

Abstract
The phase IIb, double-blind, placebo-controlled PILLAR trial investigated the efficacy and safety of two different simeprevir (SMV) doses administered once-daily (QD) with pegylated interferon (Peg-IFN)-α-2a and ribavirin (RBV) in treatment-naïve patients with HCV genotype 1 infection. Patients were randomized to one of five treatments: SMV (75 or 150 mg QD) for 12 or 24 weeks or placebo, plus Peg-IFN and RBV. Patients in the SMV arms stopped all treatment at week 24 if response-guided therapy (RGT) criteria were met; patients not meeting RGT continued with Peg-IFN and RBV until week 48, as did patients in the placebo control group. Sustained virologic response (SVR) rates measured 24 weeks after the planned end of treatment (SVR24) were 74.7%-86.1% in the SMV groups versus 64.9% in the control group (P < 0.05 for all comparisons [SMV versus placebo], except SMV 75 mg for 24 weeks). Rapid virologic response (HCV RNA <25 IU/mL undetectable at week 4) was achieved by 68.0%-75.6% of SMV-treated and 5.2% of placebo control patients. According to RGT criteria, 79.2%-86.1% of SMV-treated patients completed treatment by week 24; 85.2%-95.6% of these subsequently achieved SVR24. The adverse event profile was generally similar across the SMV and placebo control groups, with the exception of mild reversible hyperbilirubinemia, without serum aminotransferase abnormalities, associated with higher doses of SMV.
Conclusion
SMV QD in combination with Peg-IFN and RBV significantly improves SVR rates, compared with Peg-IFN and RBV alone, and allows the majority of patients to shorten their therapy duration to 24 weeks.
The availability of direct-acting antiviral agents has recently transformed the treatment of chronic hepatitis C (CHC).1,2 Triple-therapy regimens that include nonstructural protein (NS)3/4A protease inhibitors, such as boceprevir and telaprevir, combined with pegylated interferon (Peg-IFN) and ribavirin (RBV) significantly improve the rate of sustained virologic response (SVR) for patients with genotype 1 CHC infection, compared with Peg-IFN and RBV alone.3,4 Furthermore, many patients may qualify for a shortened duration of therapy by incorporating a response-guided therapy (RGT) algorithm that determines the duration of therapy according to on-treatment virologic response milestones.5
However, these regimens have also increased the complexity of treatment for patients and amplified the adverse events (AEs) associated with hepatitis C therapy.6,7 Strict adherence to three-times-daily dosing is required for boceprevir and telaprevir, along with recommendations to be administered with food (with a specific fat content for telaprevir) to enhance absorption of medications.6 Anemia is more frequent and severe when either of these agents is used with Peg-IFN and RBV, whereas skin rash is more common with telaprevir-containing regimens.3,4 Thus, effective treatments with simplified dosing schedules and improved AE profiles would benefit patients with CHC.
Simeprevir (SMV; TMC435) is an oral, once-daily (QD), investigational hepatitis C virus (HCV) NS3/4A macrocyclic protease inhibitor with potent antiviral activity in patients infected with genotype 1 as well as antiviral activity demonstrated against isolates of genotypes 2, 4, 5, and 6.8,9 In preclinical studies, the replicon half-maximal effective concentration (EC50) for SMV ranged from 8 to 28 nM, and the liver-to-plasma concentration ratio was high (ratio of 39).10 In a phase I study, patients with hepatitis C genotype 1 treated with a 5-day course of SMV monotherapy exhibited a median maximal reduction of HCV RNA of 3.9 log10, which compares favorably to that observed with boceprevir (~2.45 log10 over 7 days) and telaprevir (~4.4 log10 over 14 days).8,11,12 Manns et al. administered triple therapy with SMV (dose range: 25-200 mg QD) plus Peg-IFN-α-2a and RBV in a phase IIa study for up to 28 days.13 The majority of patients, both treatment naïve and treatment experienced, had HCV RNA below the lower level of quantification (<25 IU/mL) of the HCV RNA assay by day 28 of therapy.13
The aim of the current study was to assess the efficacy and safety of two different doses of SMV administered QD for two different durations in combination with Peg-IFN and RBV in treatment-naïve patients infected with HCV genotype 1.
Patients and Methods
Patients and Study Design
The Protease Inhibitor TMC435 study assessing optimaL dose and duration as once daiLy Antiviral Regimen (PILLAR) study (NCT00882908; www.clinicaltrials.gov) was a phase IIb, randomized, double-blind, placebo-controlled clinical trial designed to test the efficacy and safety of SMV in combination with Peg-IFN and RBV, compared with Peg-IFN and RBV alone, for the treatment of genotype 1 CHC. The study was performed in 13 countries in North America, Europe, and Asia-Pacific regions. Enrollment began in May 2009, and the study was completed in April 2011. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institutional review boards of participating institutions. All patients provided written informed consent.
Adult patients with CHC were eligible for participation if they had plasma HCV RNA >100,000 IU/mL, were infected with HCV genotype 1, had never received (Peg)IFN, RBV, or other approved or investigational agents for chronic HCV infection, and were deemed eligible to be treated with Peg-IFN-based regimens according to standard criteria.14 Patients were excluded if they had cirrhosis on liver biopsy (required within 24 months of enrollment), coinfection with human immunodeficiency virus or hepatitis B, platelet count <90,000/mm3, or hemoglobin <12 g/dL for females and 13 g/dL for males.
Participants were randomly assigned in equal proportions, using a centralized, interactive voice/web-response randomization system, to one of five treatment regimens with SMV at doses of either 75 or 150 mg administered orally QD for 12 or 24 weeks or matched placebo, in combination with Peg-IFN-α-2a 180 μg/week and RBV 1,000-1,200 mg/day (Fig. 1). Participants who were randomized to 12 weeks of SMV therapy received an additional 12 weeks of placebo plus Peg-IFN and RBV. Randomization was stratified by hepatitis C genotype (1a, 1b, versus other) and by race (black, Caucasian, and other).
Fig. 1.
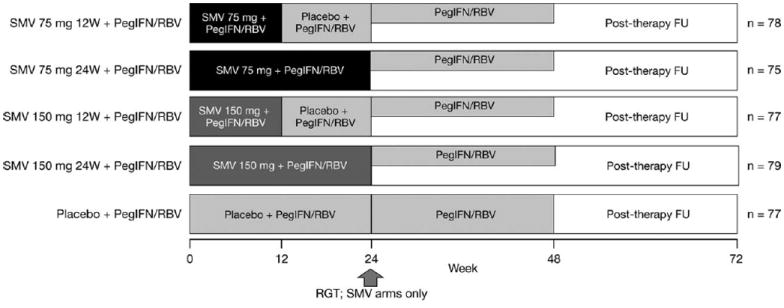
PILLAR study design.
FU, follow-up; PegIFN/RBV, peginterferonα-2a (180 μg/wk) + ribavirin (1000-1200 mg/day); RGT, response-guided therapy; SMV, simeprevir; W, weeks.
