

Abstract
Silk fibroin is a useful protein polymer for biomaterials and tissue engineering. In this work, porogen leached scaffolds prepared from aqueous and HFIP silk solutions were reinforced through the addition of silk particles. This led to about 40 times increase in the specific compressive modulus and the yield strength of HFIP-based scaffolds. This increase in mechanical properties resulted from the high interfacial cohesion between the silk matrix and the reinforcing silk particles, due to partial solubility of the silk particles in HFIP. The porosity of scaffolds was reduced from ≈90% (control) to ≈75% for the HFIP systems containing 200% particle reinforcement, while maintaining pore interconnectivity. The presence of the particles slowed the enzymatic degradation of silk scaffolds.
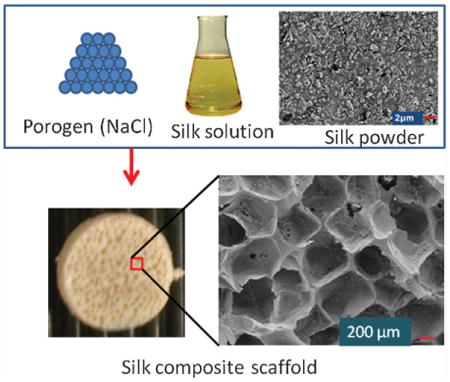
Keywords: biodegradable, composites, particles, scaffolds, silk
Introduction
Biomaterial scaffolds are an integral part of tissue engineering, which is an alternative approach to designing implant systems capable of restoring function to diseased or damaged tissues, in lieu of the fundamental limitations of autograft and allograft tissue as a clinical resource.[1] Autografts have problems such as restricted amounts of material for use in other sites in the body, donor site morbidity, and suboptimal clinical outcomes. Allografts carry the risk of transmission of disease and adverse immunological reactions.[2,3] Conversely, three-dimensional porous scaffolds have proven effective temporary substrates to template engineered tissue and have shown biological compatibility and integration. Meanwhile, there have been numerous attempts to optimize scaffold function toward improved remodeling characteristics, cell–material interactions, host integration, and structural stability. Aside from tissue engineering standpoints, scaffolds can also be used for the study of diseases due to the three-dimensional approximation to in vivo micro-environments compared to the two-dimensional monolayer cell culture systems.[4]
The frequency and types of degenerative skeletal tissue disorders in a progressively aging human population makes musculoskeletal tissue repair prominent among tissue-engineering research.[1,3] Skeletal tissue engineering scaffolds could supplant the role of artificial non-degradable prostheses, which do not interface seamlessly with surrounding host tissues and offer no remodeling potential. Conversely, biocompatible and osteoconductive scaffolds are further tasked with stringent mechanical requirements and improved integration demands.[3,5] Scaffolds formed from soft materials must resist handling during surgery and post-implantation mechanical and hydrodynamic stresses and strains.[6] The ability to control pore size in the scaffold is also important in terms of in vitro and in vivo tissue formation.[7] Large pores support vascularization and enhance osteogenesis, but can diminish the implant's in vivo load-bearing capacity. Furthermore, slow degradation is important, e.g., to ensure that the supportive functions are maintained until adequate mineralization takes place during resorption and new bone formation.[8]
Silk fibroin has emerged as an important biomaterial to meet scaffold design specifications due to the protein's good mechanical properties,[9] biocompatibility,[8,10–12] and biodegradability.[8,10,13–16] In previous works, 3-D silk-based scaffolds were able to support bone and cartilage development in vitro[8,17–20] and were used for healing critical size femur defects in rats.[5] They were useful in bridging large defects with new bone after 8 weeks and exhibited good load-bearing capabilities and torque when compared to other experimental and control groups.[5] The scaffolds also exhibited good biocompatibility, biodegradability, and vascularization in vivo.[8,20] However, 3-D silk scaffolds still need substantial improvement to meet the requirements of musculoskeletal tissue repair. For instance, it was reported that after 12 weeks of bone tissue ingrowth, despite increase in strength, they remained much weaker[17] (wet compressive modulus <200 kPa and yield strength <40 kPa) than natural bone.
Bone is an apatite–collagen composite material at the ultra-structural level and hence, a polymer matrix composite appears a natural choice for supporting bone growth.[21] Moreover, a composite system may offer synergistic material features and thus be able to meet the multiple design requirements. Existing composite design approaches include coating a bioactive layer of one material over a bioinert material[22,23] to improve cell adhesion and growth, and adding particles to a polymer matrix to enhance mechanical properties and/or cellular interactions.[22,24–32] The published reports suggest that improved mechanical properties and bioactivity could be achieved using composite systems versus single components alone. However, despite the progress and opportunities with composites, no approach to date has established the capacity to develop mature bone, both structurally and functionally, nor demonstrated sufficient efficacy of a relevant implant to warrant human clinical trials.
