

Abstract
Brain derived neurotrophic factor (BDNF) signaling through its receptor TrkB plays a crucial role in neurodevelopment and plasticity. Stress and glucocorticoids have been shown to alter TrkB signaling in neurons, and defects in TrkB expression have been reported in the prefrontal cortex of suicide subjects. Glucocorticoid treatment has been shown to induce deleterious effects on the neuronal maturation. However, the mechanisms involved in the regulation of TrkB by glucocorticoid during neurodevelopment are not clear. Here we show that acute corticosterone exposure induced posttranslational upregulation of TrkB in primary cortical neurons (days in vitro 4, DIV4), which was blocked by the proteasome inhibitors. Acute corticosterone-induced increase in TrkB protein levels was dependent on glucocorticoid receptor (GR). At the cellular level, ubiquitin E3 ligase c-Cbl mediates TrkB stabilization and corticosterone-induced TrkB levels. Moreover, the tyrosine kinase binding domain in c-Cbl plays a critical role in corticosterone-induced TrkB levels. Chronic treatment of neurons with corticosterone induced significant decreases in both TrkB and c-Cbl protein levels. Acute corticosterone treatment failed to induce any significant change in TrkB and c-Cbl protein levels in mature neurons (DIV 12), where as chronic corticosterone exposure reduced TrkB levels. Under an in vivo condition, chronic corticosterone exposure induced down-regulation of c-Cbl in mouse frontal cortex and hippocampus. Importantly, we demonstrate for the first time a significant decrease in c-Cbl mRNA levels in the prefrontal cortex of suicide subjects indicating the possible role of c-Cbl in the pathophysiology of suicidal behavior. Thus, ubiquitin-proteasome-mediated TrkB regulation may be an important mechanism for improving BDNF signaling and maintaining neuroplasticity in stress-related neuropsychiatric disorders.
Keywords: c-Cbl, suicide, TrkB, ubiquitination, neurons, glucocorticoid
Introduction
Stress and glucocorticoids are implicated in the pathophysiology of many neuropsychiatric disorders including anxiety, depression and schizophrenia (McEwen, 2005). Chronic glucocorticoid treatments have been shown to alter neuronal signaling and cognitive functions in rodents (Zafir and Banu, 2009; Chen et al., 2012; Tang et al., 2013). Glucocorticoids utilize a variety of brain-specific molecules to mediate their effects in the central nervous system (Joëls et al., 2012). Among these shared molecules are BDNF and its receptor, TrkB, which are expressed in both the developing and adult brain (Jeanneteau and Chao, 2013). BDNF via TrkB signaling exerts important actions on the neurodevelopment, maintenance, and function of the peripheral and central nervous system, and also in cognitive functions (Pillai, 2008). Moreover, several evidence indicate decreased expression of TrkB in different post-mortem brain areas, including hippocampus and frontal cortex of suicide completers (Dwivedi, 2009, Ernst et al., 2009; Dwivedi et al., 2009).
BDNF signaling is important for neuronal maturation, neurite outgrowth and synaptic connections during the developmental stage (Tartaglia et al., 2001; Miller and Kaplan, 2003). Moreover, glucocorticoid treatment has been shown to induce deleterious effects on the neuronal maturation (Kamphuis et al., 2003; Gross and Hen, 2004). Although chronic glucocorticoid treatment has been shown to cause detrimental effects on neuroplasticity, acute glucocorticoid exposure activates TrkB signaling in neurons (Jeanneteau et al., 2008). However, the mechanisms involved in the regulation of TrkB by glucocorticoid during neurodevelopment are not clear. It is known that protein posttranslational modifications play important roles in the regulation of tyrosine kinase receptors (Feng and Derynck, 2005; Kang et al., 2008). Ubiquitin proteasomal system (UPS) is an important posttranslational modification that regulates a number of cellular functions. Ubiquitination is a process involving three enzymes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) (Joazeiro and Hunter, 2000). Interactions between an E3 ligase and its target molecule are considered a key step in determining the selectivity of UPS for a target molecule and its subsequent proteasomal degradation, a process that is subject to intracellular modulation by various upstream regulators (Ardley and Robinson, 2005). Although ubiquitination often results in protein degradation, a number of studies have reported its role in the activation of signaling pathways (Pickart, 2001). It is known that ubiquitination through Lys63, but not Lys48 of the ubiquitin chain mediates cellular signaling, endocytosis, and protein kinase activation (Habelhah et al., 2004; Chen, 2005; Clague and Urbé, 2006; Jin et al., 2010). Casitas B-lineage lymphoma (c-Cbl) is an E3 ubiquitin ligase and is a regulator of receptor tyrosine kinases (RTKs) (Mohapatra et al., 2013). The tyrosine kinase binding (TKB) domain of c-Cbl is required for its binding with RTKs, and the RING finger domain is essential for its ubiquitin E3 ligase activity (Thien and Langdon, 2001). Interestingly, a recent study has shown that c-cbl promotes TGF-β signaling by neddylating and stabilizing the type II receptor (TβRII) (Zuo et al., 2013).
Prompted by studies that ubiquitination stabilizes the protein levels (Jin et al., 2010; Radovanac et al., 2013), and c-Cbl activates TGF-β signaling by stabilizing the receptor (Zuo et al., 2013), we hypothesized that glucocorticoid activates TrkB signaling via a c-Cbl dependent mechanism. We report that c-Cbl inhibition suppressed glucocorticoid-induced increase in TrkB levels in primary cortical neurons. We found that specific inhibition of Cbl interaction with RTK using a Cbl mutant (G306E) inhibits TrkB levels indicating the role of TKB domain in c-Cbl-mediated TrkB stabilization.
Materials and Methods
Human Postmortem Samples
Brain Tissue samples (Brodmann’s area 10) from 15 suicide completers and 13 psychiatrically normal controls were used in the current study. The samples were obtained from the Quebec Suicide Brain Bank (QSBB; Douglas Institute; www.douglas.qc.ca/suicide). Approval for Brain Bank was granted by the Douglas Hospital Institutional Review Board in accordance with the 1964 Declaration of Helsinki. A description of the Quebec Suicide Brain Bank and more detailed demographics and samples’ quality were previously reported (Freemantle et al., 2013a, b). Demographic and subject characteristics of the samples used in the present study are listed in Table 1.
Table 1.
