determined using a kit (BioRad DC Protein Assay) according to the manufacturer's recommendation.
Western Blotting
Western blotting was performed as previously described [11], in which equal amounts of protein (50 μg from tissue lysates or 30 μg from cell lysates) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and blotted onto polyvinylidene fluoride (PVDF) transfer membranes. Densities of the protein bands were quantified using ImageQuant software (GE Healthcare Biosciences, Piscataway, NJ). All blots were stripped and probed with GAPDH antibody to confirm equal loading. Densities from the phosphoprotein bands were normalized to total protein.
Adenovirus infection
For adenoviral infection of NRVM, cells were infected with replication-defective adenoviruses. HAB56α adenovirus (100 multiplicity of viral infection [MOI]) was provided as a gift from Dr. Terry B. Rogers (University of Maryland School of Medicine, Baltimore, MD). SERCA2a (100 MOI) was provided as a gift from Dr. David Rosenbaum (Case Western Reserve University, Cleveland, OH). Dominant-negative (DN)-PLB (100 MOI) was provided by Dr. Wolfgang Dillmann (Division of Endocrinology and Metabolism, University of California San Diego, La Jolla). SR-targeted Akt (100 MOI) and the empty vector virus control (25-50 MOI) were provided as gifts from Dr. Gianluigi Condorelli (Institute of Biomedical Technologies, Milan, Italy). Constitutively-active (CA)-MEK7 (100 MOI) was a gift from Dr. Yibin Wang (University of California, Los Angeles). DN-JNK1 (50 MOI) was purchased from Cell Biolabs Inc. (San Diego, CA), and DN-JNK2 (100 MOI) was purchased from Seven Hills Bioreagents (Cincinnati, OH). The adenovirus expressing empty vector was used to control for nonspecific effects of adenoviral infection. Levels of expressed proteins were determined by Western blot analysis. For each adenovirus used, NRVM were titrated with 25, 50, 100, 150 and 200 MOI virus to maximize expressed protein, but prevent viral toxicity. At 24 h after plating, cardiac myocytes were infected with each adenovirus diluted in DMEM/medium 199. The medium was replaced with virus-free DMEM/medium 199 after 24 h adenoviral infection, and cells were cultured for an additional 12 h prior to experiments. The viral MOI was determined by dilution assay in HEK-293 cells grown in 6-well clusters, as previously described [11, 12].
Phosphatase assays
PP2A and PP1 enzymatic activities were determined using the RediPlate 96 Enzchek Serine/Threonmine Phosphatase Assay Kit obtained from Invitrogen (Carlsbad, CA), according to the manufacturer's instructions. PP2A and PP1 enzymatic activities were calculated as a ratio of the fluorescence value (based on fluorescence values at 30 min) to the band intensity of PP2A catalytic subunit (determined by Western blot analysis), which is a measure of specific PP2A activity.
Immunofluorescence
Frozen LV tissues were sectioned (6 μm) and blocked with 5% milk in 1X automation buffer (Biomedia Corp, Foster City, CA) at 4°C for 1 h, then incubated in either troponin-I anti-rabbit or PLB anti-mouse IgG primary antibody at a dilution of 1:125 in automation buffer for 3 h at 4°C. Sections were washed with sterile PBS 3 times and incubated 2 h with Alexa Fluor 488 secondary antibody (chicken anti-rabbit or goat anti-mouse IgG) diluted 1:125 in automation buffer at 4°C. Afterwards, sections were washed 1 time with sterile PBS, incubated 10 min with 1:100 dilution of propidium iodide, and incubated 7 min with a 1:10000 dilution 6,8-difluoro-4-methylumbelliferyl phosphate (Di-FMUP) diluted in 1 mM NiCl2. Sections were rinsed in PBS and imaged using an Olympus Fluoview FV1000 confocal microscope at 40× magnification. NRVM seeded on microscope slides were fixed in 4% paraformaldehyde, permeabilized in 0.05% Triton X-100, and blocked with 5% milk in automation buffer 20 min at 37° C. Cells were then incubated 1 h with anti-rabbit hemagglutinin (HA), anti-rabbit SERCA2a, anti-mouse PLB or anti-mouse Akt (1:125) antibodies. Cells were washed in 5% milk and incubated 45 min at 37°C with Alexa Fluor 488 secondary antibody (goat anti-mouse IgG) or Alexa Fluor 350 secondary antibody (goat anti-rabbit) diluted 1:125 in automation buffer at 4°C. Nuclei were stained with Sytox Orange (Invitrogen, Carlsbad, CA) 10 min and mounted with anti-fade mounting media. Cells were viewed with an Olympus Fluoview FV1000 confocal microscope at 60× (oil).
Co-immunoprecipitation experiments
Freshly harvested NRVM lysates (300 μg protein) were co-incubated at a 1:50 dilution of either anti-MEK7 antibody or anti-pan-Akt antibody (Cell Signaling Technology) with end-over-end mixing at 4°C overnight. Subsequently, the antibody/lysate mixture was incubated with 20 μL Protein G beads (Santa Cruz Biotechnology) for 3 h at 4°C, with end-over-end mixing. Immunoprecipitates were washed with BupH TBST buffer (Pierce, Thermo Scientific, Rockford, IL) and eluted through a spin-column (Pierce, Thermo Scientific). Co-immunoprecipitates were dissociated in reducing sample buffer (IgG Elution Buffer, Pierce, Thermo Scientific) and separated on 10% and 20% Tris-HCL gels using SDS-PAGE.
Pacing and Ca2+i measurements with Fura-2-AM
NRVM plated on round glass coverslips (25 mm, #1 thickness) were incubated with MEM containing 1 μM Fura 2-AM (Molecular Probes) for 40 min at 37°C and washed with fresh MEM to remove excess dye. Fluorescence measurements were recorded with a dual-excitation fluorescence photomultiplier system (IonOptix Corporation, Milton, MA). Myocytes were imaged through a 40× Olympus objective. Cells were exposed to light emitted by a 75 Watt lamp and passed through either a 360 or a 380 nm filter (± 15 nm bandwidths), while being field stimulated to contract at 1.0-3.0 Hz (5 ms duration). Fura-2-loaded cells were excited at 360±6.5 nm and 380±6.5 nm with an ultraviolet xenon lamp. Emission fluorescence was measured at 510±15 nm. IonWizard 6.3 software (IonOptix Corporation) was utilized to perform monotonic transient analysis of Ca2+ peaks, including calculations of the single exponential tau, velocities of Ca2+ release and re-uptake and peak height.
Intracellular calcium measurements via FACS
NRVM treated with LT were co-incubated with Fluo-4 AM and Fura-red cell-permeant calcium dyes (1 μmol/L final concentration, Invitrogen) for 1 h at 37°C [13]. Cells were lifted from the 6-well plate using TrypLE and PBS and collected in a 1.5-mL centrifuge tube. Cells were centrifuged 30 sec at 1000 rpm, room temperature, resuspended in 500 μL serum-free medium and analyzed on a BD FACSCalibur flow cytometer.
Statistical analysis
Blood chemistry data, densitometry, phosphatase enzymatic activities and Ca2+ transient data were analyzed by one-way ANOVA and Tukey's post hoc test. Echocardiographic parameters were analyzed by paired t-test of baseline values compared to values post- LT treatment for each treatment group. Otherwise, comparisons between two groups were analyzed by unpaired t-test. Data are reported as mean ± SEM. Probability values <0.05 were considered significant.