Composite design faces a challenge of compatibility between components. Poor compatibility between components can result in inhomogeneous mixtures and phase separation, and also adverse tissue reactions.[21] Furthermore, non-synchronous degradation rates can lead to other complications, such as premature pore collapse. We hypothesized, therefore, that a biocompatible, biodegradable and mechanically superior composite scaffold could be formed from a single polymer, by incorporating silk particles in a regenerated silk matrix. This new type of composite system, wherein both components are the same protein, could potentially exploit the interfacial compatibility between the continuous and particle phases and thus eliminate or at least reduce some of the complications associated with the composite materials.
We have recently developed a fabrication method to prepare ultrafine particles from silk fibers through a combination of standard milling processes.[33,34] These particles retain substantial crystallinity from the parent fibers and were considered useful for scaffold reinforcement. Previously, commercial silk particles[35,36] were used for scaffold reinforcement. Co-precipitation and freeze drying techniques were adapted for scaffold fabrication and non-silk polymers were used as the continuous matrix. In the present work, we followed the porogen-leaching process to fabricate the novel silk/silk macro-porous composite scaffolds from aqueous and 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP) silk solutions through the addition of crystalline silk particles. Measurements of their morphology, mechanical properties and degradation behavior are reported. As properties of composites may be affected by composition, size distribution, and volume/weight percentage of particles,[21] the effects of such variants were also studied.
Experimental Part
Preparation of Milled Silk Powder
Bombyx mori (B. mori) and Philosamia cynthia ricini (P. c. ricini) silk cocoons were purchased from North Eastern India. B. mori cocoons were cut to remove pupae. P. c. ricini cocoons were open mouthed and were free from pupae. They were degummed in a laboratory dyeing machine (Thies Corp.) using 0.02 m sodium carbonate and 0.6g · L−1 sodium dodecyl sulfate (Sigma–Aldrich) at 100 °C with a fiber (kg) to liquor (L) ratio of 1:25. The batch size of cocoons was 1 kg. Degumming time was 20 min for B. mori and 120 min for P. c. ricini cocoons. Degummed fibers were milled as previously described.[33] Briefly, fibers were chopped into snippets using a cutter mill (Pulverisette 19, Fritsch). A stirred ball mill (1S Attritor, Union Process Inc.) was used for wet grinding of the chopped snippets using 20 kg yttrium treated zirconium oxide grinding media (5 mm) in a 9.5 L tank. The stirrer speed was 280 rpm. Time of milling was 6 h for P. c. ricini and 10 h for B. mori. Distilled water was used in the wet grinding operation. Dry powders from the wet milled slurry were recovered using a laboratory spray dryer (B-290, Buchi Labortechnik AG). A Sturtevant laboratory air jet mill with a grinding air pressure of 110 kg · cm−2 was used to further reduce the particle size. Pre-processed silk powder was fed with a powder hopper (K-Tron) at a feed rate of 200 g · h−1 and processed continuously by the air jet mill.
Preparation of Regenerated Silk Solution
Bombyx mori (B. mori) silk fibroin solutions were prepared as described previously.[16,37] Briefly, cocoons were degummed at 100 °C for 40 min in an aqueous solution of 0.02 m Na2CO3 with a fiber (kg) to liquor (L) ratio of 1:400 and then rinsed thoroughly with deionized water. The degummed silk was dissolved in 9.3 m LiBr solution at 60 °C yielding a 20% w/v solution. This solution was dialyzed against water using Slide a-Lyzer dialysis cassettes (Pierce, MWCO 3500) for three days with frequent change of water. The final concentration of the aqueous silk fibroin solution was about 7% w/v. Part of the silk solution was frozen at −80 °C and then lyophilized. The lyophilized silk was added to hexafluoroisopropanol (HFIP) (Sigma–Aldrich) to prepare 17% w/v solvent-based silk solution.
Preparation of Composite Scaffolds
Aqueous-Derived Composite Scaffolds
Aqueous-derived composite scaffolds were prepared by modifying the method of porogen leached aqueous-derived silk scaffolds previously described.[16] A schematic diagram illustrating aqueous-derived composite scaffold fabrication is shown in Figure 1. Silk powder was suspended in deionized water and then ultrasonically dispersed using a Branson 450 Sonifier (Branson Ultrasonics Co.) to achieve a stable aqueous suspension of powder. To achieve different mixture ratios in the composites, the silk powder suspensions were added to aqueous silk solutions at varying concentrations so that % powder loaded varied from 0 to 25% based on the “matrix” weight (derived from the silk solution). Final matrix concentration was kept constant at 6% w/v (i.e., 60 mg silk per 1 mL solution). As an example, for a 25% composite, the final matrix mass being 120 mg (from a 2mL solution), 30 mg of powder (in suspension) was mixed with the silk solution of an appropriate concentration. The mixture was added to a cylindrically-shaped container (18 mm in diameter) and 4 g of granular NaCl (particle size 500–600 μm) was added slowly to the container. Then, the container was covered and left at room temperature for 2 d followed by leaching out the NaCl particles in deionized water over 3 d.