Demographic data for post mortem brain samples.
| Variable | Control (N=15) | Suicide (N=13) |
|---|---|---|
| Age (years, mean±SE) | 40.73±3.52 | 28.73±4.05 |
| PMI (h, mean±SE) | 17.14±1.80 | 22.50±3.55 |
| Brain pH (mean±SE) | 6.63±0.06 | 6.43±0.11 |
Animal experiments
The acute in vivo experiments were conducted in CD-1 mice (Charles River Laboratories, Wilmington, MA, USA) at postnatal day 4 (PND4) where as the chronic in vivo experiments were conducted in 2-month old male CD1 mice (25–30 g). Adult animals were housed 4 mice per cage with water and food available ad libitum. Mice were maintained on a 12-h light–dark cycle with the lights on at 0700 hours. All experimental procedures were performed during the light cycle. All in vitro experiments were done in cerebral cortical neuronal cultures from embryonic day 16 (E16) mouse fetuses. Animal use procedures were performed after being reviewed and approved by Georgia Regents University, Committee on Animal Use for Research. Procedures were consistent with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines as per Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Cerebral Cortical Neuronal Cultures
Mouse cortical neurons were cultured as described previously (Howell et al., 2011). Cerebral cortices from CD-1 mouse embryos (E16) were aseptically dissected and plated at 3.5 × 105 cells per well on polyethyleneimine-coated plates. Neurons were cultured in Neurobasal medium supplemented with B27, 2 mM L-glutamine, and antibiotics (Invitrogen). The media was replaced with Neurobasal supplemented with B27 minus antioxidants, glutamine, and antibiotics on third day in vitro (DIV3). Neurons were used for glucocorticoid treatments at DIV 4 or DIV12. Following treatments, neurons were washed in Phosphate Buffered Saline (PBS) and collected in ice-cold RIPA buffer. Protein concentration was determined by the bicinchoninic acid method (BCA Protein Assay Kit, Sigma, USA).
Drug treatment
In vivo study
Corticosterone (Sigma, St Louis, MO) was dissolved in 0.45% hydroxypropyl-β-cyclodextrin (Sigma). In acute glucocorticoid treatment studies, mice at PND4 were given a single injection of corticosterone (10 mg/kg) or vehicle subcutaneously, and the forebrain samples were collected at 6 h following the injection. In chronic studies, mice were treated for 5 or 7 weeks with vehicle or corticosterone (35 ug/ml equivalent to 5 mg/kg/day; David et al., 2009; Kutiyanawalla et al., 2011). The vehicle as well as drug solution were delivered ad libitum in the drinking water. At the end of the treatment, the animals were sacrificed by cervical dislocation, and frontal cortex and hippocampus samples from vehicle as well as drug-treated mice were collected (Paxinos and Franklin, 2001). The tissue samples were lysed in RIPA buffer supplemented with protease inhibitor cocktail (Sigma). The supernatants were collected following centrifugation at 14,000 rpm for 15 min at 4 °C and stored at −70°C. Protein concentrations were determined by the BCA method.
In vitro study
Primary cortical neurons were treated with corticosterone (1 μM) or vehicle (DMSO). The above dose of corticosterone has been shown to induce neuroprotective effects in primary cortical neurons (Jeanneteau et al., 2008). In proteasomal inhibitor studies, the neurons were treated with lactacystin (20 μM; Tocris Bioscience) or MG132 (20 μM; Tocris) 5 h prior to corticosterone stimulation. In glucocorticoid receptor inhibitor studies, cells were treated with RU586 (10 μM; Tocris) or spironolactone (10 μM; Tocris) 30 min before corticosterone treatment.
c-Cbl plasmid
Plasmids encoding c-Cbl and Cbl-G306E were kindly provided by Jannie Borst (The Netherlands Cancer Institute) (de Melker et al., 2004). The transfection in DIV4 neurons was performed using Effectene Transfection Reagent (Qiagen). After 48 hours, cells were treated with corticosterone for 3 h.
Small Interfering RNA (siRNA)
The control as well as c-Cbl siRNA were purchased from Dharmacon Research Inc. Transfection of both siRNAs (final 50 nM) was performed using Effectene Transfection Reagent. We carried out the siRNA transfection 48 h before the corticosterone treatment.
Western blot analysis
Protein samples were resolved in SDS-polyacrylamide gels and transferred onto a nitrocellulose membrane (Bio-Rad). Membranes were blocked for 1 h in TBST (10 mM Tris-HCl, pH 8.0, 138 mM NaCl, 2.7 mM KCl, and 0.05% Tween-20) and 5% non-fat milk and incubated overnight with the indicated antibodies. The primary antibodies used were anti-TrkB (1:400; Cell Signaling, MA, USA), anti-c-Cbl (1:400; Millipore, MA, USA), anti-ubiquitin (1:400; Santa Cruz Biotech, CA, USA), anti-cullin-3 (Cul3; 1:500; Cell Signaling), anti-Nedd4 (1:500; Santa Cruz Biotech), anti-Parkin (1:1000; Cell Signaling), anti-phosphoERK1/2 (1:1000; Cell Signaling), anti-ERK1/2 (1:1000; Cell Signaling), anti-β-actin (1:10,000; Sigma, MO, USA) or anti-β-tubulin (1:10,000; Cell Signaling). After washing, the membranes were incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit or anti-mouse anti-sera in TBST and 3% non-fat milk. Following washing, the proteins were visualized by enhanced chemiluminescence. TrkB antibody shows two bands in the western blots, one at ~98 KDa for truncated TrkB and the other at ~145 KDa for full-length TrkB receptor. We have selected the full-length receptor band for our analyses. For immunoprecipitation, 300 μg of proteins were pre-cleared for 1 h with 30 μl of PureProteome Protein A and G Magnetic Beads (Millipore), followed by incubation for 2 h with the primary antibody. The immunoprecipitated proteins were subjected to western blot analysis as described above.
Glucocorticoid Receptor Transactivation Assay
Primary cortical neurons at DIV-4 were transiently transfected with the plasmids pHHLUC (Addgene) and pCMVβgal (Promega) with Effectene (Qiagen) according to the manufacture’s protocol. Three hundred nanograms of each DNA construct were used, and the total amount of DNA was adjusted with empty pcDNA vector. Twenty-four hours after transfection, medium was changed and cells were treated with 1 μM corticosterone for 3h. Whole-cell extracts, prepared using the Luciferase Assay Buffer (Promega) were assayed for luciferase activity. Where indicated, cells were pretreated with 20 μM lactacystin or 20 μM MG132 for 5h before a 3-h treatment with 1 μM corticosterone was initiated. To correct the variation between independent experiments, values were normalized to the corresponding β-galactosidase (Promega) activity.