Patients receiving SMV were eligible for shortened treatment duration using an RGT algorithm (all therapy completed at week 24 if HCV RNA <25 IU/mL at week 4 and undetectable at weeks 12, 16, and 20). SMV-treated participants who did not meet these virologic milestones continued therapy with Peg-IFN and RBV for up to 48 weeks. All participants initially randomized to the placebo control arm were treated for up to 48 weeks; RGT criteria were not applied (Fig. 1).
SMV/placebo was discontinued if viral breakthrough (VBT) occurred (confirmed increase in plasma HCV RNA of >1 log10 IU/mL, compared with the lowest recorded on-treatment value, or a confirmed plasma HCV RNA >100 IU/mL if previously below the level of quantification [<25 IU/mL]). All medications were discontinued if there was a <2 log10 decline in HCV RNA, compared with baseline at week 12, or detectable HCV RNA at week 24 or 36. Investigators and participants were blinded to HCV RNA results through week 48 of treatment. An external physician monitored individual HCV RNA results and informed investigators regarding protocol-directed treatment discontinuation.
The primary outcome measure was the proportion of patients with HCV RNA <25 IU/mL undetectable at week 72 (SVR W72; i.e., all patients assessed at the same time point irrespective of treatment duration) in each of the SMV regimens, compared to placebo control. Secondary measures included SVR at 12 and 24 weeks after planned end of treatment (SVR12 and SVR24, respectively), comparisons of AEs and quality-of-life measures between groups, an assessment of HCV NS3 sequence in patients not achieving SVR, and an assessment of SMV pharmacokinetics. The influence of interleukin-28 (IL28)B genotype on efficacy was explored in a subset of patients for whom genomic DNA was available.
Assessments
Blood samples were taken at each study visit. Plasma HCV RNA was quantified using the Roche COBAS TaqMan v2.0 assay (lower limit of quantification: 25 IU/mL; values below this limit reported as HCV RNA <25 IU/mL detectable or undetectable; Roche Molecular Diagnostics, Pleasanton, CA). HCV NS3/4A sequencing was also performed at baseline and at relevant time points throughout the study, based on changes in plasma HCV RNA for individual patients. AEs and laboratory data were recorded at each study visit. Patients completed the Fatigue Severity Scale (FSS), a fatigue symptom and impact questionnaire,15 at baseline and weeks 0, 4, 12, 24, 36, 48, 60, and 72.
Statistical Analysis
Statistical analyses were performed by SGS Life Science Services (Geneva, Switzerland) using SAS statistical software (version 9.1; SAS Institute Inc., Cary, NC).
Sample Size Determination and Analysis of Baseline Characteristics
Eighty patients were randomized to each treatment group, providing 77% power to detect a difference of 25% in SVR24 rates between groups (70% for SMV groups and 45% for placebo control). Sample-size calculation was based on SVR24 rates observed in previous studies because, at the time of protocol writing, no reference SVR W72 rates were available. Patient baseline demographics and disease characteristics were summarized descriptively.
Efficacy
SVR rates were compared between groups using a logistic regression model (closed testing procedure), with factors baseline HCV RNA, race, and genotype subtype. Combined SMV groups of the same dose (different duration) were first compared versus the control group and, if significant, the two individual groups compared to control. The 95% confidence intervals (CIs) were constructed around both the observed response rates and the differences in rates between groups. Subgroup analyses, including application of a multivariate model, were performed to assess correlation between baseline characteristics or early response criteria and SVR.
Resistance Analysis
Population sequencing of the HCV NS3/4A region was conducted on samples collected at baseline and time of treatment failure as previously described.16 The effect of the naturally occurring NS3 Q80K polymorphism, which confers low-level resistance to SMV (fold change in EC50 value of 7.7 as a single amino acid substitution in a genotype 1b replicon)16 on SVR as well as the emergence of viral variants carrying NS3 amino acid substitutions in patients not achieving SVR, were investigated.
Safety
Incidence of AEs and related parameters, clinical laboratory data, and vital-sign parameters (actual values and changes from baseline) were summarized per study arm. Laboratory abnormalities were determined according to the World Health Organization grading table and in accord with the normal ranges of the clinical laboratory.
IL28B Genotype
IL28B genotype (locus rs1297860) was determined for a subset of patients who consented to this analysis. Hardy-Weinberg’s equilibrium of the IL28B polymorphism was tested for the study population, and the distribution of genotypes, according to baseline characteristics and efficacy parameters, was summarized.
FSS
A total FSS score was calculated as the mean of nine items (range, 1-7), with higher scores indicating increased fatigue. Area under the curve from baseline to week 72 was compared between SMV and placebo control groups using a piecewise linear mixed model, with treatment group and time as factors, and assuming data missing at random (Supporting Document 1).
Results
Five hundred and six patients were screened for eligibility, of which 388 were randomized to treatment. Two patients withdrew consent before therapy (one in an SMV group and one in the placebo control group). Thus, a total of 386 patients, or 75-79 patients per treatment arm, received at least one dose of study medication and are included in the intent-to-treat analyses (Supporting Fig. A). Study completion rates were high; 92.5% of patients overall completed the study, whereas 7.5% discontinued study participation prematurely. Discontinuation of SMV and placebo and Peg-IFN and RBV occurred in 8.0%-15.6% of SMV-treated patients and 13% of the control group (Supporting Table A) and occurred because of AEs in 1.3%-5.2% in SMV groups and 5.2% in the control group.
Patient Characteristics
Demographics and baseline disease characteristics of participants within each treatment arm are shown in Table 1. Most patients were Caucasian males with a median age of 46.5 years and a body mass index (BMI) of 25.0 kg/m2. Approximately 68% of patients were enrolled in Europe, 21% in North America, and 11% in Australia and New Zealand. HCV RNA levels at baseline were greater than 800,000 IU/mL in over 80% of participants (between 81.8% and 91.1% in each treatment arm), and 50%-62% of patients were infected with HCV genotype 1b. METAVIR stage F3 was present on pre-treatment liver biopsies in 9.1%-22.7% of patients. The NS3 Q80K polymorphism was present at baseline in 10.4% of patients (22.0% in HCV genotype 1a-and 1.0% in 1b-infected patients).
Table 1. Baseline Demographic and Disease Characteristics for Patients Enrolled in the PILLAR Study.