Figure 1.
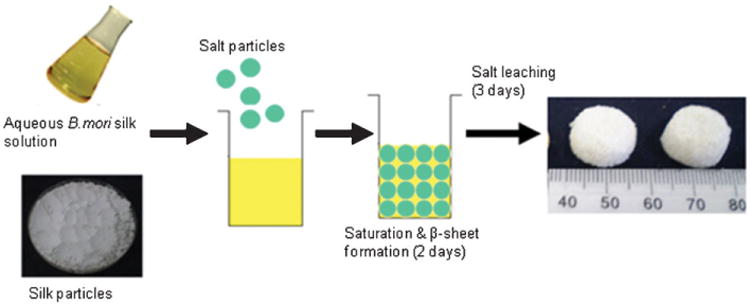
Schematic diagram of formation of aqueous based silk-silk composite scaffolds.
1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP)-Derived Composite Scaffolds
1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP)-derived composite scaffolds were prepared by modifying methods used to prepare HFIP-based silk scaffolds as previously described.[5,16,37,38] Figure 2 shows the schematic diagram of HFIP-derived composite scaffold fabrication. 1 mL of 17% w/v HFIP-based silk stock solution was poured into a glass vial (15 mm diameter) containing 3.4 g NaCl particles premixed with a desired amount of silk power. The % of silk powder to silk matrix w/w was controlled up to 200%. For example, for a 200% HFIP composite, the final matrix mass was estimated to be 170 mg, and thus 340 mg of powder was premixed with NaCl particles and put in a glass vial before the 1 mL HFIP solution was added. Pore size of the scaffolds was controlled by using NaCl particles of different size (210–300, 500–600, and 850–1000 μm). The glass vials were covered and centrifuged at 365 × g rcf. to help the viscous silk solution permeate the porogen-particle bed, and were kept covered overnight to allow for complete saturation. Subsequently, the solvent was evaporated at room temperature for 2 d. The matrices were then treated in 80% methanol for 3 h to induce a protein conformational transition to β-sheet, followed by drying for two days in a fume hood to remove the methanol. A porous composite was derived after leaching out the NaCl particles in deionized water over 3 d.
Figure 2.
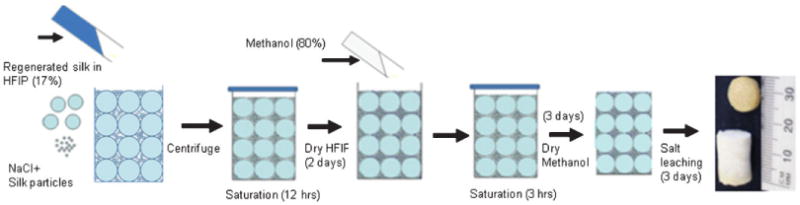
Schematic diagram of formation of HFIP-based silk-silk composite scaffolds.
Particle Characterization
Particle Size
A Mastersizer 2000 (Malvern Instruments, Worcestershire, UK), which is a laser diffraction-based particle size analyzer, was used to measure size distribution of silk particles. A Hydro 2000S side feeder was used to disperse particles for measurement and propan-2-ol (Sigma–Aldrich) was used as the dispersion medium. A refractive index of 1.561 and 1.542 were used for B. mori and P. c. ricini fibers, respectively, and were determined from the refractive indexes perpendicular and parallel to silk fiber axial direction.[39] An imaginary refractive index of 0.01 was used for necessary calculations with the software, Mastersizer 2000 (ver 5.21). The size and shape of the particles were also examined, after gold sputter coating, with a scanning electron microscope (SEM) (LEO 1530 FEG-SEM) at 2 kV accelerated voltage and 2–4 mm working distance.
Particle Solubility
An experiment was performed to evaluate the extent of silk particle dissolution in HFIP and the ability of the HFIP/silk particle solutions to form homogeneous films. 5% w/v silk particles were mixed with water or HFIP and kept at room temperature for up to 9 d, while being visually monitored. To study the presence of crystalline particles, films prepared from stored mixtures were subsequently scanned by Fourier transform infrared (FTIR) Spectrophotometry (Vertex 70 from Bruker) using attenuated total reflectance (ATR). Each spectrum was obtained in absorbance mode in the range 4000–600 cm−1. The films were also gold sputter coated and then examined under SEM (Leica S440 W-SEM) at 10 kV accelerated voltage and 8 mm working distance.
Composite Scaffold Characterization
Scanning Electron Microscope (SEM)
Scaffolds were fractured in liquid N2 using a surgical blade. Morphologies of gold sputter-coated scaffolds were observed with a Leica S440 W-SEM at 10 kV accelerated voltage and 15 mm working distance.