RNA quantification
Total RNA was isolated from postmortem tissues or cortical neurons using a commercially available kit (SV RNA Isolation, Promega, Madison, WI), according to the manufacturer’s instructions. qRT-PCR was performed on a SmartCycler (Cepheid, Sunnyvale, CA) using a SuperScript III Platinum SYBR Green One-Step qRT-PCR kit (Invitrogen, Carlsbad, CA). Master mixes were prepared and used in the PCR amplifications. A typical reaction of a total volume of 25 μl consisted of 0.5 μl Superscript III RT/Platinum Taq mix, 12.5 μl 2X SYBR Green Reaction Mix (includes 0.4 mM of each dNTP and 6 mM MgSO4), 12.5 pMol of each of forward or reverse primers and 5 μl DEPC-treated water. PCR amplification was done with an initial incubation at 55°C for 1200 sec, then at 95°C for 120 sec followed by 35 cycles of 95°C for 15 sec, 50°C for 30 sec, 72°C for 30 sec and final melting curve from 55°C to 95°C with 0.2C/sec. Primer specificity was confirmed by melting curve analysis and electrophoresis of PCR products on a 2% agarose gel to confirm the presence of a single band of the predicted size. All measurements were performed in triplicates. For TrkB mRNA from mouse cortical neurons, the mRNA values were normalized to housekeeping gene, ribosomal protein S3 (RPS3). The primers utilized were as follows: TrkB FL (forward 5′-GACAATGCACGCAAGGACTT-3′; reverse 5′-AGTAGTCGGTGCTGTACACA-3′) and housekeeping gene, ribosomal protein S3 (RPS3) (forward 5′-AATGAACCGAAGCACACCATA-3′; reverse 5′-ATCAGAGAGTTGACCGCAGTT-3′). For c-Cbl mRNA from postmortem samples, the data were normalized to two control genes (glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and ß-actin) and a geometric mean of these genes. Primers utilized were as follows: c-Cbl (forward, 5′-CGCTAAAGAATAGCCCACCTTAT-3′; and reverse, 5′-ATGGCCTCCAGCCCAGAACTGAT-3′), GAPDH (forward,5′-GAGTCAACGGATTTGGTCGT-3′ and reverse, 5′-TTGATTTTGGAGGGATCTCG-3′) and ß-actin (forward,5′-GGACTTCGAGCAAGAGATGG–3′ and reverse, 5′-AGCACTGTGTTGGCGTACAG-3′).
Statistical analysis
One way ANOVA followed by post-hoc Dunnett test or Student’s t-test was used in the statistical analysis. In postmortem studies, the strength of the association of confounding variables with c-Cbl expression was measured with Pearson’s product moment correlation for variables such as age, PMI and pH. The statistical significance in c-Cbl mRNA from postmortem samples was calculated using Mann-Whitney test. Differences in means between groups were considered significant if p<0.05. SPSS version 21.0 software was used for statistical analyses.
Results
Corticosterone increases TrkB levels in vitro and in vivo
Primary cortical neurons at DIV4 were treated with corticosterone (1 uM) for 3 h. We found a significant increase in TrkB protein levels (Fig 1A). To determine the effect of acute corticosterone exposure on TrkB levels in vivo, mice at postnatal day 4 (PND4) were treated subcutaneously with a single injection of corticosterone and forebrain tissues were collected 6 h following corticosterone treatment. Immunoblot analysis showed a significant increase in TrkB protein levels in the forebrain of mice treated with corticosterone (Fig 1B). Next, we examined the chronic treatment effects of corticosterone on TrkB levels in primary cortical neurons. Neurons were treated with corticosterone for 48 h and cell lysates were examined for TrkB protein levels at 0, 12, 24 or 48 h following corticosterone treatment. We found a significant decrease in TrkB protein levels at 48 h following corticosterone treatment (Fig. 1C). To determine whether TrkB up-regulation occurred at the mRNA level, we examined the TrkB mRNA expression (RT-PCR) in primary cortical neurons under the same condition of corticosterone exposure. The TrkB mRNA level did not differ from the baseline when examined at 3 h (Fig 1D) and 48 h (Fig 1E) of corticosterone exposure. These results indicate that corticosterone exposure induced TrkB protein levels both in vitro and in vivo.
Figure 1.
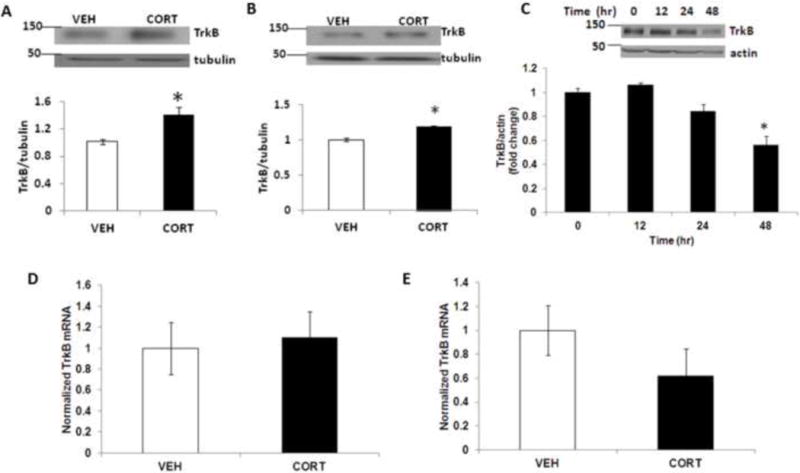
Acute corticosterone treatment increases TrkB protein levels. (A) TrkB protein levels in primary cortical neurons (DIV 4) following corticosterone (CORT) treatment for 3 h as determined by immunoblot analysis. The upper panel shows representative autoradiogram of TrkB and tubulin, and the lower panel represents fold change in normalized TrkB protein levels. Values are mean ± SE for at least three independent preparations. *p<0.05 vs vehicle control (VEH). Student’s t test. (B) TrkB protein levels in mouse forebrain at 6h following a single CORT administration as determined by immunoblot analysis. Mice at postnatal day 4 (PND4) were treated subcutaneously with VEH or CORT, and forebrain samples were analyzed for TrkB levels. The upper panel shows representative autoradiogram of TrkB and tubulin, and the lower panel represents fold change in normalized TrkB protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH. Student’s t test. (C) TrkB protein levels in primary cortical neurons (DIV 4) following corticosterone (CORT) treatment for 0–48 h as determined by immunoblot analysis. The upper panel shows representative autoradiogram of TrkB and actin, and the lower panel represents fold change in normalized TrkB protein levels. Results are mean ± SE for at least three independent preparations. *p < 0.05 vs 0 h. One-way ANOVA followed by post-hoc Dunnett test. TrkB mRNA levels at (D) 3h and (E) 48 h following corticosterone treatment in primary cortical neurons (DIV 4) as determined by qRT-PCR, and the values were normalized to housekeeping gene, ribosomal protein S3 (RPS3). Values are expressed as mean±SE for at least three independent preparations.