| Characteristics | SMV 75 mg 12W + Peg-IFN/RBV, n = 78 | SMV 75 mg 24W + Peg-IFN/RBV, n = 75 | SMV 150 mg 12W + Peg-IFN/RBV, n = 77 | SMV 150 mg 24W + Peg-IFN/RBV, n = 79 | Placebo + Peg-IFN/RBV, n = 77 |
|---|---|---|---|---|---|
| Male/female, n (%) | 40/38 (51.3/48.7) | 47/28 (62.7/37.3) | 43/34 (55.8/44.2) | 44/35 (55.7/44.3) | 39/38 (50.6/49.4) |
| Age, years, median (range) | 47 (19-66) | 46 (18-67) | 47 (18-69) | 47 (19-69) | 45 (21-67) |
| BMI, kg/m2, median (range) | 25.9 (16.8-39.6) | 24.2 (17.4-37.8) | 24.7 (18.1-38.2) | 24.9 (17.6-37.3) | 25.6 (17.5-42.2) |
| Ethnicity, n (%) | |||||
| Caucasian | 70 (89.7) | 71 (94.7) | 74 (96.1) | 73 (92.4) | 74 (96.1) |
| Black/African American | 3 (3.8) | 2 (2.7) | 3 (3.9) | 3 (3.8) | 2 (2.6) |
| Asian | 5 (6.4) | 1 (1.3) | 0 | 2 (2.5) | 0 |
| Native Hawaiian/other Pacific Islander | 0 | 1 (1.3) | 0 | 1 (1.3) | 1 (1.3) |
| HCV subtype 1a (NS5B), n (%) | 36 (46.8) | 34 (45.3) | 37 (48.7) | 37 (46.8) | 29 (38.2) |
| Baseline HCV RNA log10 IU/mL, mean (SD) | 6.49 (0.68) | 6.51 (0.59) | 6.53 (0.54) | 6.55 (0.59) | 6.41 (0.63) |
| Baseline HCV RNA ≥800,000 IU/mL, n (%) | 64 (82.1) | 63 (84.0) | 69 (89.6) | 72 (91.1) | 63 (81.8) |
| Baseline HCV RNA <400,000 IU/mL, n (%) | 6 (7.7) | 8 (10.7) | 4 (5.2) | 5 (6.3) | 9 (11.7) |
| METAVIR score, n (%) | |||||
| F0 | 9 (11.5) | 7 (9.3) | 12 (15.6) | 8 (10.1) | 9 (11.7) |
| F1 | 33 (42.3) | 25 (33.3) | 32 (41.6) | 33 (41.8) | 35 (45.5) |
| F2 | 26 (33.3) | 26 (34.7) | 26 (33.8) | 25 (31.6) | 26 (33.8) |
| F3 | 10 (12.8) | 17 (22.7) | 7 (9.1) | 12 (15.2) | 7 (9.1) |
| F4 | 0 | 0 | 0 | 1 (1.3)* | 0 |
| IL28B genotype | |||||
| Patients with data, n† | 58 | 51 | 55 | 52 | 46 |
| CC, n (%) | 13 (22.4) | 18 (35.3) | 22 (40.0) | 13 (25.0) | 12 (26.1) |
| CT, n (%) | 36 (62.1) | 28 (54.9) | 27 (49.1) | 33 (63.5) | 28 (60.9) |
| TT, n (%) | 9 (15.5) | 5 (9.8) | 6 (10.9) | 6 (11.5) | 6 (13.0) |
One patient was inadvertently randomized despite a METAVIR score of F4 on biopsy. The patient was discontinued from treatment 13 days after starting therapy and discontinued the trial. These data are included in efficacy and safety analyses.
IL28B data only available for patients who signed the separate consent form.
Abbreviation: SD, standard deviation; W, week.
Virologic Response
The primary outcome measure, SVR W72, ranged between 70.7% and 84.8% for SMV regimens, compared with 64.9% of those treated with Peg-IFN and RBV alone (Table 2). The differences between SMV 150-mg groups and control were statistically significant (P < 0.05; Table 2). SVR24 was achieved in 74.7%-86.1% of those treated with SMV regimens, compared to 64.9% of those treated with placebo. The differences in response rates between SVR W72 and SVR24 were largely the result of missing HCV RNA data at week 72 from participants who were lost to follow-up between those post-treatment time points. All SVR24 comparisons between SMV treatment groups and placebo controls were statistically significant (P < 0.05 or 0.005), except for SMV 75 mg for 24 weeks (Table 2).
Table 2. Rates of Undetectable HCV RNA at Treatment Weeks 4, 12, and 24, and Rates of SVR and Treatment Failure, for Each Treatment Group.
| Rate, n/N (%) | SMV 75 mg 12W + Peg-IFN/RBV | SMV 75 mg 24W + Peg-IFN/RBV | SMV 150 mg 12W + Peg-IFN/RBV | SMV 150 mg 24W + Peg-IFN/RBV | Placebo + Peg-IFN/RBV |
|---|---|---|---|---|---|
| Week 4 | 59/78 (75.6) | 51/75 (68.0) | 58/77 (75.3) | 59/79 (74.7) | 4/77 (5.2) |
| Week 12 | 71/78 (91.0) | 70/75 (93.3) | 72/77 (93.5) | 75/79 (94.9) | 43/77 (55.8) |
| Week 24 | 72/78 (92.3) | 70/75 (93.3) | 65/77 (84.4) | 69/79 (87.3) | 60/77 (77.9) |
| SVR12 | 65/78 (83.3) | 57/75 (76.0) | 62/77 (80.5) | 68/79 (86.1) | 51/77 (66.2) |
| SVR24 | 64/78 (82.1)* | 56/75 (74.7) | 62/77 (80.5)* | 68/79 (86.1)* | 50/77 (64.9) |
| SVR W72 | 63/78 (80.8) | 53/75 (70.7) | 60/77 (77.9)* | 67/79 (84.8)* | 50/77 (64.9) |
| VBT | 5/78 (6.4) | 2/75 (2.7) | 6/77 (7.8) | 2/79 (2.5) | 4/77 (5.2) |
| Viral relapse | 8/72 (11.1) | 14/72 (19.4) | 6/69 (8.7) | 6/75 (8.0) | 11/62 (17.7) |
Statistically significant difference versus placebo and Peg-IFN/RBV; P < 0.05.
Abbreviation: W, week.
Among patients treated with SMV, 68.0%-75.6% achieved rapid virologic response (RVR; HCV RNA undetectable at week 4), of whom 87.9%-94.9% subsequently achieved SVR24. Only 5.2% of placebo-treated patients had RVR. Based on RGT criteria, 79.2%-86.1% of SMV-treated patients were eligible to complete treatment by week 24. SVR24 was subsequently achieved in 85.2%-95.6% of these patients (Fig. 2).
Fig. 2.
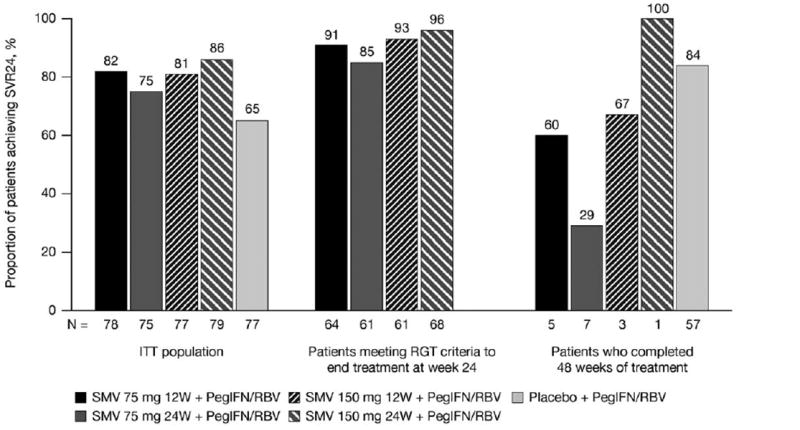
Rates of SVR for the ITT population and for patients who met virologic criteria for shortened duration of therapy.
ITT, intent-to-treat; PegIFN/RBV, peginterferon α-2a + ribavirin; RGT, response-guided therapy; SMV, simeprevir; SVR24, sustained virologic response (HCV RNA <25 IU/ml undetectable) 24 weeks after planned end of treatment; W, week.