Results
Anthrax LT intoxication causes a temporal increase in vascular permeability and acute diastolic dysfunction in vivo
Although recent animal studies identified the heart as a target of anthrax toxicity [14], the signaling mechanisms responsible for LT-induced cardiac dysfunction remain to be determined. We previously established that PA is internalized within 20 min post-LT injection [4], therefore we sought to elucidate the acute response of LT toxicity on cardiac function and MAPK signaling. LF metalloprotease activity cleaves MEKs within the N-terminal docking site [15]; therefore, we hypothesized that MAP kinases p38, JNK1/2 and/or ERK may be directly affected in the heart, resulting in contractile dysfunction. To test this hypothesis, we performed a time course (2, 4, 8 and 24 h LT) of anthrax toxicity in SD rats. A bolus i.v. injection of 20 ng/mL PA + 10 ng/mL LF, a dose corresponding to low LT toxicity [4], was administered through the lateral tail vein. We chose a low dose of LT to determine whether LT toxicity has a cardiac-specific functional effect, in contrast to higher doses of LT that may produce cardiac dysfunction secondary to systemic toxicity. We observed a 68% survival rate at the conclusion of the 24 h time-course (Figure 1A).
Figure 1. LT-treated SD rats develop diastolic dysfunction.
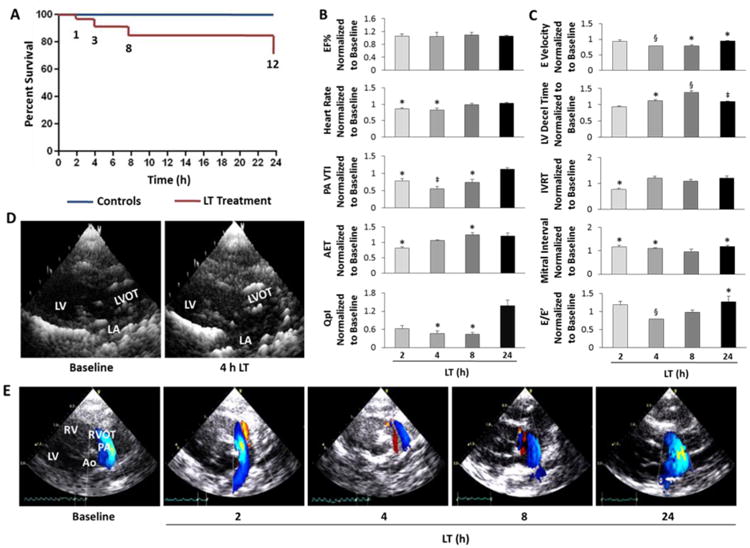
A, Kaplan-Meier survival curve for controls and LT-treated SD rats. B, Cardiac indices of systolic function, including percent ejection fraction (EF%), blood flow through the pulmonary artery (PA) measured as the PA velocity time integral (VTI), aortic ejection time (AET), and cardiac output normalized to body weight (QpI). C, Cardiac indices of diastolic function, including early (E) LV diastolic filling velocities, LV deceleration (decel) time, and isovolumic relaxation time (IVRT) of LT-treated SD rats normalized to individual baseline values for each rat. E/E′ indicates early diastolic filling velocities measured by continuous wave Doppler divided as a ratio to the tissue Doppler E velocity. D, Representative parasternal long-axis views of the left atrium (LA), LV outflow tract (LVOT) and LV at baseline and 4 h post-LT. 2-D images were rendered as 3-D images using Gwyddion software. E, Representative color flow Doppler images of pulmonary artery blood flow during the time-course of LT toxicity. Red indicates blood flow towards the probe and blue indicates blood flow away from probe. All values are reported as mean ± SEM. n = 6-10 rats per group. Statistical significance is depicted as *P<0.05, ‡P<0.01 and §P<0.001 by paired t-test to individual baseline values for each animal.
Rats with acute LT toxicity (2-4 h post-LT) had diminished heart rates and pulmonary blood flow (normalized to body weight, QpI), although ejection fraction (EF %) remained relatively constant during the time-course (Figure 1B). At 4-8 h post-LT, the aortic ejection time (AET) was prolonged and the early diastolic filling velocity was slowed. Left ventricular deceleration time was also significantly slowed at 4-8 and 24 h post-LT, whereas the mitral interval was prolonged both acutely (2-4 h) and at 24 h post-LT (Figure 1C), indicating impaired ventricular relaxation. At 24 h post-LT, significantly elevated E/E′ values further established the presence of diastolic dysfunction. Pulmonary vein flow velocities occurring during systole, diastole and atrial contraction (S, D and A waves, respectfully) indicated reduced diastolic flow and increased atrial flow velocities at 2 and 4 h post-LT injection (Supplementary Figure 1). In addition, left atrium chamber dilation at 4 h LT (Figure 1D) and pulmonary regurgitation (2-8 h LT) were observed (Figure 1E), suggesting increased pulmonary flow and LV pressures resulting from LV diastolic dysfunction. Taken together, the cardiac functional data reflect an acute (2-4 h) decline in diastolic function with compensation occurring at 24 h post-LT.
Pulmonary effusion observed during human anthrax toxicity [2] suggests that anthrax toxin increases vascular permeability. In validation of our animal model and LT dosage, our rat model also demonstrated pulmonary effusion with associated temporal changes in concentrations of specific plasma proteins and electrolytes. Diagnostic analysis of plasma collected from LT-treated rats revealed significant reductions in concentrations of plasma albumin, globulin and total protein at 4 h (Supplementary Table 1), consistent with observed increases in abdominal fluid and pulmonary edema (data not shown). Although changes in plasma proteins may indicate hepatic or renal etiologies [16, 17], the lack of significant changes in alanine aminotransferase (ALT), blood urea nitrogen (BUN) or creatine levels suggests that neither acute kidney nor liver injury were responsible. The significant increase in plasma calcium and protein levels at 4 h coincided with decreased blood flow (Figure 1B), therefore the protein and electrolyte abnormalities may be explained by the systemic effects of LT causing vascular permeability or cardiovascular etiologies.
LT diminishes JNK, Akt and PLB phosphorylation and enhances PP2A activity in rat hearts
LT-mediated MEK cleavage inhibits MEK-MAPK binding affinity, not MEK phosphorylation [18], thus we examined activities (phosphorylation) of ERK, JNK and p38 MAPKs in LV tissue lysates from LT-treated rats (Figure 2A). ERK phosphorylation was not significantly altered, JNK activity was significantly decreased during the early stages (2 to 8 h post-LT), and p38 phosphorylation was diminished at later times (8 and 24 h) (Figure 2A).
Figure 2. MAPK, Akt and Ca2+ihandling signaling proteins are dysregulated in LV tissue of LT-treated SD rats.
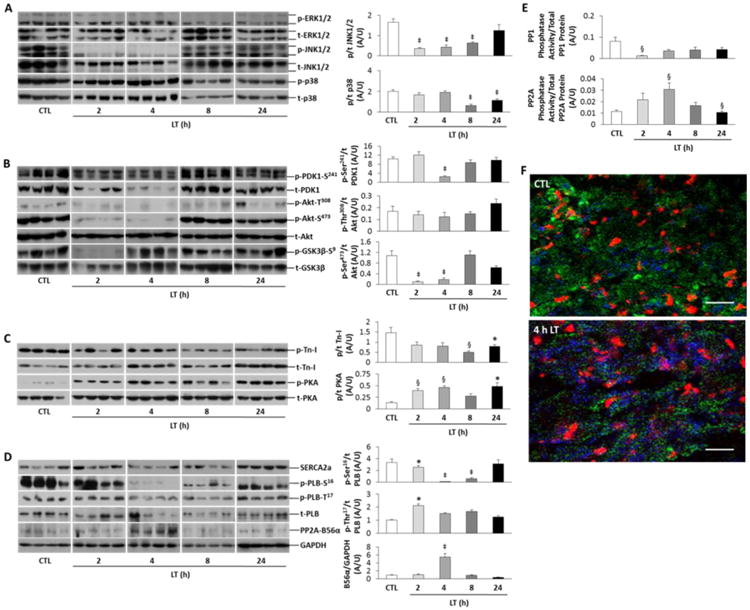
A, Representative immunoblots of phosphorylated (p) and total (t) ERK, JNK and p38 MAPK. Densitometry of p/t is shown at right for JNK and p38. B, Immunoblots for PDK1, Akt phosphorylation at Thr308 and Ser473 and GSK3β. Densitometry at right shows p/t for PDK1 and Akt. C, Immunoblots and densitometry (p/t) for troponin-I (Tn-I) and PKA. D, Immunoblots of phospholamban (PLB, p-Ser16 and p-Thr17), SERCA2a, PP2A-B56α and GAPDH total protein. Densitometry shows p/t for B56α/GAPDH and PLB (p-Ser16 and p-Thr17/t). E, PP1 and PP2A specific phosphatase activities are represented as a ratio of PP1 or PP2A enzymatic activity to total PP1 or PP2Ac (PP2A catalytic subunit) protein. F, Representative confocal microscopic images (60×, oil) of frozen LV tissue sections from a control and 4 h LT-treated rat showing immunofluorescence of Tn-I (green), propidium iodide (red)staining and PP2A activity (Di-FMU fluorescence, blue). Scale bar is 200 μm. All values are reported as mean ± SEM. For all analyses, n = 6-10 rats per group. Statistical analysis: *P<0.05, ‡P<0.01 and §P<0.001 versus Controls in one-way ANOVA.