Porosity Measurement by Liquid Displacement
Porosity of the scaffolds was determined via a liquid displacement method with hexane, as previously reported.[37] The scaffolds were lyophilized and then immersed in a graduated cylinder of known volume of hexane (v1). A series of quick evacuation–repressurization cycles were performed to completely evacuate entrapped air and to impregnate the scaffold with hexane; thereafter, the volume in the cylinder was recorded (v2). The hexane impregnated scaffold was removed and the volume was recorded again (v3). Any change of volume due to evaporation during the evacuation cycles was checked using another cylinder without the scaffold. The porosity of the scaffold is expressed as:
Porosity Measurement by Density
The bulk density of the scaffolds (ρb) was determined by dividing the weight of the dry scaffolds by measured scaffold volume (v2 – v3) from the hexane method above. The density of silk (ρ) (matrix + particles) was calculated from known density of silk from a previous study[39] (1.355 g·cm−3 for B. mori and 1.314 g·cm−3 for P. c ricini). The relative density and the porosity were calculated by the following equations:
Mechanical Tests
Unconfined compression tests were performed on an Instron (Norwood, MA, USA) 3366 testing frame equipped with a 100 N capacity load cell, and smooth impermeable stainless steel compression test platens. Tests were conducted in a 37 °C temperature controlled 0.1 m PBS bath (Biopuls, Instron Corp.). These samples were soaked in PBS at 20 °C for more than 24 h to ensure equilibration in wet conditions, followed by at least 2 min of temperature equilibration in the 37 °C bath prior to initiating the tests. All tests were performed using a displacement control mode at a rate of 5 mm · min−1, and all other conditions followed ASTM standard D1621-04a (Standard Test Method for Compressive Properties of Rigid Cellular Plastics) with the following alterations. After the tests, the compressive stress and strain were graphed based on the measured cross-sectional area (diameter of 12 mm) and sample height (nominal ≈4–5 mm, measured automatically at 0.02 N tare load), respectively. The yield strength as well as the compressive modulus and standard deviation were determined after testing was complete, based on previously reported methods.[17,19,40] Briefly, the elastic modulus was calculated based on a linear regression fitting of a 5% strain section that precedes an identifiable plateau region. The compressive yield strength was determined using an offset-yield approach. A line was drawn parallel to the modulus line, but offset by 0.5% of the initial sample gauge length. The corresponding stress value at which the offset line crossed the stress–strain curve was defined as the compressive yield strength of the scaffold, and is an estimate of the linear elastic and collapse plateau transition point. The compressive modulus and yield strength were divided by the bulk density to calculate the specific modulus and specific yield strength.
In vitro Enzymatic Degradation
The degradation of the silk fibroin scaffolds was evaluated using protease XIV from Streptomyces griseus (Sigma–Aldrich) with an activity of 4.5 U · mg−1. Each sample (12 mm diameter, 2 mm height) was lyophilized and divided into two parts and weighed. One part was treated with 1 mg of the protease per 1 mL phosphate buffer (pH 7.4) and the other part only in phosphate buffer as control. The material to liquid ratio was 1:150 and the samples were agitated at 37 °C on a shaker (New Brunswick Scientific, NJ, USA). Enzyme solutions were replaced every 2 d to maintain enzyme activity. Scaffolds were collected at different time points, washed gently and thoroughly with deionized water, lyophilized, and weighed. Percentage loss in weight at respective points was calculated.
Statistics
Statistical analysis was performed using Student's t-test (two tails). Differences were considered significant when p ≤ 0.05.
Results and Discussion
Particle Characteristics
Table 1 contains the silk particle specifications in terms of silk species as source material (B. mori or P. c. ricini), particle preparation method, and median particle size [d(0.5)] of a volume-based size distribution. SEM images of silk particles are shown in Figure 3. Attritor milled (AM) particles were nearly spherical aggregates (images “a” and “b” in Figure 3). Air jet milling (AJM) separated the aggregates and further reduced the particle size (image “c” in Figure 3) to less than 1 μm. Table 1 and Figure 3(a, b) suggest that the size and morphology of B. mori AM (BM-AM) and P. c. ricini AM (P.C.R-AM) silk particles were similar.
Table 1.
Specification of silk particles for composites.
| Particle code | Type of species | Particle fabrication method | Volume d(0.5) |
|---|---|---|---|
|
| |||
| μm | |||
| BM-AM | B. mori | Wet attritor milling for 10 h | 7.5 |
| PCR-AM | P. c. ricini | Wet attritor milling for 6 h | 5.6 |
| PCR-AM-AJM | P. c. ricini | PCR-AM + air jet milling | 0.77 |
Figure 3.

Scanning electron microscope (SEM) image of silk particles; (a) BM-AM, (b) PCR-AM, and (c) PCR-AM-AJM.