Corticosterone induces TrkB ubiquitination in cortical neurons
It has been shown that ubiquitination is a critical mechanism for the posttranslational regulation of proteins (Li and Ye, 2008). Given that the TrkB upregulation after corticosterone exposure occurred at the posttranslational level, we examined whether ubiquitination would play a major role in this process. We found a significant increase in TrkB ubiquitination in primary cortical neurons following corticosterone treatment (Fig 2A). Next, we examine the role of proteasomal pathway on corticosterone-induced changes in TrkB levels. The corticosterone-induced increase in TrkB protein levels was completely blocked by co-treatment of corticosterone with the selective proteasome inhibitor lactacystin (Fig 2B) or MG132 (Fig 2C), when lactacystin or MG132 was added into the incubation for 5 h before the initiation of corticosterone exposure. Next, we examined GR transactivation in neurons treated with corticosterone in the presence or absence of proteasome inhibitor. We found that lactacystin as well as MG132 significantly inhibited GR transactivation in neurons (Fig 2D). Together, these results indicate that corticosterone induced the ubiquitination-mediated TrkB upregulation.
Figure 2.
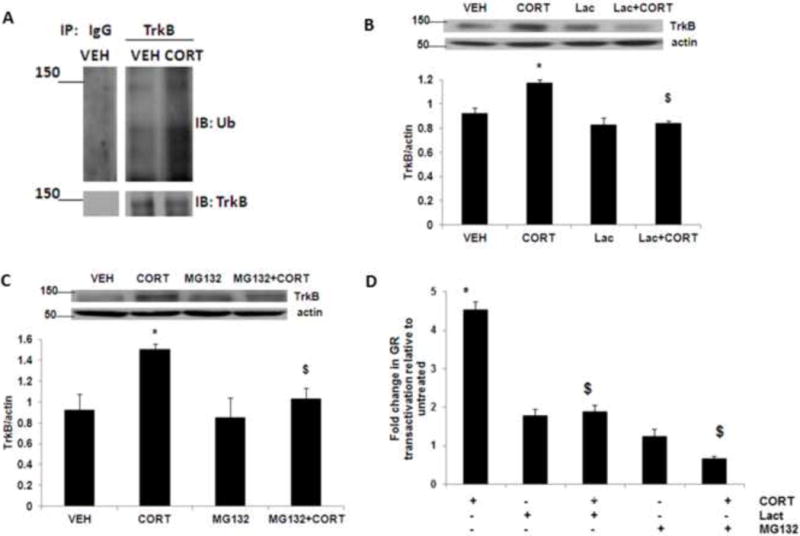
Acute corticosterone treatment induces TrkB ubiquitination. (A) Corticosterone treatment induces TrkB ubiquitination in neurons. Primary cortical neurons (DIV 4) were treated with vehicle (VEH) or corticosterone (CORT) for 3 h and lysates were immunoprecipitated (IP) with an anti-TrkB antibody, followed by immunoblotting (IB) with an anti-ubiquitin antibody (Ub) or anti-TrkB antibody. IgG, IgG control. Results are representative of three independent experiments. Primary cortical neurons (DIV 4) were treated with CORT or CORT plus the selective proteasome inhibitor (B) lactacystin (Lac) or (C) MG132, and lysates were subjected to immunoblot analysis. The upper panel shows representative autoradiogram of TrkB and actin, and the lower panel represents fold change in normalized TrkB protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH; $p<0.05 vs CORT. One-way ANOVA followed by post-hoc Dunnett test. (D) Primary cortical neurons at DIV-4 were transiently transfected with the plasmids pHHLUC and pCMVβgal. Twenty-four hours after transfection, cells were treated with CORT or CORT plus the selective proteasome inhibitor lactacystin (Lac) or MG132, and luciferase activity was measured. Results are mean ± SE for at least three independent preparations expressed as fold change in GR transactivation relative to untreated cells. *p<0.05 vs VEH; $p<0.05 vs CORT. One-way ANOVA followed by post-hoc Dunnett test.
c-Cbl mediates corticosterone-induced increase in TrkB levels
It is known that the specificity of ubiquitination is determined by E3 ligases, which interact with their selective protein substrates. We sought to identify the E3 ligase partner of TrkB responsible for the TrkB ubiquitination and its upregulation. We examined possible TrkB conjugation with several candidate E3 ligases, including c-cbl, Parkin, Cullin-3, and Nedd4, in forebrain samples from mice at PND4 and each of these immune complexes was screened for copurification of TrkB by immunoblot. TrkB was detected only in c-Cbl immunoprecipitates, but not in the control IgG, Nedd4, Parkin, or Cullin-3 complex (Fig. 3A). Using a reciprocal approach, we found c-Cbl coprecipitated with TrkB but not with a control IgG (Fig. 3B). These data provide evidence for the interaction between TrkB and c-Cbl.
Figure 3.
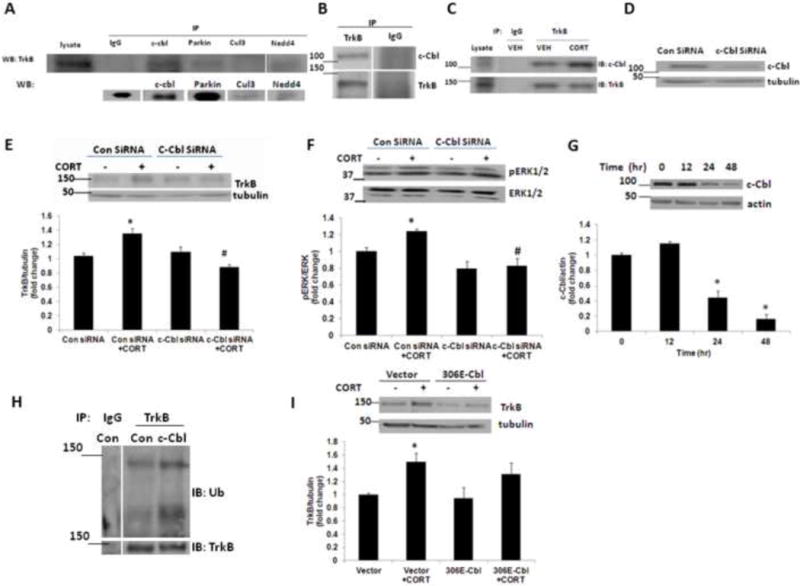
The ubiquitin E3 ligase c-Cbl interacts with TrkB. (A) c-Cbl coprecipitated with TrkB. Forebrain samples from PND4 mice were subjected to immunoprecipitation (IP) using antibodies against several ubiquitin E3 ligases followed by western blotting (WB) with TrkB or ubiquitin E3 ligase. TrkB coprecipitated with c-Cbl, but not with a control IgG, Parkin, Cullin-3 or Nedd4. Lysate (lane 1) represents 10% of the amount used in the IP. (B) c-Cbl coprecipitated with TrkB. Forebrain samples from PND4 mice were immunoprecipitated with an anti-TrkB antibody. C-Cbl was coprecipitated in TrkB precipitates, but not in control IgG precipitates. (C) Acute corticosterone induces the interaction between c-Cbl and TrkB. Primary cortical neurons (DIV 4) were treated with vehicle (VEH) or corticosterone (CORT) for 3 h and lysates were immunoprecipitated (IP) with an anti-TrkB antibody, followed by immunoblotting (IB) with an anti-c-Cbl antibody or anti-TrkB antibody. IgG, IgG control. Results are representative of three independent experiments. (D) c-Cbl was markedly knocked down with siRNA. Primary cortical neurons (DIV 4) were transfected with control siRNA or c-Cbl siRNA, and the c-Cbl expression was examined at 48 h after transfection. (E) c-Cbl knockdown abolished the corticosterone (CORT)-induced TrkB upregulation. Primary cortical neurons (DIV 4) transfected with control siRNA or c-Cbl siRNA were treated with vehicle or CORT, and TrkB expression was examined. The upper panel shows representative autoradiogram of TrkB and tubulin, and the lower panel represents fold change in normalized TrkB protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH; #p<0.05 vs CORT. One-way ANOVA followed by post-hoc Dunnett test. (F) c-Cbl knockdown inhibited the corticosterone (CORT)-induced ERK1/2 activation. Primary cortical neurons (DIV 4) transfected with control siRNA or c-Cbl siRNA were treated with vehicle or CORT, and phospho ERK1/2 (pERK1/2) and total ERK1/2 protein levels were examined. The upper panel shows representative autoradiogram of pERK1/2 and total ERK1/2, and the lower panel represents fold change in pERK1/2 to ERK1/2 ratio. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH; #p<0.05 vs CORT. One-way ANOVA followed by post-hoc Dunnett test. (G) c-Cbl protein levels in primary cortical neurons (DIV 4) following corticosterone (CORT) treatment for 0–48 h as determined by immunoblot analysis. The upper panel shows representative autoradiogram of c-Cbl and actin, and the lower panel represents fold change in normalized c-Cbl protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH. One-way ANOVA followed by post-hoc Dunnett test. (H) c-Cbl increases TrkB ubiquitination. Primary cortical neurons (DIV 4) were transfected with control or c-Cbl plasmid and the lysates were immunoprecipitated (IP) with an anti-TrkB antibody, followed by immunoblotting (IB) with an anti-ubiquitin antibody (Ub) or anti-TrkB antibody. IgG, IgG control. (I) Primary cortical neurons (DIV 4) transfected with vector control or 306E Cbl mutant (306E-Cbl) were treated with vehicle or CORT, and TrkB expression was examined. The upper panel shows representative autoradiogram of TrkB and tubulin, and the lower panel represents fold change in normalized TrkB protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH. One-way ANOVA followed by post-hoc Dunnett test.
Next, we examined whether acute corticosterone treatment induces increase in association between TrkB and c-Cbl in neurons. Immunoprecipitation of TrkB followed by immunoblotting for c-Cbl or TrkB showed a significant increase in c-Cbl levels in TrkB immunoprecipitates from corticosterone-treated neurons (Fig 3C). We also examined whether knockdown of c-Cbl using siRNA would prevent the TrkB upregulation after corticosterone exposure. We found a significant reduction in c-Cbl protein levels in primary cortical neurons transfected with c-Cbl siRNA when examined at 48 h after the transfection (Fig. 3D). The siRNA knockdown of c-Cbl successfully prevented corticosterone-induced TrkB upregulation as compared with the control siRNA transfectants (Fig. 3E). Furthermore, corticosterone-induced increase in ERK activation, a downstream molecule in TrkB signaling, was also inhibited in c-Cbl siRNA-treated neurons (Fig. 3F). Next, we examined the chronic treatment effects of corticosterone on c-Cbl protein levels. We found a significant reduction in c-Cbl protein levels at 24 h as well as 48 h following corticosterone treatment (Fig. 3G). Taken together, these results indicate that c-Cbl played a critical role as an E3 ligase in TrkB regulation.
TKB domain is involved in c-Cbl-mediated TrkB stabilization
Primary cortical neurons were transfected with WT or c-Cbl plasmid and the lysates were examined for TrkB ubiquitination. We found a significant increase in TrkB ubiquitination in c-Cbl overexpressing cells (Fig. 3H). To further examine role of c-Cbl in TrkB regulation, neurons were transfected with vector control or tyrosine kinase binding domain (TKB) mutant c-Cbl (306 E c-Cbl), and the cells were treated with corticosterone for 3 h. We found a significant reduction in TrkB protein levels in neurons transfected with 306 E c-Cbl suggesting an important role of tyrosine kinase binding domain in c-Cbl mediated TrkB stabilization (Fig. 3I).
GR mediates corticosterone-induced increases in TrkB and c-Cbl protein levels
To examine the glucocorticoid receptor mediating corticosterone-induced changes in TrkB as well as c-Cbl protein levels, primary cortical neurons were treated with RU486, a GR antagonist or spironolactone, a MR antagonist before corticosterone exposure. Immunoblotting analysis showed that RU486 significant attenuated corticosterone-induced increase in TrkB protein levels in neurons (Fig 4A). In addition, the increase in c-Cbl protein levels induced by corticosterone exposure was significantly blocked by RU486 treatment (Fig 4B). We did not find any significant effect of spironolactone on corticosterone-induced TrkB or c-Cbl protein levels (data not shown). These data indicate that GR mediates acute corticosterone-induced increase in TrkB as well as c-Cbl protein levels in neurons.
Figure 4.
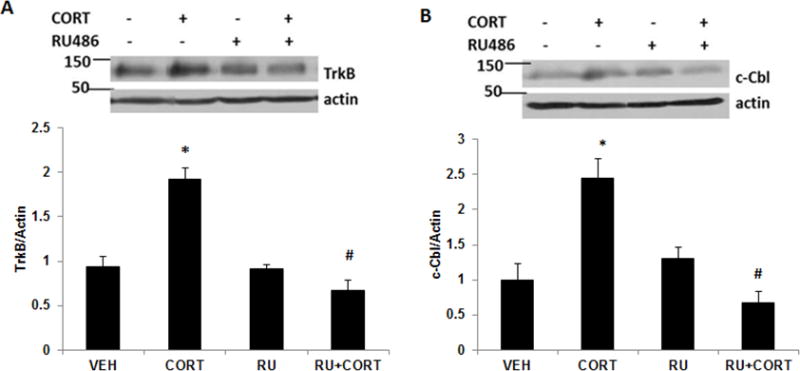
GR mediates acute corticosterone-induced increase in TrkB and c-Cbl levels. Primary cortical neurons (DIV 4) were treated with CORT or CORT plus GR antagonist, RU486 and lysates were subjected to immunoblot analysis for (A) TrkB and (B) c-Cbl levels. The upper panel shows representative autoradiogram of TrkB or c-Cbl and actin, and the lower panel represents fold change in normalized TrkB or c-Cbl protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH; #p<0.05 vs CORT. One-way ANOVA followed by post-hoc Dunnett test.