Because no differences in SVR were noted between the 12- or 24-week durations of SMV, the two SMV 75-mg dose groups and the two 150-mg groups were pooled for exploratory subgroup analyses. For patients treated with SMV 75 mg, the SVR24 rates were lower for genotype 1a, compared with genotype 1b (66.2% and 88.9%, respectively), although the rates in genotype 1a and 1b patients were similar with SMV 150 mg (82.4% and 83.8%, respectively). SVR rates for those treated with placebo were 66.7% for genotype 1a and 63.8% for genotype 1b.
Multivariate regression analysis was performed for factors associated with SVR, including baseline HCV RNA level, METAVIR score (F3 versus F0-F2), genotype (1a versus 1b), race (Caucasian versus black), and gender (Fig. 3). Treatment with SMV was associated with higher rates of SVR for all of these factors. For patients with METAVIR scores of F0-F2 (n = 332), SVR rates were 81.7% and 84.6% with SMV (75 and 150 mg, respectively) and 64.3% with placebo control. For patients with scores of F3 (n = 53), rates were 63.0% and 75.0% with SMV (75 and 150 mg, respectively), compared to 71.4% with placebo control.
Fig. 3.
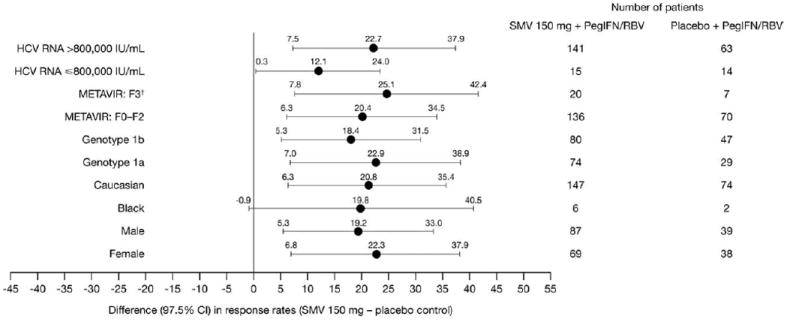
Difference in SVR24 between SMV 150 mg and placebo groups by demographic parameters.
CI, confidence interval; PegIFN/RBV, peginterferon α-2a + ribavirin; SMV, simeprevir; SVR24, sustained virologic response (HCV RNA <25 IU/mL undetectable 24 weeks after planned end of treatment.
†Patients with cirrhosis were not eligible.
Among SMV-treated patients infected with HCV genotype 1a, those without a Q80K polymorphism at baseline experienced higher SVR24 rates (SMV 150 mg: 53 of 62 [85.5%]; 75 mg: 36 of 51 [70.6%]), compared with patients with the Q80K polymorphism (SMV 150 mg: 8 of 12 [66.7%]; 75 mg: 11 of 20 [55.0%]).
On-Treatment Failure and Relapse
In SMV treatment arms, the frequency of VBT was low, with 15 of 309 (4.9%) patients experiencing VBT and, consequently, ceasing treatment with SMV (Table 2). VBT was evenly distributed between the 75-mg (7 of 15) and 150-mg (8 of 15) arms, and each of these 15 patients subsequently met a stopping criterion at week 12 or 24, ending all treatment. No additional SMV patients met stopping criteria. In contrast, 14.3% of those in the placebo control group met stopping criteria at week 12, 24, or 36 and ceased all treatment.
The majority of VBTs (9 of 15) occurred during the first 12 weeks of SMV dosing (Supporting Table B), in line with previous reports. In SMV 12-week arms, 5 of 11 instances of VBT occurred after the end of SMV dosing; however, initial evidence of failure emerged before week 12. VBT was more frequent among patients with genotype 1a, compared with genotype 1b, although this difference was less apparent in the SMV 150-mg dose group (6.8% and 3.8% with genotypes 1a and 1b, respectively).
Among patients who had undetectable HCV RNA at the end of treatment, viral relapse occurred less frequently among those treated with SMV regimens than with placebo control (11.8% versus 17.7%; Table 2). As with VBT, viral relapse was less common with the SMV 150-mg dose, particularly in HCV genotype 1a-infected patients, of whom 9% relapsed.
All 15 SMV-treated patients with sequence information available who experienced VBT and 27 of 33 (81.8%) with viral relapse had emerging mutations at NS3 amino acid positions 80, 155, and/or 168 at the time of VBT or relapse.
Differences in the type of emerging mutations were observed between genotype 1a- (mainly emerging R155K alone or in combination with other mutations at NS3 positions 80 and/or 168) and 1b (mainly D168V)-infected patients. The majority (10 of 12; 83.3%) of patients with baseline Q80K polymorphism experiencing VBT or relapse had emerging mutations (mainly R155K) at time of failure.
IL28B Genotype and Response
Approximately two thirds of patients consented to IL28B genotype testing and, of these, 60%-78% had a non-CC IL28B genotype (Table 1). Among patients with non-CC genotypes, rates of SVR24 were generally higher for patients treated with SMV 75 and 150 mg, compared to placebo control. SVR rates with SMV 75 mg were 83.9%, 78.1%, and 50.0%, and with SMV 150 mg were 97.1%, 80.0%, and 66.7% (CC, CT, and TT, respectively). In contrast, in the placebo control group, SVR rates were 100% with CC and 50% with both CT and TT (Supporting Table D). VBT was observed exclusively in the non-CC genotype (data not shown).
Safety
The most frequent AEs—fatigue, influenza-like illness, pruritus, headache, and nausea—were those typically associated with Peg-IFN and RBV therapy and were similar across SMV and placebo treatment groups (Table 3). AEs were the most common reason for discontinuation of at least one study drug, ranging between 4% and 10.4% of participants treated with SMV, compared to 13% in the placebo control group. Serious AEs (SAEs) occurred with similar frequency among those treated with SMV (3.8%-11.5%) or placebo control (13%). There were no deaths in the study.
Table 3. Incidence of AEs Overall, Including Most Common AEs*.