Formability of Composite Scaffolds
It is known that NaCl induces chain folding and chain–chain interactions in silk fibroin, leading to gelation, and formation of a water stable β-sheet structure.[16] In order to prepare porous scaffold from aqueous silk solution, slow and homogeneous gelation is required to form the silk network around the porogens.[16] An increase in concentration of silk solution resulted in a failure of scaffold formation and excessive gelling is thought responsible for this failure.[16] In the aqueous scaffold composite protocol presented here, the presence of the silk particles did not induce gelation of silk fibroin solution prior to the addition of NaCl and did not inhibit scaffold formation with up to 25% particle addition. Hence, the particle-reinforced aqueous-based silk porous scaffolds from a 6% silk solution were successfully obtained through the salt leaching method. However, higher loading of the silk particles (50%) in the aqueous silk solution did not result in the formation of free standing porous scaffolds. It was reported that insoluble silk microspheres induced β-sheet during silk film preparation.[41] Hence, an accelerated rate of NaCl-induced hydrogel formation might occur with a high density of silk particles in the silk solution. Therefore, the initial silk solution concentration as well as particle loading amount should be the key factors for forming aqueous composite scaffolds. Conversely, HFIP does not induce ionic dissolution of NaCl, resulting in no β-sheet formation of silk molecules in the HFIP process. Hence, methanol treatment was required to stabilize porous scaffolds prior to NaCl leaching.[16,37] These differences led to differing optimization strategies for each system, and showed that the HFIP-based process offered the widest range of initial concentrations of silk solution as well as higher particle loading for composite scaffolds.
In the present work, shrinkage of the scaffolds was restricted as indicated by the visual observation during the methanol treatment. It is known that, depending on the process employed during silk scaffold formation, methanol treatment presents a problem of dimensional stability. For example, 35.7% shrinkage during methanol treatment with freeze-dried silk scaffolds was reported, while reinforcement by chitin whiskers reduced this effect substantially.[6] The good dimensional stability of the scaffolds in the present study is due to the porogen approach and the use of the silk particles. Another problem cited previously with HFIP-based scaffolds was the lack of homogeneity due to non-uniform rate of drying.[16,37] However, HFIP-based composite scaffolds with high particle loading (100–200%) in this study were less heterogeneous (Figure 2).
Gross Pore Morphology of Composite Scaffolds
Aqueous-Derived Scaffolds
Scanning electron microscope (SEM) images of interconnected porous structures of the aqueous-derived scaffolds without particles and with 24% BM-AM, PCR-AM, and PCR-AM-AJM particles are presented in Figure 4. Based on previous findings on formability and optimum mechanical properties of aqueous based silk scaffolds, porogen size was restricted to 500–600 μm.[16,42] The surface of the control scaffolds without particles was smooth [Figure 4(a, b)]. Images of scaffolds reinforced with submicron scale particles of PCR-AM-AJM are shown in Figure 4(d–f). Pore interconnectivity was not affected by the particles according to Figure 4(d). The embedded particles are clearly seen in Figure 4(e), while the fracture image in Figure 4(f) shows no evidence of phase separation, demonstrating good miscibility of PCR-AM-AJM particles with the matrix. Comparing “c” and “g” with “d” in Figure 4, it is clear that roughness of pore walls increased due to the larger particles (PCR-AM and BM-AM). Greater roughness due to larger particles is particularly evident from the enlarged image, Figure 4(h). Despite the rough walls, smooth fracture image in Figure 4(i) further demonstrates excellent miscibility between the matrix and the BM-AM particles. It was observed that both PCR-AM and BM-AM reinforced scaffolds were brittle when dry and were difficult to handle compared to the no particle control and the composite scaffold reinforced with ultrafine particles (PCR-AM-AJM).
Figure 4.

Scanning electron microscope (SEM) images of aqueous-derived scaffolds (6% w/v matrix, 500–600 μm NaCl particles); (a) with no reinforced silk particles, (b) magnified image from “a,” (c) reinforced with 24% PCR-AM particles, (d) reinforced with 24% PCR-AM-AJM particles, (e) magnified image of pore wall of “d,” (f) magnified image of fracture surface of “d,” (g) reinforced with 24% BM-AM particles, (h) magnified image of pore wall of “g,” (i) magnified image of fracture surface of “g.” Black arrows show the self assembled silk particles formed due to salt-matrix silk interactions. White arrows show the location of the embedded reinforced silk particles.
1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP)-Derived Scaffolds
Compared to the aqueous-based process, HFIP allows a wider range of porogen sizes to be used in scaffold processing.[42] Hence, a greater range of porogens (210–300, 500–600, and 850–1 000 μm) were examined. The HFIP protocol was also used to understand the effects of particle volume/weight fraction as more particles could be used effectively through this process. To minimize the influence of other particle variants during this study and also based on initial test results which suggest more homogeneous composites from BM-AM particles, only BM-AM particles were used for all combinations of porogen size and particle load. However, for screening the effects of silk species and particle size, PCR-AM and PCR-AM-AJM were also used, but restricted to composites prepared with 500–600 μm porogens and 100% particle reinforcements only.