Akt (protein kinase B) activity plays a dynamic role in influencing both survival and ventricular contractile performance [19, 20]. We further hypothesized that cardiac Akt activity may be affected by LT. Examination of PDK1, the upstream activator of Akt-Thr308, revealed diminished phosphorylation at 4 h post-LT treatment with decreased total PDK1 protein at 2 and 4 h LT. Interestingly, Akt-Thr308 phosphorylation and total protein levels were not significantly reduced compared to controls; however, phosphorylation of Akt-Ser473 was significantly diminished during the early time points (2 and 4 h LT). At 8 h LT, Akt-Ser473 phosphorylation normalized (Figure 2B). These results suggest that an upstream activator of Akt-Ser473 may be a target of LT, and that the Akt-Thr308 site maintains phosphorylation possibly through a PDK1-independent mechanism.
To determine whether prolonged relaxation time is indicative of impaired myofilament Ca2+ cycling dynamics, LT-exposed LV tissues were probed for troponin-I (Tn-I) phosphorylation (Ser23/24). Tn-I activity was significantly decreased compared to controls at all time-points post-LT treatment. Interestingly, PKA activity was increased at all time-points post-LT, implying β-adrenergic receptor activation occurred secondary to reduced cardiac function. Impaired myocardial relaxation is often also associated with dysregulated sarcoplasmic reticulum (SR) Ca2+ handling. Therefore, we further hypothesized that molecular machinery regulating diastolic Ca2+ levels, such as PLB phosphorylation or SERCA2a expression, may act as downstream targets of LT. SERCA2a protein levels were not significantly different between control and LT-treated groups (Figure 2D), however, both PLB-Ser16 phosphorylation and total protein levels declined at 2 h and were dramatically decreased at 4 and 8 h post-LT. Phosphorylation of PLB-Thr17, though significantly higher at 2 h post-LT, was not different through the remainder of the time-course. Because dephosphorylation of SR Ca2+ handling proteins may be attributable to PP2A-B56α targeting [21], we also measured PP2A-B56α total protein, which was significantly enhanced at 4 h post-LT.
The activities and phosphorylation of several cardiac Ca2+-handling proteins, including PLB, Tn-I, ryanodine receptor, and the L-type Ca2+ channel are regulated by PP1 and PP2A protein phosphatases [22]. We hypothesized that dephosphorylation of PLB at the 4-8 h LT time-points may be due to PP1 and/or PP2A-B56α activation. PP1 and PP2A protein levels were invariable (data not shown); however, PP2A enzymatic activity was elevated at 2 h post-LT and significantly increased at 4 h (Figure 2E), as confirmed by confocal microscopy of frozen LV tissue sections (Figure 2F). Enzymatic activity of PP1 significantly decreased at 2 h post-LT (Figure 2E), consistent with high β1-adrenergic stimulation. Acute β-adrenergic stimulation is reported to indirectly inhibit PP1, whereas chronic β-adrenergic stimulation increases PP2A phosphatase activity [23]. Based on the diastolic dysfunction observed during acute (4-8 h) LT toxicity, we theorized that LT-mediated loss in cardiac function is a result of targeted PLB dephosphorylation by PP2A in the myocardium.
LT dysregulates cardiomyocyte signaling in vitro and in creases diastolic Ca2+i levels
Phosphorylation of cardiac PKA in vivo and the compensatory changes in LV function observed at 24 h suggest that catecholamines played a role in modulating the signaling effects of LT observed in the LV tissue samples. As isolated neonatal rat ventricular myocytes (NRVM) are readily cultured and amenable to gene transfer methods, we used NRVM as a model in vitro system to determine the direct effects of anthrax LT on cardiac signaling and Ca2+i handling.
Based on our in vivo studies, we expected to observe LT-induced dephosphorylation of MAPK and Akt within 4 h LT treatment. Therefore, we chose a time-course of 15 min, 30 min, 1 h, 2 h and 4 h LT treatment to determine the temporal effects of LT on NRVM signaling. A dose of 0.05 ng/ml PA and 0.025 ng/ml LF was used, which induced JNK dephosphorylation in dose-response studies (Supplementary Figure 2A). Within 30 min to 1 h of LT treatment, phosphorylation of JNK1/2, ERK1/2, and Akt-Thr308/Ser473 were significantly diminished (Figure 3A).
Figure 3. Characterization of temporal signaling and Ca2+ihandling effects of LT in isolated CMs.
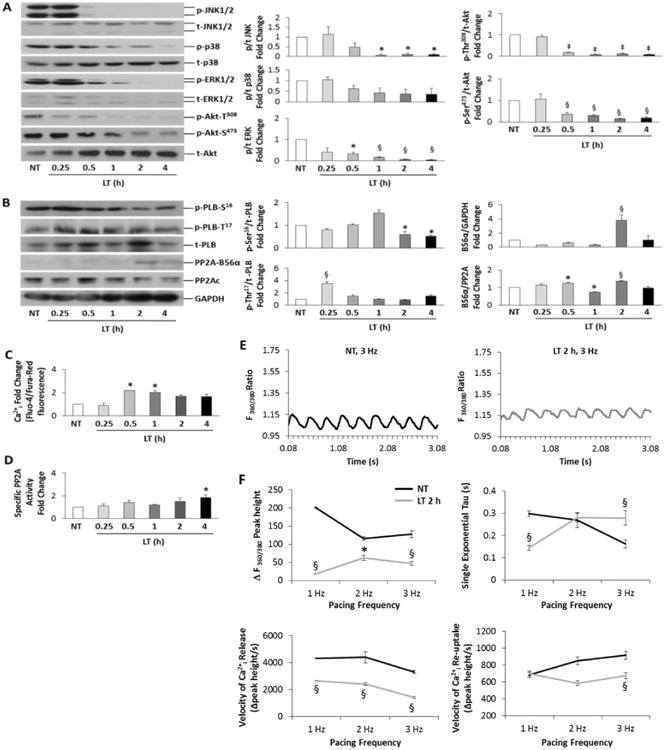
A, Western blot analysis (left) of MAPKs and Akt phosphorylation (p) and total (t) protein during timecourse of LT (0.05 ng/mL PA + 0.025 ng/mL LF) toxicity. Densitometry (p/t) fold change is shown at right. B, Immunoblotting (left) of PLB, PP2Ac (catalytic subunit) and PP2A-B56α. Densitometry (p/t) fold change is shown at right. GAPDH was used as a loading control. C, Ratiometric Ca2+I determination by Fluo-4/Fura-Red fluorescence, normalized to control (dye only). D, Specific PP2A activity fold change represented as PP2A enzymatic activity/PP2Ac total protein. n = 6-8 per group Densitometry and phosphatase enzymatic data were analyzed by one-way ANOVA; *P<0.05, ‡P<0.01 and §P<0.001 versus Controls. All values are reported as mean ± SEM. E, Representative Ca2+ transients monitored by Fura-2 ratiometic (360/380 nm) fluorescence imaging for CMs field-stimulated at 3.0 Hz; control (NT, non-treated) CMs (top) versus CMs treated with LT for 2 h (bottom). F, Group data for Ca2+ transient peak height [ΔF(360/380)], tau, velocities of Ca2+ release and re-uptake for NT and LT-treated CMs field-stimulated at 1, 2 and 3 Hz. Data are averaged from n=5 dispersions, averaging 4 groups of CMs and 5-10 transients per group. *P<0.05 and §P<0.001 versus NT in Student's t-test.