| AEs, n (%) | SMV 75 mg 12W + Peg-IFN/RBV, n = 78 | SMV 75 mg 24W + Peg-IFN/RBV, n = 75 | SMV 150 mg 12W + Peg-IFN/RBV, n = 77 | SMV 150 mg 24W + Peg-IFN/RBV, n = 79 | All SMV n = 309 | Placebo + Peg-IFN/RBV n = 77 |
|---|---|---|---|---|---|---|
| Any AE | 77 (98.7) | 75 (100.0) | 76 (98.7) | 79 (100.0) | 307 (99.4) | 75 (97.4) |
| Any grade 1 or 2 AE | 75 (96.2) | 75 (100.0) | 76 (98.7) | 79 (100.0) | 305 (98.7) | 75 (97.4) |
| Grade 1 | 75 (96.2) | 74 (98.7) | 75 (97.4) | 79 (100.0) | 303 (98.1) | 74 (96.1) |
| Grade 2 | 55 (70.5) | 58 (77.3) | 56 (72.7) | 57 (72.2) | 226 (73.1) | 56 (72.7) |
| Any grade 3 or 4 AE | 21 (26.9) | 22 (29.3) | 28 (36.4) | 28 (35.4) | 99 (32.0) | 27 (35.1) |
| Grade 3 | 21 (26.9) | 22 (29.3) | 27 (35.1) | 28 (35.4) | 98 (31.7) | 25 (32.5) |
| Grade 4 | 4 (5.1) | 8 (10.7) | 4 (5.2) | 2 (2.5) | 18 (5.8) | 3 (3.9) |
| Any death | 0 | 0 | 0 | 0 | 0 | 0 |
| Any SAE | 9 (11.5) | 4 (5.3) | 4 (5.2) | 3 (3.8) | 20 (6.5) | 10 (13.0) |
| Any AE leading to permanent stop of: | ||||||
| SMV/placebo and Peg-IFN/RBV | 4 (5.1) | 1 (1.3) | 4 (5.2) | 2 (2.5) | 11 (3.6) | 4 (5.2) |
| SMV/placebo only | 3 (3.8) | 1 (1.3) | 3 (3.9) | 4 (5.1) | 11 (3.6) | 2 (2.6) |
| Peg-IFN/RBV only | 0 | 1 (1.3) | 1 (1.3) | 1 (1.3) | 3 (1.0) | 4 (5.2) |
| Most common AEs: | ||||||
| General disorders and administration-site conditions | 65 (83.3) | 68 (90.7) | 70 (90.9) | 69 (87.3) | 272 (88.0) | 68 (88.3) |
| Fatigue | 26 (33.3) | 35 (46.7) | 32 (41.6) | 38 (48.1) | 131 (42.4) | 37 (48.1) |
| Influenza-like illness | 21 (26.9) | 32 (42.7) | 18 (23.4) | 27 (34.2) | 98 (31.7) | 29 (37.7) |
| Pyrexia | 18 (23.1) | 15 (20.0) | 15 (19.5) | 16 (20.3) | 64 (20.7) | 13 (16.9) |
| Asthenia | 20 (25.6) | 12 (16.0) | 18 (23.4) | 13 (16.5) | 63 (20.4) | 17 (22.1) |
| Irritability | 10 (12.8) | 7 (9.3) | 14 (18.2) | 11 (13.9) | 42 (13.6) | 8 (10.4) |
| Skin and subcutaneous tissue | 59 (75.6) | 47 (62.7) | 53 (68.8) | 54 (68.4) | 213 (68.9) | 55 (71.4) |
| Pruritus | 25 (32.1) | 17 (22.7) | 30 (39.0) | 24 (30.4) | 96 (31.1) | 35 (45.5) |
| Rash | 21 (26.9) | 10 (13.3) | 16 (20.8) | 18 (22.8) | 65 (21.0) | 18 (23.4) |
| Dry skin | 12 (15.4) | 12 (16.0) | 17 (22.1) | 22 (27.8) | 63 (20.4) | 14 (18.2) |
| Alopecia | 20 (25.6) | 11 (14.7) | 11 (14.3) | 11 (13.9) | 53 (17.2) | 16 (20.8) |
| Nervous-system disorders | 52 (66.7) | 42 (56.0) | 43 (55.8) | 44 (55.7) | 181 (58.6) | 49 (63.6) |
| Headache | 41 (52.6) | 34 (45.3) | 35 (45.5) | 32 (40.5) | 142 (46.0) | 40 (51.9) |
| Gastrointestinal disorders | 48 (61.5) | 43 (57.3) | 41 (53.2) | 45 (57.0) | 177 (57.3) | 51 (66.2) |
| Nausea | 26 (33.3) | 16 (21.3) | 20 (26.0) | 24 (30.4) | 86 (27.8) | 21 (27.3) |
| Diarrhea | 12 (15.4) | 14 (18.7) | 11 (14.3) | 10 (12.7) | 47 (15.2) | 12 (15.6) |
| Psychiatric disorders | 42 (53.8) | 34 (45.3) | 37 (48.1) | 42 (53.2) | 155 (50.2) | 40 (51.9) |
| Insomnia | 19 (24.4) | 14 (18.7) | 23 (29.9) | 13 (16.5) | 69 (22.3) | 23 (29.9) |
| Depression | 8 (10.3) | 4 (5.3) | 9 (11.7) | 11 (13.9) | 32 (10.4) | 14 (18.2) |
| Musculoskeletal and connective-tissue disorders | 36 (46.2) | 35 (46.7) | 35 (45.5) | 36 (45.6) | 142 (46.0) | 39 (50.6) |
| Myalgia | 17 (21.8) | 12 (16.0) | 16 (20.8) | 10 (12.7) | 55 (17.8) | 17 (22.1) |
| Arthralgia | 11 (14.1) | 12 (16.0) | 14 (18.2) | 16 (20.3) | 53 (17.2) | 11 (14.3) |
| Respiratory, thoracic, and mediastinal disorders | 30 (38.5) | 29 (38.7) | 30 (39.0) | 30 (38.0) | 119 (38.5) | 32 (41.6) |
| Cough | 18 (23.1) | 9 (12.0) | 12 (15.6) | 13 (16.5) | 52 (16.8) | 15 (19.5) |
| Dyspnea | 12 (15.4) | 8 (10.7) | 7 (9.1) | 6 (7.6) | 33 (10.7) | 6 (7.8) |
| Blood and lymphatic-system disorders | 27 (34.6) | 35 (46.7) | 28 (36.4) | 27 (34.2) | 117 (37.9) | 30 (39.0) |
| Neutropenia | 15 (19.2) | 23 (30.7) | 19 (24.7) | 18 (22.8) | 75 (24.3) | 16 (20.8) |
| Anemia | 15 (19.2) | 16 (21.3) | 17 (22.1) | 15 (19.0) | 63 (20.4) | 16 (20.8) |
| Infections and infestations | 25 (32.1) | 23 (30.7) | 23 (29.9) | 25 (31.6) | 96 (31.1) | 32 (41.6) |
| Investigations | 19 (24.4) | 16 (21.3) | 21 (27.3) | 20 (25.3) | 76 (24.6) | 18 (23.4) |
| Metabolism and nutrition disorders | 30 (25.6) | 17 (22.7) | 22 (28.6) | 16 (20.3) | 75 (24.3) | 19 (24.7) |
| Anorexia | 10 (12.8) | 12 (16.0) | 11 (14.3) | 9 (11.4) | 42 (13.6) | 11 (14.3) |
| Eye disorders | 13 (16.7) | 12 (16.0) | 16 (20.8) | 12 (15.2) | 53 (17.2) | 17 (22.1) |
| AEs of clinical interest: | ||||||
| Discontinuation of SMV/placebo because of anemia | 0 | 0 | 0 | 0 | 0 | 0 |
| Grade 3 or 4 anemia | 0 | 0 | 0 | 0 | 0 | 1 (1.3) |
| Discontinuation of SMV/placebo because of rash (any type) | 0 | 0 | 1 (1.3) | 1 (1.3) | 2 (0.6) | 1 (1.3) |
| Grade 3 or 4 rash (any type) | 0 | 0 | 1 (1.3) | 0 | 1 (0.3) | 0 |
| Discontinuation of SMV/placebo because of hyperbilirubinemia | 1 (1.3) | 0 | 0 | 0 | 1 (0.3) | 0 |
| Grade 3 or 4 hyperbilirubinemia | 1 (1.3) | 0 | 1 (1.3) | 0 | 2 (0.6) | 0 |
Occurring in >10% of patients in the pooled SMV groups during the overall treatment period.