Figure 5 shows the morphology of HFIP-based composite scaffolds containing interconnected pores. However, the interconnectivity is less widespread compared to aqueous-derived scaffolds (Figure 4) irrespective of the content of silk particles. This result could be explained by both the high concentration of silk solution (17% silk) used in HFIP processing and the lack of ionic dissolution of NaCl. Hence there was no salt induced microsphere formation from the NaCl fibroin interface to create the gaps in pore walls. Formation of such microspheres from aqueous silk solution has been reported[43] and can also be seen in aqueous composite scaffolds (Figure 4). However, it did not happen during HFIP-based scaffold processing. A previous study also reported less interconnected porous structure from a 17% HFIP silk solution.[37]
Figure 5.

Scanning electron microscope (SEM) images of HFIP-derived scaffolds (17% w/v matrix with BM-AM particle reinforcement); first two columns (a–f) NaCl 210–300 μm, third column (g–i) NaCl 850–1 000 μm; top row (a), (d), and (g) no particles, middle row (b), (e), and (h) 100% silk particles, bottom row (c), (f), and (i) 200% silk particles, (d) magnified fracture image of “a,” (e) magnified fracture image of “b,” (f) magnified fracture image of “c.”
Scanning electron microscope (SEM) images (Figure 5) show the effect of BM-AM particle mass loading on gross porous structure. We estimated that the thickness of the pore walls increased and distance between the pores widened while increasing particle content from 0 to 200% for HFIP-based scaffolds. Roughness of pore walls and fracture surfaces are shown in Figure 5(d–f). Excellent mixing of the particles with the matrix is evident from the lack of phase separation. Comparing “d” with “e and f” in Figure 5, the presence of particles is clear. Morphologies of scaffolds with P. c. ricini silk particles of similar nature (PCR-AM) were found to be same (results not included). Together with our observation during scaffold preparation, these fracture images encouraged us to look for evidence of partial solubility of silk particles in HFIP.
Silk Particle Interaction with 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP)
To test silk solubility in HFIP, particles (“BM-AM” or “PCR-AM”) alone were mixed with HFIP and then cast into films. Allowing the mixture to stand longer increased the total dissolution in HFIP. There was no separation of particles from HFIP at any point of time during storage. These effects can be seen from the images as well as photographs of the films at different time points [Figure 6(a–d)]. A granular structure in Figure 6(a) was formed when the mixture was immediately cast into a film (day 0). It was observed that the film was fragile and could easily be crushed into powder.
Figure 6.
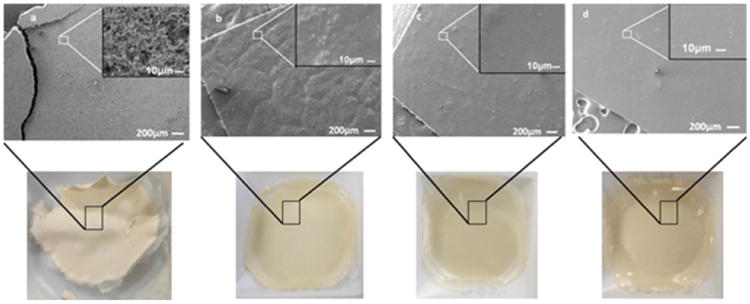
Films from silk particles (BM-AM) in HFIP (5% w/v); (a) day 0, (b) day 2, (c) day 6, and (d) day 9.
The FTIR scan “a” in Figure 7 has a peak at 1626 cm−1 (amide I). However, another peak developed at 1653 cm−1 in films prepared from the particles treated with HFIP for extended periods of time. To represent the β-sheet fraction in amide I, Tretinnikov and Tamada used 1626 cm−1, while peaks in the range 1641 to 1659 cm−1 were taken to represent random coil and α-helix fractions in B. mori silk fibroin.[44] Thus, FTIR scan results suggest that silk particles (BM-AM) retained their crystalline β-sheet features. Scan “d” represents an amorphous film without β-sheet prepared from silk solution. Comparing scans “b, c” with scans “a” and “d,” the film from particles mixed with HFIP and stored for some time seems to have characteristics between crystalline silk particles and amorphous lyophilized silk. The PCR-AM particles behaved similarly (results not shown). On the other hand, both BM-AM and PCR-AM were totally insoluble in water and no films formed. The FTIR scans of HFIP-based films prepared solely from particles suggest that partial dissolution of silk particles occurred during scaffold formation while the silk solution in HFIP was allowed to saturate the silk particles. This resulted in increased miscibility between the matrix and particles. The partial solubility of silk particles could be due to high surface area of the particles and defects created in the β-sheet structure during silk milling. In addition to good miscibility due to partial solubility, particulates still generated a rougher surface which might be useful toward improved cell attachment.[28]
Figure 7.