Consistent with in vivo data, PLB-Ser16 phosphorylation was significantly diminished at 2 and 4 h LT, and PLB-Thr17 phosphorylation was enhanced during the early (15 min post LT) time-course (Figure 3B). B56α levels were significantly elevated at 2 h LT when normalized to GAPDH, and significantly increased from 30 min to 2 h when normalized to total PP2Ac (PP2A catalytic subunit) protein. Since B56α protein levels are negatively regulated by JNK activity in NRVM [24], we hypothesized that LT may indirectly increase basal Ca2+i levels through dephosphorylation of PLB by PP2A-B56α. Ratiometric Ca2+i levels measured via flow cytometry of Fluo-4/Fura-red fluorescence revealed significantly elevated Ca2+i at 30 min and 1 h LT, although Ca2+i levels remained elevated approximately 2-fold for the remainder of the time-course (Figure 3C). Since the effects of LT on PLB-Ser16 were observed at 2-4 h LT, and B56α total protein was increased at 30 min - 4 h LT, the observance of elevated Ca2+i at 30 min may be due to targeting of PP2A-B56α to other proteins of the cardiac contractile apparatus, such as the ryanodine receptor or sodium pump. This observation is currently under investigation in our laboratory. Furthermore, NRVM treated with LT were characterized by elevated PP2A activity at 2 h, which was significantly enhanced at 4 h LT (Figure 3D).
To confirm that LT increases Ca2+i levels, we measured ratiometric Fura-2 Ca2+i fluorescence (excitation 360/380 nm, emission 510 nm) of paced NRVM at frequencies of 1, 2 and 3 Hz during 2 h of LT toxicity (Figure 3E). Compared to control cells (NT), LT-treated NRVM displayed higher diastolic Ca2+i levels at a pacing frequency of 3 Hz (Figure 3E), although the Ca2+ transients were characterized by significantly smaller changes in peak height (peak height-baseline) and slower velocities of Ca2+ release at all pacing frequencies (Figure 3F). Importantly, a significantly slower rate of Ca2+ release and re-uptake was observed for LT-treated NRVM at 3.0 Hz pacing, corresponding to an increased Tau, or slower ventricular recovery time. These findings suggest that both Ca2+ release and reuptake are directly affected by LT, resulting in dysregulated Ca2+i levels.
MEK7 protects against LT-mediated PP2A activation and Ca2+i dysregulation via JNK1 and Akt activation in vitro
To test the hypothesis that B56α overexpression would simulate the effects of LT toxicity with regard to PP2A activation and Ca2+i dysregulation, we transfected NRVM with HA-B56α1-expressing adenovirus and measured PP2A activity and Ca2+i levels. A 6-fold over-expression of HA-B56α1 in NRVM resulted in significantly higher levels of Ca2+i compared to virus control, which was enhanced when cells were co-treated with LT for 2 h (Supplementary Figure 2B, E). Although HA-B56α1 over-expression did not increase global PP2A activity levels (Supplementary Figure 2C), co-immunoprecipitation and co-localization of PLB, PP2Ac and Akt protein with HA-B56α revealed direct interactions between these proteins (Supplementary Figure 2D, F). These results suggest that PP2A activation and targeting by B56α to the SR are distinct molecular signaling events.
B56α over-expression alone was insufficient to induce PP2A activity; therefore, we tested whether JNK1/2 may serve as a functional regulator of Ca2+i handling through PP2A activation as well as through regulation of B56α protein levels. Previous studies support a lower level of binding efficiency of JNK to cleaved MEK7, compared to the other MAPK substrates and their respective MEKs [8]. We over-expressed constitutively active MEK7 (CA-MEK7) in NRVM by adenoviral infection (100 MOI) as an approach to sustain JNK1/2 activity during LT toxicity, and determine whether JNK1 or JNK2 may modulate Ca2+i levels and PP2A activity by co-expression with dominant-negative (DN)-JNK1 and DN-JNK2 viral vectors. These gain-of-function studies revealed a significant improvement in Ca2+i handling in NRVM during CA-MEK7 expression after 2 h LT toxicity compared to the empty vector (EV) virus control after LT treatment (Figure 4A). Furthermore, the protective effect of CA-MEK7 expression was lost with co-expression of DN-JNK1, both in the presence and absence of LT. These results indicate that JNK1, but not JNK2, plays a role in modulating Ca2+i levels in NRVM. The CA-MEK7 and DN-JNK1/DN-JNK2 co-expression studies further revealed a role for JNK1 in regulation of PP2A activity, as the significant increase in PP2A activity caused by LT was attenuated with CA-MEK7 expression (Figure 4B). Similar to the Ca2+i studies, the protection afforded by CA-MEK7 on PP2A activity was lost with co-expression of Ca-MEK7+DN-JNK1, but not during inhibition of JNK2, suggesting that Ca2+i levels are modulated by JNK1 through regulation of PP2A activity in NRVM.
Figure 4. MEK7 over-expression protects against LT-induced Ca2+idysregulation and PP2A activation.
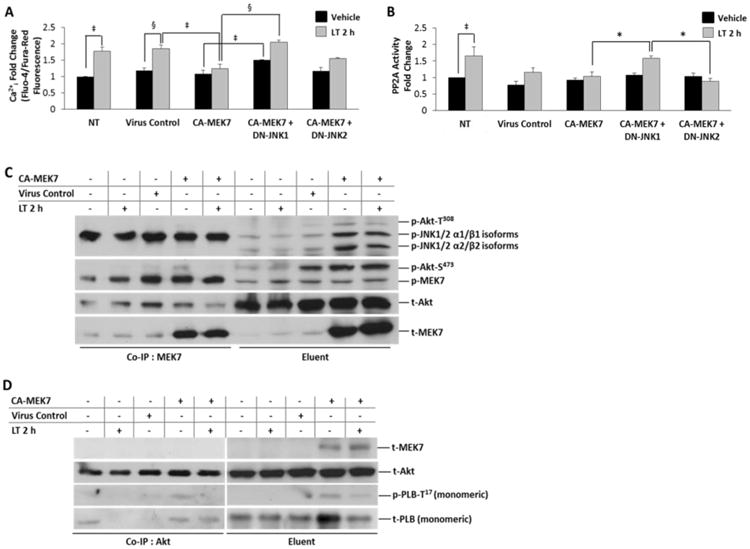
A, Ratiometric Ca2+i determination by Fluo-4/Fura-Red fluorescence, normalized to baseline fluorescence (dye only). NRVM were infected with the indicated adenoviral constructs (CA-MEK7, CA-MEK7+DN-JNK1 or CA-MEK7+DN-JNK2 for 36 h prior to either vehicle (distilled water) or 2 h LT (0.05 ng/mL PA + 0.025 ng/mL LF) treatment. Lysates harvested from NRVM infected with an empty expression vector were used as the virus control. B, Specific PP2A activity fold change represented as PP2A enzymatic activity/PP2Ac total protein. Adenovirus-infected NRVM were treated with either vehicle (distilled water) or LT (0.05 ng/mL PA + 0.025 ng/mL LF) 2 h prior to fluorometric determination of PP2A enzymatic activity. C, Co-immunoprecipitation of MEK7 with phosphorylated MEK7, JNK, Akt-T308 and Akt-S473 from NRVM lysates expressing CA-MEK7. Co-immunoprecipitated protein is shown at left, and the input lysate (eluent) is shown at right. D, Co-immunoprecipitation of p- PLB-T17 and t-Akt from NRVM lysates with anti-Akt (pan) antibody after expression of CA-MEK7 adenovirus. Co-immunoprecipitated protein is shown at left, and the input lysate (eluent) is shown at right. MEK7 protein is found in the lysate (eluent); CA-MEK7-induced p-PLB-T17/Akt interactions are revealed as co-immunoprecipitated (Co-IP) by Akt. N = 4.