Abbreviation: W, week.
Anemia was reported as an AE in 19.0%-22.1% of patients treated with SMV (all grade 1-2) and in 20.8% of those receiving placebo, and did not lead to discontinuation of SMV or placebo (Table 3). The median maximal decrease in hemoglobin was similar for SMV and placebo regimens (data not shown). Of note, the use of erythropoietin was not permitted during the study. Skin rash of any type (considered by the investigator to be at least possibly related to the study medication) was reported in 17.3%-30.8% of patients treated with SMV and in 23.4% of those treated with placebo. Rash AEs led to discontinuation of SMV or placebo in only 3 patients (2 in the SMV and 1 in the placebo control groups) occurred with comparable frequency between SMV study arms, and the majority were grade 1-2 in severity (Table 3).
A mild, isolated, and reversible increase in serum bilirubin was noted in patients treated with SMV. Most elevations in bilirubin, both direct and indirect, were grade 1, and hyperbilirubinemia led to discontinuation of SMV or placebo in only 1 patient (Table 3). The mean increase in bilirubin was most pronounced in the SMV 150-mg dose groups. Bilirubin decreased to baseline levels promptly after patients completed SMV dosing. Of note, there were no concomitant increases in serum aspartate aminotransferase, alanine aminotransferase (ALT), or alkaline phosphatase (ALP) associated with bilirubin elevations, and most patients with increased serum amino-transferases at baseline elicited a biochemical response during therapy (Fig. 4).
Fig. 4.
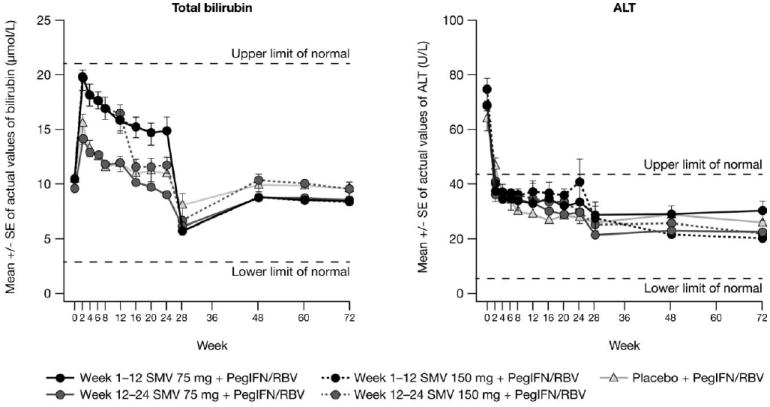
Change in bilirubin and ALT concentration up to week 72.
ALT, alanine aminotransferase; PegIFN/RBV, peginterferonα-2a + ribavirin; SE, standard error; SMV, simeprevir.
Patient-Reported Fatigue
Mean (± standard error) total FSS score increased from baseline (indicating more debilitating fatigue) in a similar manner in all groups by week 24. Mean score then returned to values comparable to baseline by week 36 and by week 60 in SMV and control groups, respectively (Fig. 5). Results of the piecewise linear mixed model confirmed that fatigue scores for patients receiving SMV were sig-nificantly lower, compared to placebo control patients, over the 72-week study period (P < 0.001), likely resulting from the shorter mean treatment duration in these groups.
Fig. 5.
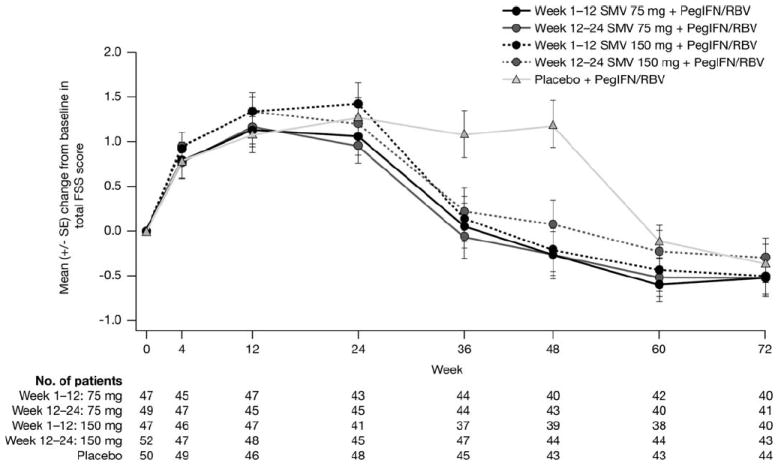
Change from baseline in total FSS score up to week 72.
FSS, Fatigue Severity Scale; PegIFN/RBV, peginterferon α-2a and ribavirin; SE, standard error; SMV, simeprevir; W, week.
Discussion
SMV administered QD in combination with Peg-IFN and RBV significantly improved rates of SVR, compared with the Peg-IFN and RBV and placebo control groups. The study was designed to evaluate two doses of SMV and two different durations of triple combination therapy. Results from this study indicated that all four SMV treatment arms accomplished similar rates of SVR, ranging between 75% and 86%, which were statistically significantly higher than placebo control in all but one treatment group. Higher response rates were observed with SMV 150 mg, compared with the 75-mg dose, in several patient subgroups, including those infected with HCV genotype 1a. Importantly, the majority (79%-86%) of patients treated with SMV were able to shorten the total duration of therapy to only 24 weeks. Of these patients, SVR rates of 93%-96% were observed among those who completed 24 weeks of treatment with SMV 150 mg.
The current study was not powered to provide definitive data across subpopulations, and observations must be confirmed in larger phase III trials. However, exploratory analyses of several subpopulations, some associated with lower response to Peg-IFN and RBV, such as high levels of viremia at baseline and advanced fibrosis, indicated that triple therapy with SMV provided benefit, compared to control. The influence of unfavorable IL28B genotype, which is reported as being strongly associated with response to Peg-IFN and RBV,17-22 was ameliorated by the addition of SMV. An increase in SVR24 was generally evident in SMV treatment regimens, compared to placebo control, for non-CC genotypes. Whereas VBT was infrequent, it was noted only among non-CC genotypes. For patients infected with HCV genotype 1a, higher SVR rates were observed in patients without the Q80K polymorphism, compared with those with the polymorphism. In the presence of Q80K, higher SVR rates were noted with the 150-mg dose (the dose selected for ongoing phase III trials), compared with the 75-mg dose. However, because the number of patients in the study with this polymorphism was small (SMV 150 mg: n = 12; 75 mg: n = 21), no firm conclusions can be drawn. The results of larger, ongoing phase III trials will allow further investigation of this effect.