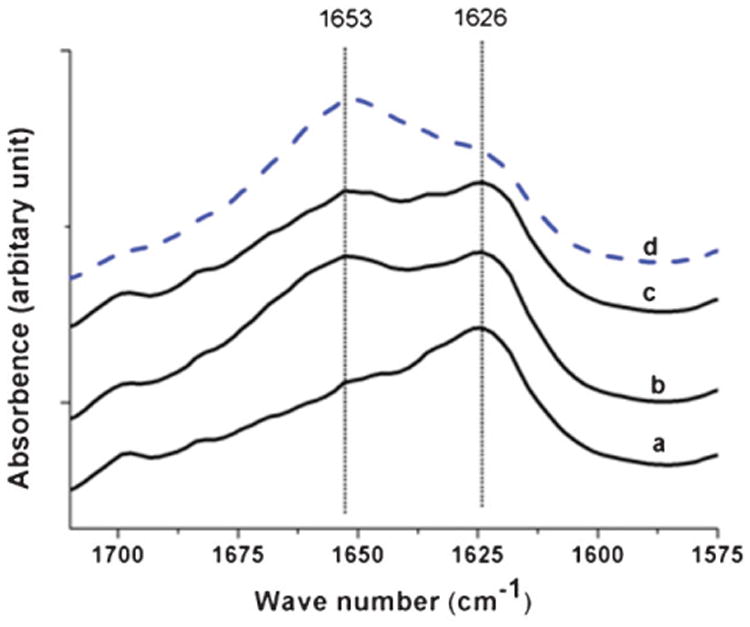
Fourier transform infrared (FTIR) scans of films prepared from 5% silk particles (BM-AM) in HFIP; (a) day 0, (b) day 4, (c) day 9, and (d) film prepared from lyophilized silk dissolved in HFIP without particles.
Porosity of Scaffolds
Table 2 shows the porosity of the HFIP-derived composite scaffolds measured by the two methods. The bulk density of a scaffold takes into account the volume of pores. The percentage porosity was estimated from the ratio between the bulk density of a scaffold and the density of polymer matrix, silk. However, this measurement does not distinguish close and open porous structures. On the other hand, the liquid displacement method measures the porosity by taking into account the diffused liquid into the pores. As the presence of closed pores is not accounted for, porosity measured by liquid displacement is usually less than the density method. Hexane was used for porosity measurement as it does not change the volume of silk.[37]
Table 2.
Percentage porosity of HFIP-derived scaffolds.
| Silk particles | Porogen size | ||
|---|---|---|---|
|
|
|
||
| % | μm | ||
| Density method | |||
| 210–300 | 500–600 | 850–1000 | |
| 0 | 94.3 ± 0.7 | 93.5 ± 0.6 | 91.3 ± 0.8 |
| 50a) | 90.1 ± 0.6 | – | – |
| 100 | 81.9 ± 0.9 | 84.4 ± 1.4 | 81.5 ± 0.9 |
| 200 | 78.2 ± 0.4 | 79.2 ± 2.4 | 77.8 ± 0.1 |
| Liquid displacement method | |||
| 210–300 | 500–600 | 850–1000 | |
| 0 | 93.5 ± 1.1 | 92.3 ± 1.2 | 90.9 ± 0.8 |
| 50a) | 89.6 ± 1.3 | – | – |
| 100 | 80.1 ± 1.0 | 83.3 ± 1.2 | 78.8 ± 1.4 |
| 200 | 66.7 ± 2.1 | 77.2 ± 2.3 | 78.6 ± 1.3 |
Porosity values are average ± standard deviation; N = 4.
No scaffolds were made with 50% reinforcement other than 210–300 μm pore size.