Our data shows Akt phosphorylation to follow a similar pattern as JNK phosphorylation both in vivo and in vitro during anthrax toxicity. Akt directly phosphorylates PLB at Thr17 to modulate SR Ca2+ handling in rat ventricular myocytes [25]; therefore, we extended our hypothesis to integrate Akt activation and targeting to the SR as an effector of MEK7-mediated JNK1 regulation. The effects of CA-MEK7 expression on JNK and Akt phosphorylation monitored by Western blot analysis revealed significantly enhanced phosphorylation of Akt at both the Thr308 and Ser473 sites with concomitant increases in JNK1/2 phosphorylation during CA-MEK7 expression (Supplementary Figure 3). Although phosphorylation Akt-Thr308 was unaffected during CA-MEK7 expression by DN-JNK1 or DN-JNK2, phosphorylation of the Akt-Ser473 site was significantly increased during DN-JNK2 expression. Furthermore, expression of CA-MEK7 maintained significantly higher phosphorylation levels of JNK1/2 and Akt-Ser473 during 2 h LT treatment compared to LT treatment alone (Supplementary Figure 3), which suggests that activation of MEK7 may protect against LT-induced Ca2+i dysregulation through sustained phosphorylation of JNK1/2 and Akt. To further support this hypothesis, we observed that total PP2A-B56α protein levels were significantly lowered during MEK7-CA + LT 2h treatment compared to LT 2h.
To determine whether CA-MEK7 expression results in complex formation between JNK and Akt-Ser473 we performed co-immunoprecipitation experiments using anti-MEK7 antibody and NRVM lysates prepared from cells expressing CA-MEK7 with or without LT 2h treatment. We found that MEK7 coimmunoprecipitated with JNK1/2 heavy isoforms (α1β1) and Akt-Ser473 but not Akt-Thr308 (Figure 4C). Interestingly, coimmunoprecipitated T-Akt and Akt-Ser473 were reduced during LT treatment. We further investigated whether CA-MEK7, with or without LT treatment, would induce an interaction between Akt and PLB-Thr17. Interestingly, CA-MEK7 induced interactions between Akt and monomeric p-PLB-Thr17, which were diminished during LT toxicity (Figure 4D). These results suggest that LF-mediated MEK7 cleavage may be responsible for reduced MEK7-JNK1/2-Akt-Ser473 interactions and Akt-mediated PLB-Thr17 phosphorylation, and that JNK1-mediated regulation of PP2A activity and Ca2+i levels can be rescued by CA-MEK7 expression during LT toxicity in vitro.
SR-Targeted Akt Functionally Improves Diastolic Ca2+i Handling during LT Toxicity through phosphorylation of PLB-T17
To establish whether the LT-mediated increase in Ca2+i levels can be rescued by strategies aimed to enhance diastolic Ca2+i handling, we infected NRVM with adenoviral vectors that would over-express SERCA2a, reduce PLB functional inhibition of SERCA2a (DN-PLB) or phosphorylate (inactivate) PLB-Thr17 (SR-targeted Akt adenovirus, denoted as Akt→SR). Over-expression of HA-B56α was used as a negative control. Compared to the virus control, both the DN-PLB and the Akt→SR adenoviral constructs protected against elevated Ca2+i levels during 2 h LT toxicity (Figure 5A). The Akt→SR adenoviral construct, however, was the only strategy that maintained Ca2+i levels during LT toxicity compared with its baseline Ca2+i in the absence of LT. While over-expression of SERCA2a did not produce an effect different from the virus control, the HA-B56α adenovirus enhanced LT-induced Ca2+ dysfunction. Immunoblotting confirmed that Akt→SR adenoviral expression improved Ca2+i handling through phosphorylation of PLB-Thr17 (Figure 5B). We further tested whether expression of Akt→SR or CA-MEK7 could improve diastolic Ca2+i handling in paced myocytes in the presence and absence of LT. Although we had hypothesized that CA-MEK7 expression may enhance Ca2+i handling, only Akt→SR expression exhibited the ability to maintain baseline diastolic Ca2+i after 2 h LT, compared to either virus control (EV) or CA-MEK7 at 3 Hz pacing frequency (Figure 5C). Furthermore, Akt→SR expression allowed for a significantly more efficient release and re-uptake of Ca2+i during LT toxicity, although peak Ca2+ levels were higher both at 1 Hz and 3 Hz pacing frequencies (Figure 5D). Importantly, the efficient re-uptake of Ca2+i at 3 Hz correlated with a significantly lower Tau. CA-MEK7 expression, however, appeared to function similar to the virus control at 1 Hz, but at faster pacing frequencies, velocities of Ca2+i release and reuptake declined to values lower than the control, resulting in a higher tau. These results suggest that direct phosphorylation of PLB-T17 may function as a therapeutic target in acute LT-induced diastolic dysfunction.
Figure 5. Akt→SR adenoviral expression improves SR Ca2+ihandling, release and re-uptake during acute LT toxicity.
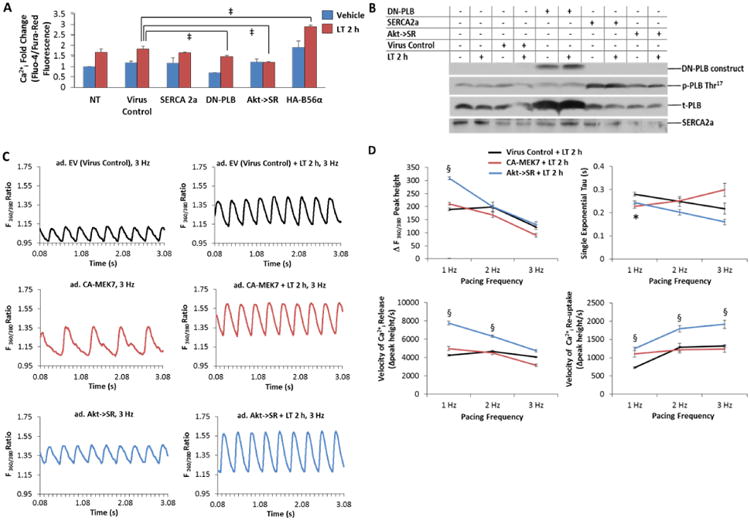
A, NRVM were infected with the indicated adenoviral constructs (SERCA 2a, DN-PLB, Akt→SR or HA-B56α) for 36 h prior to either vehicle (distilled water) or 2 h LT (0.05 ng/mL PA + 0.025 ng/mL LF) treatment. Ratiometric Ca2+i determination by Fluo-4/Fura-Red fluorescence was normalized to baseline fluorescence (dye only). Lysates harvested from NRVM infected with an empty expression vector were used as the virus control. B, Representative immunoblots of DN-PLB, SERCA 2a. Akt→SR or empty vector adenoviral expression in the presence or absence of LT treatment reveals that both SERCA 2a and Akt→SR expression induces phosphorylation of PLB-T17 . C, Representative Ca2+ transients monitored by Fura-2 ratiometic (360/380 nm) fluorescence imaging for CMs field-stimulated at 3.0 Hz are depicted for adenovirus-treated (ad.) CMs in the absence of LT (left) and in the presence of LT 2 h treatment (right). Expression of empty vector (EV) adenovirus serves as virus control (top) for CA-MEK7 (middle) and Akt→SR (bottom). D, Group data for Ca2+ transient peak height [ΔF(360/380)], tau, velocities of Ca2+ release and re-uptake for EV (virus control), CA-MEK7 and Akt→SR after 2 h LT-treatment of CMs field-stimulated at 1, 2 and 3 Hz. Data are averaged from n=5 dispersions, averaging 4 groups of CMs and 5-10 transients per group. *P<0.05 and §P<0.001 versus virus control.