The magnitude of SVR in the control group treated with Peg-IFN and RBV alone was unexpected, although not unprecedented.23,24 Progressively higher rates of SVR with Peg-IFN and RBV have been noted as the importance of adherence has been recognized for patients, and clinicians have become more adept at managing AEs. Adherence to treatment is essential for optimization of SVR rates, and participation in well-controlled, prospective clinical trials may increase adherence and, ultimately, SVR rates (17-21).17-21 The high rates of treatment completion (over 90%) in the current study are consistent with this observation. The exclusion of patients with cirrhosis and the enrollment of few black patients may have also been contributing factors to the high SVR rate in the control group. However, no single factor was identified in post-hoc analyses to explain the higher than anticipated response rate in the control group. Nevertheless, statistical superiority of treatment with SMV-containing regimens over Peg-IFN and RBV alone was established for all dose and duration regimens, with the exception of SMV 75 mg administered for 24 weeks.
The high rates of SVR in SMV dosing groups were achieved without exacerbating the well-defined safety pro-file of Peg-IFN and RBV. Treatment-emergent AEs among those receiving SMV were generally similar to Peg-IFN and RBV alone. Specifically, there was no increase in anemia, neutropenia, or adverse skin manifestations (rash and pruritus), in contrast to the increased frequency of these side effects reported with currently available triple-therapy regimens that include boceprevir or telaprevir.4,6 Hyperbilirubinemia, most evident in the SMV 150-mg dosing arms, plateaued early during treatment, was not progressive, and was rapidly reversible at the end of SMV dosing. There were no concomitant increases in serum aminotransferases or ALP during the SMV dosing intervals, further supporting the absence of hepatotoxicity. Studies in vitro have demonstrated that SMV is an inhibitor of bilirubin transporters organic anion-transporting polypeptide 1B1 and multidrug resistance-associated protein 2, suggesting a likely mechanism for hyperbilirubinemia during SMV treatment.25
Incidence and severity of fatigue AEs were comparable for all treatment groups. Fatigue severity measured using the FSS, a generic self-report fatigue measure that patients completed during clinical visits throughout the study, clarified the relationship between treatment and fatigue. Fatigue severity increased comparably during therapy in all groups and then returned to baseline when treatment ended; the shortened treatment duration experienced by the majority of patients receiving SMV led to a reduced overall duration of fatigue in those groups, without significantly increasing fatigue severity during treatment.
However, it should be noted that the exclusion of patients with cirrhosis (METAVIR score F4) from this study precludes any firm conclusions from being made regarding the SMV safety profile in patients with cirrhosis.
The ultimate aim of this phase IIb trial was to identify the optimal dose and duration of SMV to be used for phase III registration trials. In the current study, there were no substantial differences across SMV treatment regimens in the proportion of patients qualifying for RGT, achieving SVR, or in safety analyses, with which to select the dose for phase III studies. Thus, other, more subtle information derived from this study informed dosing decisions for the phase III trials. First, results indicate that a 12-week treatment duration is most appropriate. VBT, although uncommon in patients treated with SMV, was usually evident within the first 12 weeks of therapy and viral relapse rates were similar between the 12- and 24-week groups, suggesting that extending SMV to 24 weeks would not provide additional benefit. Second, data support the selection of the SMV 150-mg dose. In the SMV 150-mg dose group, higher SVR and lower viral relapse rates were observed with the 1a genotype, compared to the 75-mg groups. Response rates in patients with IL28B non-CC genotypes were also slightly higher with 150 mg, compared with 75 mg. Furthermore, in patients infected with HCV genotype 1a, higher response rates were observed with the SMV 150-mg group (compared with 75 mg) in patients with higher BMI as well as in those with METAVIR F3 scores. Thus, SMV 150 mg administered QD for 12 weeks in combination with Peg-IFN and RBV, followed by Peg-IFN and RBV for an additional 12 weeks in an RGT algorithm, was selected for ongoing phase III trials in treatment-naïve and -experienced populations.
Supplementary Material
Acknowledgments
The authors thank the patients and their families as well as the PILLAR study investigators and their study staff.
Potential conflict of interest: This study was funded by Janssen Research & Development, LLC. Editorial support was provided by Dr. Bethan Hahn, on behalf of Complete Medical Communications, funded by Janssen Research & Development, LLC. Dr. Fried has received research grants from, and serves as an ad hoc advisor to, Merck, Genentech, Vertex Pharmaceuticals, Tibotec/Janssen, Gilead, Bristol-Myers Squibb, Abbott, and AbbVie. Dr. Buti has participated in advisory boards and/or been a speaker for Merck, Sharpe and Dohme (MSD), Gilead, Vertex, Janssen, and Bristol-Myers Squibb. Dr. Dore has participated in an advisory committee for, and/or received financial support for research, teaching, or speaking, and/or received a travel scholarship from, Roche, Merck, Bristol-Myers Squibb, Janssen, AbbVie, and Gilead. Dr. Flisiak acts as an advisor for, and/or has received lecture fees from, Bristol-Myers Squibb, Gilead, Janssen, Merck, Novartis, Roche, and Vertex Pharmaceuticals. Dr. Ferenci was a member of an advisory board and speaker’s bureau for Roche (Basel, Switzerland) and Rottapharm/Madaus (Monza, Italy), an advisor to Boehringer Ingelheim, Vertex Pharmaceuticals, Pfizer, Novartis, GlaxoSmithKline, Sanofi, and MSD, a consultant and advisor to Janssen, and receives an unrestricted research grant from Roche (Austria). Dr. Jacobson has received clinical research grants from, and/or was a consultant/advisor or member of a speaker’s bureau for, Abbott, Achillion Pharmaceuticals, Anadys Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Enanta, Gilead, GlaxoSmithKline, GlobeImmune, Inhibitex, Idenix, Kadmon, Novartis, Pfizer, Pharmasset, Presidio, Roche/Genentech, Schering/Merck, Tibotec/Janssen, Vertex Pharmaceuticals, and Zymogenetics. Dr. Marcellin has received grants and/or financial support for speaking or providing expert opinion for Roche, Schering-Plough, Gilead, Bristol-Myers Squibb, Vertex Pharmaceuticals, Novartis, Pharmasset, Tibotec, MSD, Boehringer Ingelheim, Biolex, Intermune, Alios BioPharma, Abbott, Pfizer, and Zymogenetics. Dr. Sherman consults and is on the speakers’ bureau for Janssen, Vertex, and Merck. He also consults for Gilead and Boehringer Ingelheim. Dr. Zeuzem consults, advises, and is on the speakers’ bureau for Janssen, Vertex, and Merck. Dr. Scott and Dr. De Smedt own stock in Janssen. Dr. Gilles, Dr. Peeters, and Dr. Sekar own stock in Johnson & Johnson.