Acute corticosterone treatment does not change TrkB protein levels in mature neurons
Since we found a significant increase in TrkB and c-Cbl protein levels in DIV4 neurons following acute corticosterone treatment, we next examined the effect of acute as well as chronic corticosterone on TrkB and c-Cbl protein levels in mature neurons. Primary cortical neurons at DIV12 were treated with corticosterone for 3 or 48 h, and the lysates were examined for TrkB and c-Cbl protein levels (Fig. 5A). We did not find any significant change in TrkB protein levels at 3 h following corticosterone treatment. However, a significant reduction in TrkB levels was found at 48 following corticosterone exposure. No significant change in c-Cbl protein levels was found in mature neurons following 3 h or 48 h corticosterone treatment.
Figure 5.
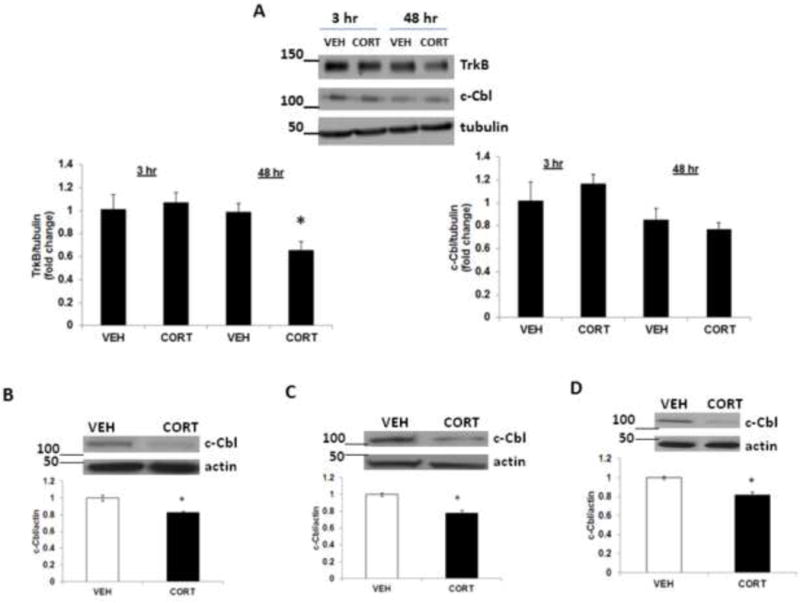
Effects of corticosterone treatment on c-Cbl protein levels in mature neurons in vitro and in vivo. (A) Primary cortical neurons (DIV 12) were treated with vehicle (VEH) or corticosterone (CORT) for 3 h or 48 h, and lysates were subjected to immunoblot analysis for TrkB and c-Cbl levels. The upper panel shows representative autoradiogram of TrkB or c-Cbl and tubulin, and the lower panels represent fold change in normalized TrkB or c-Cbl protein levels. Results are mean ± SE for at least three independent preparations. *p<0.05 vs VEH. Student’s t test. (B-D). Chronic corticosterone treatment decreases c-Cbl protein levels in vivo. CD-1 male mice were treated with corticosterone (CORT; 35 ug/ml/day) or vehicle (VEH; 0.45% hydroxypropyl-β-cyclodextrin) for 5 or 7 weeks. c-Cbl protein levels were determined by immunoblot analysis in (B) frontal cortex after 7 weeks, (C) hippocampus after 5 weeks and (D) hippocampus after 7 weeks. The upper panel shows a representative autoradiogram of c-Cbl and the lower panel represents the fold change in optical density values normalized to vehicle-treated controls. β-actin was used as a protein loading control. Values are mean ± SE (n = 5–6 mice per group). *p<0.05 versus VEH.
Chronic corticosterone treatment decreases c-Cbl protein levels in adult mice
To examine the chronic treatment effects of corticosterone on c-Cbl levels in adulthood, the expression of c-Cbl was examined in frontal cortex and hippocampus of adult mice after 5 and 7 weeks of corticosterone treatment. In the frontal cortex, treatment with corticosterone for 5 weeks did not change c-Cbl protein levels (data not shown), but 7-week treatment resulted in a significant decrease in c-Cbl protein levels (Fig. 5B). In hippocampus, c-Cbl protein levels were significantly decreased in groups treated with corticosterone for 5 (Fig. 5C) and 7 (Fig. 5D) weeks.
Decreased c-Cbl mRNA expression in prefrontal cortex of suicide subjects
Reverse-transcription real-time PCR was used for the determination of mRNA expression levels of c-Cbl for the postmortem samples. In the brain samples from Quebec Suicide Brain Bank, we found a significant decrease in mean c-Cbl mRNA levels in the suicide subjects compared to the control group (Fig. 6). We have also examined the effect of confounding factors on the mRNA levels of c-Cbl in the control and suicide groups. The information on the confounding factors available for the Quebec Suicide Brain Bank samples were age at death, PMI, pH and gender. All samples used in the current study were obtained from male subjects. However, no significant correlation was found between the mRNA expression of c-Cbl and the PMI, age or pH (data not shown).
Figure 6.
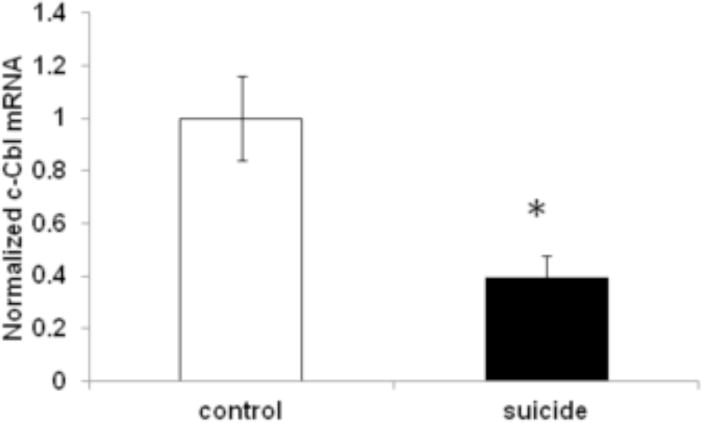
Decreased c-Cbl mRNA expression in the prefrontal cortex of suicide subjects. c-Cbl mRNA was determined by qRT-PCR in the prefrontal cortex of suicide (N=13) and control (N=15) subjects, and the values were normalized to the geometric mean of two control genes (glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and ß-actin). Values are expressed as mean±SEM. *p<0.05 versus controls.