Discussion
Our work suggests a regulatory role for JNK in cardiomyocyte relaxation, through inhibition of PP2A-B56α and activation of Akt, which influence PLB activity and SR Ca2+i handling. This is the first report, to our knowledge, that describes a mechanism of acute anthrax-induced diastolic dysfunction originating from LF-induced disruption of JNK MAPK signaling. Our findings support a model of anthrax LT toxicity, by which acute diastolic dysfunction occurs as a result of dysregulated MEK7-JNK1-PP2A-PLB signaling and SR Ca2+i handling in vivo (Figure 6). Furthermore, we provide evidence that MEK7-induced JNK1/2 activation regulates Akt-Ser473 phosphorylation and PLB-Akt interactions in myocytes. Importantly, adenoviral-mediated strategies to improve SR Ca2+i handling through phosphorylation of PLB-Thr17 by Akt resulted in significant improvements in myocyte Ca2+i handling, re-uptake and speed of diastolic recovery (Tau) during LT toxicity, establishing PLB as a potential therapeutic target for anthrax-induced cardiac dysfunction.
Figure 6. Signaling diagram depicting the direct effect of anthrax LT on MEK7, which prevents MEK7-JNK1/2-Akt complex formation, and induces PP2A-B56α activity.
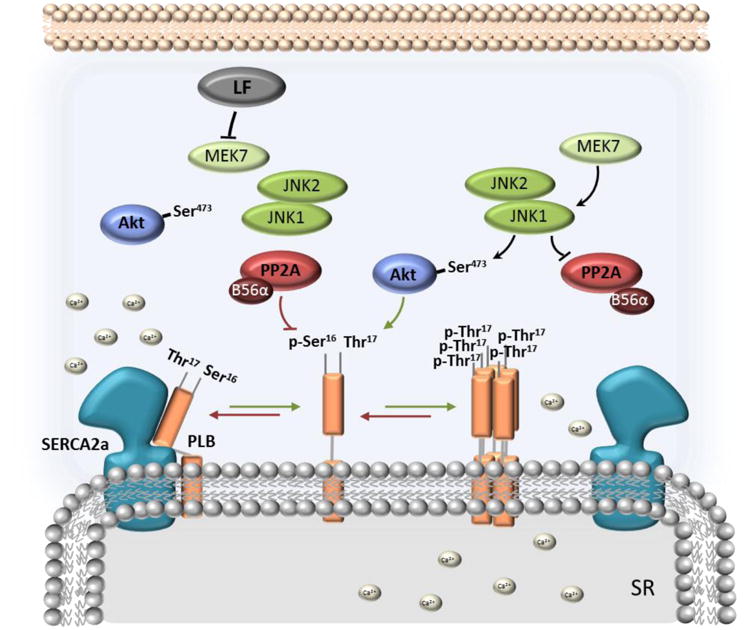
Downstream of MEK7, JNK1 activation negatively regulates PP2A activity and targeting by B56α and positively regulates Akt- PLB-T17 interactions. PP2A-B56α dephosphorylates PLB (red arrows), causing inhibitory interactions between PLB monomer and SERCA2a, preventing Ca2+i cycling from the cytoplasm inside the SR and resulting in impaired cardiac myocyte relaxation. We further propose a hypothetical model of p-PLB- Thr17-induced PLB pentamerization (green arrows), resulting from phosphorylation of PLB-Thr17 by Akt.
Bardwell, et al identified MEK7 and MEK4, the upstream activators of JNK1/2, as having the highest rate of catalytic cleavage by LF compared to MEK1/2, 3 and 6, due to the position of a second cleavage site within the docking domains of MEK4 and MEK7 [8]. In addition, the role of JNK during basal and acute stress responses, which can vary from protective to mal-adaptive, may be determined by the particular upstream MEK, the strength of the binding interaction with the docking domain [26], as well as other kinases and scaffold proteins that facilitate JNK activation during complex formation. Therefore, it was interesting to note in co-immunoprecipitation studies that over-expression of MEK7 induced complex formation with p-Akt-Ser473 and JNK1/2 α1β1 (heavy isoforms), but not p-Akt-Thr308. This complex formation, however, was diminished during LT toxicity, suggesting that cleavage of MEK7 by LT impairs MEK7/JNK/Akt-Ser473 interactions. Furthermore, co-immunoprecipitation experiments during CA-MEK7 expression revealed Akt/PLB-T17 interactions, which were also reduced during LT toxicity. Although JNK activation by MEK7 and MEK4 is reportedly synergistic in non-cardiac cells, MEK7 is required for JNK activation and responds to pro-inflammatory cytokines [26]. Our findings suggest that LT may disrupt a stress-activated pathway involving MEK7 signaling interactions with JNK1/2 (α1β1 isoforms) and p-Akt-Ser473, which functionally enhances Ca2+i cycling dynamics during conditions of acute myocardial stress through specific targeting of Akt to PLB-Thr17. Functional Ca2+i handling studies, however, revealed that MEK7 over-expression only afforded protection during low-frequency (1 Hz) pacing during LT toxicity; while direct targeting of PLB-T17 phosphorylation by Akt (Akt→SR adenoviral expression) significantly improved Ca2+i reuptake throughout the pacing challenge. Thus, MEK7 activation may play a role in fine-tuning Ca2+i handling in cardiomyocytes, as during mechanosignaling, but is probably not responsible for dramatic changes in SR Ca2+ handling and contractile function as observed during β1-adrenergic responses. Further work will reveal whether Akt-SR targeting in the heart may preserve diastolic function during LT toxicity in vivo.
We propose that the deleterious effects of LT on MAPKs and downstream signaling targets, such as Akt, are responsible for a range of pathologic effects, depending on the context and cell-specific roles of JNK, p38 and ERK in various tissues. This postulate is supported by findings from studies investigating the effects of anthrax LT in other tissue systems. For example, anthrax infection has been shown to inhibit Ecadherin-mediated cell-cell interactions in lung epithelial cells through inhibition of Akt activity [27], which may also contribute to increased endothelial barrier dysfunction and vascular permeability [28]. In immune cells, however, effects of LT are more profound in relation to p38 and ERK, while effects of JNK activity are negligible. Inhibition of p38 by LT induces apoptosis in macrophages [29], and disruption of p38 and ERK signaling impairs inflammatory-mediated superoxide production in neutrophils [30] and activation of murine T-lymphocytes [31]. Our studies, however, revealed LT-induced impairment of JNK, ERK and Akt, while p38 activity was not significantly affected. This finding may be due to the dual nature of SAP kinases in mediating protection and survival during acute stress and adaptive remodeling during chronic stress to maintain contractility [32]. Thus, the pathological effects of anthrax LT in different tissue systems may be largely dependent on the range of functions and specific signaling dynamics of individual MAPKs within that cell type.