Abbreviations
- AE
adverse event
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- BMI
body mass index
- CHC
chronic hepatitis C
- CI
confidence interval
- EC50
half-maximal effective concentration
- FSS
Fatigue Severity Scale
- HCV
hepatitis C virus
- IL28
interleukin-28
- Peg-IFN
pegylated interferon
- NS
nonstructural protein
- PILLAR
The Protease Inhibitor TMC435 study assessing optimaL dose and duration as once daiLy Antiviral Regimen
- QD
once-daily
- RBV
ribavirin
- RGT
response-guided therapy
- RVR
rapid virologic response
- SAE
serious adverse event
- SMV
simeprevir
- SVR
sustained virologic response
- SVR12
SVR 12 weeks after the planned end of treatment
- SVR24
SVR 24 weeks after the planned end of treatment
- SVR W72
SVR at week 72
- VBT
viral breakthrough
Appendix
The PILLAR study investigators and their study staff are: Australia: Paul Desmond, Greg Dore, Jacob George, Alice Lee, Graeme Macdonald, and Stuart Roberts; Austria: Peter Ferenci, Michael Gschwantler, and Hermann Laferl; Belgium: Jochen Decaestecker, Yves Horsmans, Peter Michielsen, Christophe Moreno, Frederik Nevens, Hans Orlent, Hendrik Reynaert, and Hans Van Vlierberghe; Canada: Pierre Cote, Maged Peter Ghali, Gideon Hirschfield, Sam Lee, and Morris Sherman; Denmark: Peer Christensen, Jan Gerstoft, Alex Lund Laursen, Lars Mathiesen, and Axel Møller; France: Yves Benhamou, Jean-Pierre Bronowicki, Jean-Didier Grange, Christophe Hezode, Patrick Marcellin, Albert Tran, Christian Trepo, and Jean-Pierre Zarski; Germany: Keikawus Arasteh, Thomas Berg, Peter Buggisch, Tobias Goeser, Hartwig Klinker, Michael P. Manns, Stefan Mauss, Jens Rasenack, Hans-Jürgen Stellbrink, Andreas Trein, and Stefan Zeuzem; New Zealand: Graeme Dickson, Edward Gane, and Catherine Stedman; Norway: Trond Bruun, Jon Florholmen, Kjell Hellum, Zbigniew Konopski, and Bent von der Lippe; Poland: Robert Flisiak, Waldemar Halota, Andrzej Horban, Maciej Jablkowski, Ewa Janczewska-Kazek, and Wieslaw Kryczka; Russia: Pavel O. Bogomolov, Vladimir T. Ivashkin, Olga V. Korochkina, Vyacheslav G. Morozov, Igor G. Nikitin, Vladimir V. Rafalskiy, Evgeny E. Voronin, Alexey A. Yakovlev, and N. Zakharova; Spain: Maria Buti, José-Luis Calleja, Moises Diago, Ricardo Moreno-Otero, and Manuel Romero; United States: Edwin DeJesus, Kyle Etzkorn, Michael Fried, Andrei Gasic, Nigel Girgrah, Ira M. Jacobson, Donald M. Jensen, Mark E. Jonas, Daniel Pam-bianco, Fred Poordad, Coleman Smith, Jawahar Taunk, Lawrence Wruble, and Ziad Younes.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ghany MG, Nelson DR, Strader DB, Thomas DL, Seeff LB. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:1433–1444. doi: 10.1002/hep.24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asselah T, Marcellin P. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 2012;32(Suppl 1):88–102. doi: 10.1111/j.1478-3231.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 4.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 2011;365:1014–1024. doi: 10.1056/NEJMoa1014463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barritt AS, Fried MW. Maximizing opportunities and avoiding mistakes in triple therapy for hepatitis C virus. Gastroenterology. 2012;142:1314–1323. doi: 10.1053/j.gastro.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hezode C, Dorival C, Zoulim F, Poynard T, Mathurin P, Pol S, et al. Safety of telaprevir or boceprevir in combination with peginterferon alfa/ribavirin, in cirrhotic non responders. First results of the French Early Access Program (ANRS CO20-CUPIC) J Hepatol. 2012;56:8. [abstract] [PMC free article] [PubMed] [Google Scholar]
- 8.Reesink HW, Fanning GC, Farha KA, Weegink C, Van Vliet A, van ’t Klooster G, et al. Rapid HCV-RNA decline with once daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 2010;138:913–921. doi: 10.1053/j.gastro.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 9.Moreno C, Berg T, Tanwandee T, Thongsawat S, Van Vlierberghe H, Zeuzem S, et al. Antiviral activity of TMC435 monotherapy in patients infected with HCV genotypes 2-6: TMC435-C202, a phase IIa, open-label study. J Hepatol. 2012;56:1247–1253. doi: 10.1016/j.jhep.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 10.Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, et al. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob Agents Chemother. 2009;53:1377–1385. doi: 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reesink HW, Zeuzem S, Weegink CJ, Forestier N, Van Vliet A, van de Wetering de Rooij J, et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterology. 2006;131:997–1002. doi: 10.1053/j.gastro.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Sarrazin C, Rouzier R, Wagner F, Forestier N, Larrey D, Gupta SK, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007;132:1270–1278. doi: 10.1053/j.gastro.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 13.Manns M, Reesink H, Berg T, Dusheiko G, Flisiak R, Marcellin P, et al. Rapid viral response of once-daily TMC435 plus pegylated inter-feron/ribavirin in hepatitis C genotype-1 patients: a randomized trial. Antivir Ther. 2011;16:1021–1033. doi: 10.3851/IMP1894. [DOI] [PubMed] [Google Scholar]
- 14.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleinman L, Zodet MW, Hakim Z, Aledort J, Barker C, Chan K, et al. Psychometric evaluation of the fatigue severity scale for use in chronic hepatitis C. Qual Life Res. 2000;9:499–508. doi: 10.1023/a:1008960710415. [DOI] [PubMed] [Google Scholar]
- 16.Lenz O, Verbinnen T, Lin T-I, Vijgen L, Cummings MD, Lindberg J, et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother. 2010;54:1878–1887. doi: 10.1128/AAC.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark PJ, Thompson AJ. Host genomics and HCV treatment response. J Gastroenterol Hepatol. 2012;27:212–222. doi: 10.1111/j.1440-1746.2011.06918.x. [DOI] [PubMed] [Google Scholar]
- 18.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 19.Rauch A, Kutalik Z, Descombes P, Cai T, Di Iulio J, Mueller T, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 20.Suppiah V, Moldovan M, Ahlenstiel G, Berg T, Weltman M, Abate ML, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka Y, Nishida N, Sugiyama M, Kurosaki M, Matsuura K, Sakamoto N, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 22.Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, Shianna KV, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–129. doi: 10.1053/j.gastro.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Manns MP, Gane E, Rodriguez-Torres M, Stoehr A, Yeh CT, Marcellin P, et al. Vaniprevir with pegylated interferon alpha-2a and ribavirin in treatment-naive patients with chronic hepatitis C: a randomized phase II study. Hepatology. 2012;56:884–893. doi: 10.1002/hep.25743. [DOI] [PubMed] [Google Scholar]
- 24.Sulkowski MS, Ferenci P, Emanoil C, Asselah T, Caruntu F, Lalezari J, et al. SILEN-C1: early antiviral activity and safety of BI 201335 combined with peginterferon alfa-2a and ribavirin in treatment-naive patients with chronic genotype 1 HCV infection. Hepatology. 2009;50:2A. [Google Scholar]
- 25.Huisman MT, Snoeys J, Monbaliu J, Martens M, Sekar V, Raoof A. In vitro studies investigating the mechanism of interaction between TMC435 and hepatic transporters. Poster 278 presented at the 61st Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); Boston, MA. October 29-November 2, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.