Discussion
The present study demonstrates a UPS-mediated cellular mechanism for the posttranslational regulation of TrkB. In developing cortical neurons, the UPS-mediated TrkB stabilization contributed to the corticosterone-induced TrkB regulation because inhibition of the proteasome activity abolished the corticosterone-induced increase in TrkB levels in neurons. Since the TrkB signaling pathway plays an important role in neurodevelopment (Pandya et al., 2013), UPS-mediated stabilization of TrkB may be an important mechanism to maintain TrkB levels and its downstream signaling pathway during neuronal maturation.
In the current in vitro and chronic in vivo experiments, we used corticosterone which is a natural glucocorticoid. Interestingly, an earlier study has shown that acute corticosterone treatment increases TrkB signaling in cortical as well as hippocampal neurons (Jeanneteau et al., 2008). Our study was performed using the same dose and duration of exposure (1 uM for 3 h) at which neuroprotective effects were observed in the above study. Our data demonstrate that corticosterone increased TrkB at the protein but not mRNA level. Moreover, proteasome inhibition prevented the upregulation of TrkB in corticosterone-treated cells. Our data on GR transactivation is in agreement with a previous report that inhibition of proteasomal pathway results in the reduction of GR transactivation in neurons (Wang and DeFranco, 2005). Since our findings indicate that GR mediates corticosterone-induced increase in TrkB levels in neurons, the inhibition of GR transactivation (via proteasomal inhibition) could abolish corticosterone-induced increase in TrkB levels. A recent study has shown that low and high cortisol concentrations exert differential MR- and GR-dependent effects on neurogenesis in human hippocampal cell lines (Anacker et al., 2013). The above study found that low cortisol-induced increase in progenitor cell proliferation is mediated predominantly by MR-dependent mechanism. Since BDNF/TrkB signaling is involved in neuronal proliferation, an increase in TrkB signaling following low level or acute glucocorticoid exposure could account for the increase in proliferation found in the above study. The discrepancy in the receptor involved in mediating the above effects (MR vs GR) could be due to the difference in cell types such as primary cortical neurons vs human hippocampal cell lines. Our earlier study has shown that chronic corticosterone treatment downregulates TrkB protein levels in mouse frontal cortex and hippocampus (Kutiyanawalla et al., 2011). In the present study, we observed that chronic corticosterone also induced the downregulation of c-Cbl in the above brain regions. These results indicate that TrkB may be subject to the proteasomal regulation through a c-Cbl-mediated pathway under in vivo conditions as well.
c-Cbl interacts with a number of signaling molecules including various receptors, adaptors, ubiquitin, and structural proteins via its various domains (Thien and Langdon, 2005). Full length c-Cbl has been shown to contain N-terminal TKB domain, ring finger domain, proline rich region and c-terminal ubiquitin associated domain (Lupher et al., 1998). Among these domains, TKB domain binds to phosphotyrosines on activated receptor tyrosine kinases (RTKs) and other signaling proteins (Lill et al., 2000). Our data show that c-Cbl functions as E3 ligase to stabilize TrkB. However, the mutation in the TKB domain, G306E failed to promote corticosterone-induced increase in TrkB levels in neurons. This provides direct evidence that the increase in TrkB levels caused by c-Cbl depends on an intact G306 in the TKB domain. Earlier studies have indicated that the c-Cbl TKB domain has a critical role in targeting proteins for ubiquitination (Lill et al., 2000; Severe et al., 2011). Our data report that acute corticosterone induces TrkB ubiquitination in neurons and that TKB domain of c-Cbl is critical for corticosterone-induced TrkB protein levels.
The present study was conducted using prefrontal cortex samples from suicide and normal subjects. A number of studies have used the same set of samples obtained from Quebec Suicide Brain Bank to examine various pathological markers in suicide subjects (Maussion et al., 2012; Smalheiser et al., 2012; Freemantle et al., 2013a, b). Though there are a number of potential variables (age, PMI or brain pH), which can affect the quality of RNA as well as the gene expression status, the results from the present study did not show any significant correlation between c-Cbl mRNA levels and these confounding variables. Interestingly, lower mRNA and protein levels of TrkB have been reported in the prefrontal cortex of suicide subjects (Dwivedi et al., 2003). The decrease in c-Cbl mRNA expression found in our study might be one of the possible mechanisms for the decrease in TrkB protein levels in the prefrontal cortex of suicide subjects. The intriguing question here is whether c-Cbl has any role in the regulation of TrkB at the mRNA level. Further studies are needed to establish whether decreased expression of c-Cbl is sufficient to produce the altered expression of TrkB in the prefrontal cortex of suicide subjects.
The present data suggest a possible mechanism for TrkB regulation by corticosterone in neurons. It is important to note that the acute corticosterone treatment failed to increase TrkB protein levels in mature neurons suggesting differential effects of glucocorticoid on BDNF/TrkB signaling in developing and mature neurons. Our current data does not exclude the possibility that ubiquitin-independent mechanism might also play a role in the regulation of TrkB. The exact cellular mechanisms of corticosterone in regulating c-Cbl E3 ligase expression and function will be an important step in our future studies.
Acknowledgments
Postmortem samples were donated by the Quebec Suicide Brain Bank. Dr. Pillai is thankful to Dr. Borst for the c-Cbl constructs.
Financial Disclosures
This work was supported in part by grant from NIH/NIMH to A.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare no competing financial interests.
Contributors
AP designed the study and wrote the protocol. CP and AK performed analyses. GT provided reagents. AP wrote the manuscript. All authors have approved the final manuscript.