Although depressed ventricular function is well-documented in various animal models of anthrax toxicity [5, 33-36], it is not widely accepted that direct LT-mediated cardiac dysfunction plays an integral role in anthrax-related human deaths. Our findings, however, suggest a new paradigm regarding the perception of how anthrax affects cardiac function. In particular, our studies reveal three considerations to be important: (1) the acute nature of the diastolic and compensatory effects in the heart and lack of early (acute phase) diagnostic data on anthrax patients; (2) the combination of specific multisystem effects of anthrax toxin on disease progression; and (3) the occurrence of LT-induced diastolic dysfunction due to acute, intracellular signaling-dependent Ca2+i dysregulation that distinguishes it from other cardiac disease etiologies.
The first point relates to our observation that once in the bloodstream, LT effects occur quickly, in the magnitude of hours, not days, within small animal models. In the SD rats, we observed compensatory cardiac effects within 24 h, after the acute diastolic inefficiency has passed. In many animal studies monitoring cardiac function during anthrax toxicity, experimental observations are on the magnitude of days, which would explain why depressed hemodynamics at later time points appear to correspond to systolic failure [5, 34-36]. In human patients, it is perhaps more likely that diastolic dysfunction would occur within 1-3 days, depending on the severity of bacillus anthracis contact and dependence of this bacterial strain on quorum sensing [37] for release of anthrax toxins. In three detailed patient reports [1, 2], however, the patients were admitted to the hospital 3, 5 and 7 days after development of initial symptoms. The patient reported by Mina, et al was admitted on day 3 and initially treated for congestive heart failure, but the other two patients were quickly diagnosed with anthrax toxicity [1, 2]. In each case, however, cardiac compensation had already occurred (characterized by tachycardia), and the patient succumbed to the disease. The clinical implications of our study suggest that acute diastolic dysfunction occurs due to the direct signaling effects of LT on cardiac myocytes, contributing to the vascular and respiratory effects of LT that culminate in cardiovascular failure. We did not observe tachycardia in the LT-treated SD rats in this study; however we have previously reported tachycardia in SD rats due to ET administration [4]. In human anthrax infections, the presence of both LT and ET may cause an acute form of diastolic dysfunction characterized by tachycardia, which may initially preserve cardiac function by maintaining a constant ejection fraction. It is anticipated, however, that as endothelial barrier dysfunction and capillary wall disintegration worsen, venous return will be significantly dampened and cardiac compensation lost. Because similar symptoms, such as dyspnea and pulmonary edema, accompany both diastolic dysfunction and LT-mediated respiratory distress, echocardiography is a crucial diagnostic test to detect LT-induced diastolic dysfunction in humans in conjunction with immediate analysis of blood cultures for anthrax bacilli.
The second consideration accounts for the multi-organ, systemic effects of LT and ET, which may also progressively differentiate and worsen, depending on the health of the patient prior to anthrax exposure. As the cell-specific effects of MEK cleavage and adenylate cyclase activity occur in various organ systems over the period of anthrax toxicity, the infection becomes systemic. In the above three cases, all three patients were hypoxemic and developed pericardial effusion and pulmonary edema. Pulmonary edema and pericardial effusion were observed in our SD rats and commonly accompany diastolic dysfunction; however, these symptoms may also occur due to endothelial barrier dysfunction mediated by LT in lung tissue [27]. Although it is currently unknown whether the effects of LT on the coronary vasculature contribute to anthrax-induced cardiac dysfunction, hypoxemia and capillary wall disintegration have been reported in various organ systems [38], causing respiratory, hepatic and renal insufficiency [39]. Apparently, the heart is able to functionally compensate during the initial stages of diastolic dysfunction accompanying anthrax toxicity, but as respiratory and vascular effects become more devastating, cardiac reserves eventually fail. Thus, the time of cardiac failure is probably also dependent on the cardiac health of the patient before anthrax exposure.
The last consideration proposes that the type of diastolic dysfunction we have reported herein is novel and may serve as an early marker of anthrax toxicity. Although clinical analysis of anthrax-induced diastolic dysfunction remains to be reported, our studies reveal that LT-induced diastolic dysfunction occurs through MEK7/JNK1/PP2A-B56α/Akt/PLB signaling dysregulation that differentiates anthrax effects from those associated with other cardiac disease etiologies. Moreover, the type of diastolic dysfunction we characterized diverges from the classical hypertrophic signaling, fibrosis and ECM remodeling that occurs during chronic pressure-overload and diabetic cardiomyopathies [40, 41]. We provide evidence for a class of diastolic dysfunction characterized by intracellular signaling-dependent (extracellular remodeling-independent) effects on Ca2+i handling and relaxation. Closely-related examples include alveolar hypoxia, endotoxemia and sepsis. Interestingly, PP2A activation in cardiac myocytes differentiates LT-induced diastolic dysfunction from endotoxemia [42], which is characterized by inhibition of cardiac myocyte PP2A activity. Alveolar hypoxia, however, is associated with enhanced cardiac myocyte PP2A activity and dephosphorylation of PLB-S16, through a mechanism involving amplified circulating IL-18 levels [43]. Moreover, anthrax toxicity in rats and humans is not accompanied by an inflammatory response [3], which further differentiates anthrax-related cardiac dysfunction from alveolar hypoxia and sepsis [44]. These signaling patterns provide a molecular blueprint that distinguishes and identifies the underlying cause of cardiomyopathy.
In summary, our work identified a specific pathway, the MEK7/JNK1/PP2A/PLB signaling axis to be important for regulation of Ca2+i handling, and molecular interactions such as MEK7/JNK/Akt complex formation, as signaling targets of anthrax LT in the heart. Future work will be required to determine whether SR Ca2+i handling may serve as an early therapeutic target to prevent anthrax-induced cardiac dysfunction, or to maintain functional cardiac reserves during antibiotic treatment of anthrax infection and patient recovery.
Supplementary Material
Acknowledgments
This manuscript is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas. This work was supported by grants from the Department of Veterans Affairs (1I01BX000801, to D.D.), National Institutes of Health (HL-68838, to D.D) and the Scott and White Health Care System (to H.G., L.W. and D.D.).
Footnotes
There are no potential conflicts of interest among the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Borio L, Frank D, Mani V, et al. Death due to bioterrorism-related inhalational anthrax: report of 2 patients. Jama. 2001;286:2554–9. doi: 10.1001/jama.286.20.2554. [DOI] [PubMed] [Google Scholar]
- 2.Mina B, Dym JP, Kuepper F, et al. Fatal inhalational anthrax with unknown source of exposure in a 61-year-old woman in New York City. Jama. 2002;287:858–62. doi: 10.1001/jama.287.7.858. [DOI] [PubMed] [Google Scholar]
- 3.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alphaindependent hypoxia-mediated toxicity in mice. The Journal of clinical investigation. 2003;112:670–82. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watson LE, Kuo SR, Katki K, et al. Anthrax toxins induce shock in rats by depressed cardiac ventricular function. PLoS ONE. 2007;2:e466. doi: 10.1371/journal.pone.0000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson LE, Mock J, Lal H, et al. Lethal and edema toxins of anthrax induce distinct hemodynamic dysfunction. Front Biosci. 2007;12:4670–5. doi: 10.2741/2416. [DOI] [PubMed] [Google Scholar]
- 6.Duesbery NS, Webb CP, Leppla SH, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–7. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 7.Vitale G, Bernardi L, Napolitani G, et al. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. The Biochemical journal. 2000;352 Pt 3:739–45. [PMC free article] [PubMed] [Google Scholar]
- 8.Bardwell AJ, Abdollahi M, Bardwell L. Anthrax lethal factor-cleavage products of MAPK (mitogen-activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. The Biochemical journal. 2004;378:569–77. doi: 10.1042/BJ20031382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson LE, Sheth M, Denyer RF, Dostal DE. Baseline echocardiographic values for adult male rats. J Am Soc Echocardiogr. 2004;17:161–7. doi: 10.1016/j.echo.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Golden HB, Gollapudi D, Gerilechaogetu F, et al. Isolation of cardiac myocytes and fibroblasts from neonatal rat pups. Methods Mol Biol. 2012;843:205–14. doi: 10.1007/978-1-61779-523-7_20. [DOI] [PubMed] [Google Scholar]
- 11.Lal H, Verma SK, Smith M, et al. Stretch-induced MAP kinase activation in cardiac myocytes: differential regulation through beta1-integrin and focal adhesion kinase. Journal of molecular and cellular cardiology. 2007;43:137–47. doi: 10.1016/j.yjmcc.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lal H, Verma SK, Golden HB, et al. Stretch-induced regulation of angiotensinogen gene expression in cardiac myocytes and fibroblasts: opposing roles of JNK1/2 and p38alpha MAP kinases. Journal of molecular and cellular cardiology. 2008;45:770–8. doi: 10.1016/j.yjmcc.2008.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burchiel SW, Edwards BS, Kuckuck FW, et al. Analysis of free intracellular calcium by flow cytometry: multiparameter and pharmacologic applications. Methods (San Diego, Calif. 2000;21:221–30. doi: 10.1006/meth.2000.1002. [DOI] [PubMed] [Google Scholar]
- 14.Moayeri M, Crown D, Dorward DW, et al. The heart is an early target of anthrax lethal toxin in mice: a protective role for neuronal nitric oxide synthase (nNOS) PLoS Pathog. 2009;5:e1000456. doi: 10.1371/journal.ppat.1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vitale G, Pellizzari R, Recchi C, et al. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochemical and biophysical research communications. 1998;248:706–11. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 16.Marion D. Crush-related acute kidney injury (acute renal failure) In: Basow D, editor. UpToDate. Waltham, MA: UpToDate; 2010. [Google Scholar]
- 17.Marion D. Tests of the liver's biosynthetic capacity. In: Basow D, editor. UpToDate. Waltham, MA: UpToDate; 2010. [Google Scholar]
- 18.Turk BE, Wong TY, Schwarzenbacher R, et al. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat Struct Mol Biol. 2004;11:60–6. doi: 10.1038/nsmb708. [DOI] [PubMed] [Google Scholar]
- 19.Cittadini A, Monti MG, Iaccarino G, et al. Adenoviral gene transfer of Akt enhances myocardial contractility and intracellular calcium handling. Gene therapy. 2006;13:8–19. doi: 10.1038/sj.gt.3302589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rota M, Boni A, Urbanek K, et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility. Circulation research. 2005;97:1332–41. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 21.Bhasin N, Cunha SR, Mudannayake M, et al. Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. American journal of physiology. 2007;293:H109–19. doi: 10.1152/ajpheart.00059.2007. [DOI] [PubMed] [Google Scholar]
- 22.El-Armouche A, Bednorz A, Pamminger T, et al. Role of calcineurin and protein phosphatase-2A in the regulation of phosphatase inhibitor-1 in cardiac myocytes. Biochemical and biophysical research communications. 2006;346:700–6. doi: 10.1016/j.bbrc.2006.05.182. [DOI] [PubMed] [Google Scholar]
- 23.De Arcangelis V, Soto D, Xiang Y. Phosphodiesterase 4 and phosphatase 2A differentially regulate cAMP/protein kinase a signaling for cardiac myocyte contraction under stimulation of beta1 adrenergic receptor. Molecular pharmacology. 2008;74:1453–62. doi: 10.1124/mol.108.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaser ND, Lukyanenko YO, Wang Y, et al. JNK activation decreases PP2A regulatory subunit B56alpha expression and mRNA stability and increases AUF1 expression in cardiomyocytes. American journal of physiology. 2006;291:H1183–92. doi: 10.1152/ajpheart.01162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catalucci D, Latronico MV, Ceci M, et al. Akt increases sarcoplasmic reticulum Ca2+ cycling by direct phosphorylation of phospholamban at Thr17. The Journal of biological chemistry. 2009;284:28180–7. doi: 10.1074/jbc.M109.036566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. Eur J Cell Biol. 2011;90:536–44. doi: 10.1016/j.ejcb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Popova T, Espina V, Bailey C, et al. Anthrax infection inhibits the AKT signaling involved in the E-cadherin-mediated adhesion of lung epithelial cells. FEMS Immunol Med Microbiol. 2009;56:129–42. doi: 10.1111/j.1574-695X.2009.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warfel JM, Steele AD, D'Agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am J Pathol. 2005;166:1871–81. doi: 10.1016/S0002-9440(10)62496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–51. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 30.Xu L, Fang H, Frucht DM. Anthrax lethal toxin increases superoxide production in murine neutrophils via differential effects on MAPK signaling pathways. J Immunol. 2008;180:4139–47. doi: 10.4049/jimmunol.180.6.4139. [DOI] [PubMed] [Google Scholar]
- 31.Comer JE, Chopra AK, Peterson JW, Konig R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infection and immunity. 2005;73:8275–81. doi: 10.1128/IAI.73.12.8275-8281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy:dichotomy between cultured myocytes and animal models. Journal of molecular and cellular cardiology. 2003;35:1385–94. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Cui X, Li Y, Li X, et al. Bacillus anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J Infect Dis. 2007;195:572–80. doi: 10.1086/510856. [DOI] [PubMed] [Google Scholar]
- 34.Hicks CW, Li Y, Okugawa S, et al. Anthrax edema toxin has cAMP-mediated stimulatory effects and high-dose lethal toxin has depressant effects in an isolated perfused rat heart model. American journal of physiology. 2011;300:H1108–18. doi: 10.1152/ajpheart.01128.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lawrence WS, Marshall JR, Zavala DL, et al. Hemodynamic effects of anthrax toxins in the rabbit model and the cardiac pathology induced by lethal toxin. Toxins (Basel) 2011;3:721–36. doi: 10.3390/toxins3060721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sweeney DA, Cui X, Solomon SB, et al. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J Infect Dis. 2010;202:1885–96. doi: 10.1086/657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones MB, Jani R, Ren D, et al. Inhibition of Bacillus anthracis growth and virulence-gene expression by inhibitors of quorum-sensing. J Infect Dis. 2005;191:1881–8. doi: 10.1086/429696. [DOI] [PubMed] [Google Scholar]
- 38.Moayeri M, Leppla SH. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med. 2009;30:439–55. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Firoved AM, Miller GF, Moayeri M, et al. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol. 2005;167:1309–20. doi: 10.1016/S0002-9440(10)61218-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falcao-Pires I, Hamdani N, Borbely A, et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. 2011;124:1151–9. doi: 10.1161/CIRCULATIONAHA.111.025270. [DOI] [PubMed] [Google Scholar]
- 41.Falcao-Pires I, Palladini G, Goncalves N, et al. Distinct mechanisms for diastolic dysfunction in diabetes mellitus and chronic pressure-overload. Basic Res Cardiol. 2011;106:801–14. doi: 10.1007/s00395-011-0184-x. [DOI] [PubMed] [Google Scholar]
- 42.Marshall M, Anilkumar N, Layland J, et al. Protein phosphatase 2A contributes to the cardiacdysfunction induced by endotoxemia. Cardiovascular research. 2009;82:67–76. doi: 10.1093/cvr/cvp037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larsen KO, Lygren B, Sjaastad I, et al. Diastolic dysfunction in alveolar hypoxia: a role for interleukin-18-mediated increase in protein phosphatase 2A. Cardiovascular research. 2008;80:47–54. doi: 10.1093/cvr/cvn180. [DOI] [PubMed] [Google Scholar]
- 44.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.