References
- Anacker C, Cattaneo A, Luoni A, Musaelyan K, Zunszain PA, Milanesi E, Rybka J, Berry A, Cirulli F, Thuret S, Price J, Riva MA, Gennarelli M, Pariante CM. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology. 2013;38:872–883. doi: 10.1038/npp.2012.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardley HC, Robinson PA. E3 ubiquitin ligases. Essays Biochem. 2005;41:15–30. doi: 10.1042/EB0410015. [DOI] [PubMed] [Google Scholar]
- Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci. 2012;15:1707–1714. doi: 10.1038/nn.3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbé S. Endocytosis: the DUB version. Trends Cell Biol. 2006;16:551–559. doi: 10.1016/j.tcb.2006.09.002. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and – independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melker AA, van der Horst G, Borst J. c-Cbl directs EGF receptors into an endocytic pathway that involves the ubiquitin-interacting motif of Eps15. J Cell Sci. 2004;117:5001–5012. doi: 10.1242/jcs.01354. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2009;60:804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat. 2009;5:433–449. doi: 10.2147/ndt.s5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, Ernst N, Quirion R, Gratton A, Szyf M, Turecki G. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch Gen Psychiatry. 2009;66:22–32. doi: 10.1001/archpsyc.66.1.22. [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Freemantle E, Chen GG, Cruceanu C, Mechawar N, Turecki G. Analysis of oxysterols and cholesterol in prefrontal cortex of suicides. Int J Neuropsychopharmacol. 2013a;16:1241–1249. doi: 10.1017/S1461145712001587. [DOI] [PubMed] [Google Scholar]
- Freemantle E, Mechawar N, Turecki G. Cholesterol and phospholipids in frontal cortex and synaptosomes of suicide completers: relationship with endosomal lipid trafficking genes. J Psychiatr Res. 2013b;47:272–279. doi: 10.1016/j.jpsychires.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Gross C, Hen R. The developmental origins of anxiety. Nat Rev Neurosci. 2004;5:545–552. doi: 10.1038/nrn1429. [DOI] [PubMed] [Google Scholar]
- Habelhah H, Takahashi S, Cho SG, Kadoya T, Watanabe T, Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. EMBO J. 2004;23:322–332. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell KR, Kutiyanawalla A, Pillai A. Long-term continuous corticosterone treatment decreases VEGF receptor-2 expression in frontal cortex. PLoS One. 2011;6:e20198. doi: 10.1371/journal.pone.0020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, Chao MV. Are BDNF and glucocorticoid activities calibrated? Neuroscience. 2013;239:173–195. doi: 10.1016/j.neuroscience.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, Garabedian MJ, Chao MV. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci USA. 2008;105:4862–4867. doi: 10.1073/pnas.0709102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Jin HR, Jung HS, Lee SJ, Lee JH, Lee JJ. An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J Biol Chem. 2010;285:30539–30547. doi: 10.1074/jbc.M110.129551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CA, Hunter T. Biochemistry. Ubiquitination–more than two to tango. Science. 2000;289:2061–2062. doi: 10.1126/science.289.5487.2061. [DOI] [PubMed] [Google Scholar]
- Joëls M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev. 2012;64:901–938. doi: 10.1124/pr.112.005892. [DOI] [PubMed] [Google Scholar]
- Kamphuis PJ, Gardoni F, Kamal A, Croiset G, Bakker JM, Cattabeni F, Gispen WH, van Bel F, Di Luca M, Wiegant VM. Long-lasting effects of neonatal dexamethasone treatment on spatial learning and hippocampal synaptic plasticity: involvement of the NMDA receptor complex. FASEB J. 2003;17:911–913. doi: 10.1096/fj.02-0333fje. [DOI] [PubMed] [Google Scholar]
- Kang JS, Saunier EF, Akhurst RJ, Derynck R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat Cell Biol. 2008;10:654–664. doi: 10.1038/ncb1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutiyanawalla A, Terry AV, Jr, Pillai A. Cysteamine attenuates the decreases in TrkB protein levels and the anxiety/depression-like behaviors in mice induced by corticosterone treatment. PLoS One. 2011;6:e26153. doi: 10.1371/journal.pone.0026153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Ye Y. Polyubiquitin chains: functions, structures, and mechanisms. Cell Mol Life Sci. 2008;65:2397–2406. doi: 10.1007/s00018-008-8090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill NL, Douillard P, Awwad RA, Ota S, Lupher ML, Jr, Miyake S, Meissner-Lula N, Hsu VW, Band H. The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J Biol Chem. 2000;275:367–377. doi: 10.1074/jbc.275.1.367. [DOI] [PubMed] [Google Scholar]
- Lupher ML, Jr, Andoniou CE, Bonita D, Miyake S, Band H. The c-Cbl oncoprotein. Int J Biochem Cell Biol. 1998;30:439–444. doi: 10.1016/s1357-2725(97)00075-7. [DOI] [PubMed] [Google Scholar]
- Maussion G, Yang J, Yerko V, Barker P, Mechawar N, Ernst C, Turecki G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by HsamiR-185* in frontal cortex of suicide completers. PLoS One. 2012;7:e39301. doi: 10.1371/journal.pone.0039301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol. 2003;13:391–398. doi: 10.1016/s0959-4388(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, Bailey TA, Palermo N, Borgstahl GE, Natarajan A, Raja SM, Naramura M, Band V, Band H. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta. 2013;1833:122–139. doi: 10.1016/j.bbamcr.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya CD, Kutiyanawalla A, Pillai A. BDNF-TrkB signaling and neuroprotection in schizophrenia. Asian J Psychiatr. 2013;6:22–28. doi: 10.1016/j.ajp.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. 2. Academic Press; San Diego: 2001. [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pillai A. Brain-derived neurotropic factor/TrkB signaling in the pathogenesis and novel pharmacotherapy of schizophrenia. Neurosignals. 2008;16:183–193. doi: 10.1159/000111562. [DOI] [PubMed] [Google Scholar]
- Radovanac K, Morgner J, Schulz JN, Blumbach K, Patterson C, Geiger T, Mann M, Krieg T, Eckes B, Fässler R, Wickström SA. Stabilization of integrin-linked kinase by the Hsp90-CHIP axis impacts cellular force generation, migration and the fibrotic response. EMBO J. 2013;32:1409–1424. doi: 10.1038/emboj.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sévère N, Miraoui H, Marie PJ. The Casitas B lineage lymphoma (Cbl) mutant G306E enhances osteogenic differentiation in human mesenchymal stromal cells in part by decreased Cbl-mediated platelet-derived growth factor receptor alpha and fibroblast growth factor receptor 2 ubiquitination. J Biol Chem. 2011;286:24443–24450. doi: 10.1074/jbc.M110.197525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Lugli G, Rizavi HS, Torvik VI, Turecki G, Dwivedi Y. MicroRNA expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS One. 2012;7:e33201. doi: 10.1371/journal.pone.0033201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang VM, Young AH, Tan H, Beasley C, Wang JF. Glucocorticoids Increase Protein Carbonylation and Mitochondrial Dysfunction. Horm Metab Res. 2013;45:709–715. doi: 10.1055/s-0033-1345119. [DOI] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem. 2001;276:37585–37593. doi: 10.1074/jbc.M101683200. [DOI] [PubMed] [Google Scholar]
- Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- Thien CB, Langdon WY. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem J. 2005;391:153–166. doi: 10.1042/BJ20050892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, DeFranco DB. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. 2005;19:1474–1482. doi: 10.1210/me.2004-0383. [DOI] [PubMed] [Google Scholar]
- Zafir A, Banu N. Modulation of in vivo oxidative status by exogenous corticosterone and restraint stress in rats. Stress. 2009;12:167–177. doi: 10.1080/10253890802234168. [DOI] [PubMed] [Google Scholar]
- Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, Zhang T, Lee HW, Jeong LS, Chen Y, Deng H, Feng XH, Luo S, Gao C, Chen YG. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol Cell. 2013;49:499–510. doi: 10.1016/j.molcel.2012.12.002. [DOI] [PubMed] [Google Scholar]