The results in Table 2 reveal that with the exception of scaffolds prepared with 210–300 μm porogens and 200% particle reinforcement, interconnectivity in HFIP-derived scaffolds was sufficient for liquid to pass into the pores. Though less interconnected structures were indicated by the SEM images (Figure 5), the sinking of scaffolds during porosity measurement as well as insignificant difference between porosity measured by density and liquid displacement methods in most cases (p > 0.05) suggests otherwise. Previous studies showed that HFIP-derived scaffolds from 17% silk solution (same as the controls used in the present study) supported well-distributed tissue development indicating that such scaffolds had interconnectivity required for cell ingrowth.[38,45] Data in Table 2 show that the liquid displacement porosity was lower for 850–1 000 μm 100% BM-AM particle reinforced scaffolds (p < 0.05) compared to density porosity, indicating some closed pores. In the case of 210–300 μm 200% BM-AM particle reinforced scaffolds, the difference was significant (p < 0.01). This could be the result of insufficient space between finer porogens (210–300 μm) to accommodate the silk particles and hence probably a thick coating of particles was formed over the porogens during mixing. The porosity of other scaffolds was above 75% even with 200% particles. The results are encouraging as pore size, geometry, and pore distribution are important in bone tissue engineering.[7]
Mechanical Properties
Observations and Mechanism
Preliminary tests indicated poor mechanical properties of all aqueous-derived composites compared to HFIP-based ones. Moreover, particle reinforcement was limited to only 25%. Hence, investigations focused on the HFIP-based composites in this work. Figure 8(a) shows that the compressive load–strain curves changed substantially after BM-AM particle reinforcement. Three distinct deformation regimes were identified for each compression test, in accordance with a theoretical framework defined for low-density open-cell foams,[46] used previously for studying the behavior of tissue engineering scaffold architectures.[47] The first “linear elastic” load–strain behavior of a material consisting of open-pore cells is controlled by cell edge bending and buckling resistance. Theoretically, before yielding, compressive loads induce small deformations of cell edges along the direction of load, and the walls do not buckle. During the first region, the “elastic modulus” can be measured and offset approach used to estimate the yield point at the first load–strain inflection location. A second nearly “plateau” region was identified for each specimen, following the theoretical yield point, which suggests cell wall buckling and pore collapse which continues without much additional increase in force. The third “densification” region represents total pore collapse throughout the material (cell walls pressing against each other) which ultimately causes the material to act like a solid non-porous material.[46,47] For the entire load–strain curve, local strut or pore wall characteristics impart global strength properties.
Figure 8.
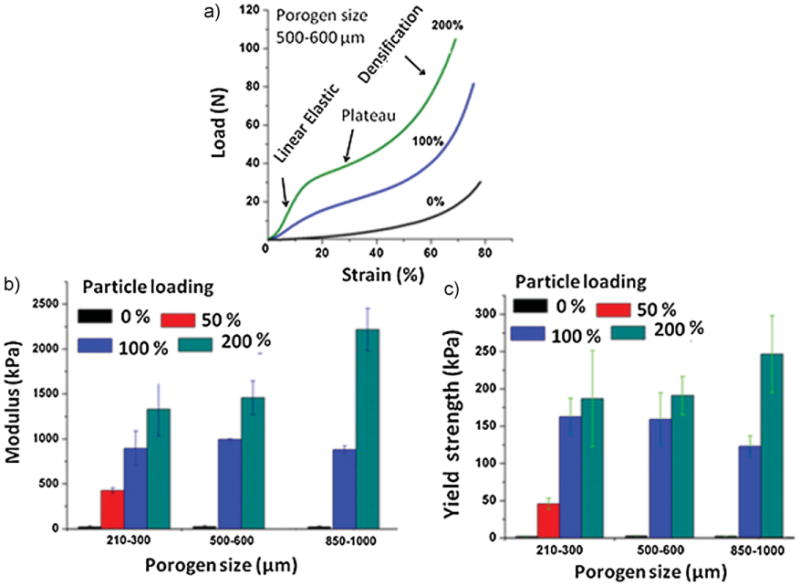
Compressive properties of HFIP-derived silk (17% w/v) scaffolds reinforced with different % BM-AM silk particles: (a) raw compression curves prior to analysis, (b) compressive modulus changes with varying porogen size, (c) compressive yield strength changes with varying porogen size; N = 5.
The increase in stiffness of scaffolds due to particles was most likely caused by the compatibility between the silk matrix and silk particles and due to the excellent interfacial cohesion due to the partial dissolution of particles. It was reported that partial dissolution of polyphosphazene could bind nano-hydroxyapatite to form stronger particulate reinforced composites.[48] Such features of a strong interlock between particles and the polymer matrix can effectively transfer the load from the matrix to the reinforcement and help eliminate stress concentrations, resulting in increased toughness and strength.[21] In the absence of similar strong interfacial bonds, binding agents such as glutaraldehyde were used in the past to improve interfacial cohesion between the particles and matrix in the case of nano-hydroxyapatite reinforced silk fibroin composites, which in turn led to increased strength.[49] These results suggest that silk–silk composite provide an important option to enhance material interactions through hydrogen bonding assisted by the partial solubility in HFIP. The role of intermolecular hydrogen bonds between silk fibroin chains in β-sheets is well known.[50]
Effects of Particle Content
Modulus and Yield Strength
The improvement in mechanical properties due to the increase in BM-AM particle content is reflected in modulus and yield strength results in Figure 8. The 50% particle reinforced scaffolds were prepared only with 210–300 μm porogens and hence data of 50% particle loaded scaffolds are absent for scaffolds prepared using other porogens in Figure 8. The highest mechanical properties of about 2 250 kPa stiffness and 250 kPa yield strength were achieved in the case of 200% particle loaded scaffolds prepared with 850–1000 μm porogens. The compressive properties of scaffolds prepared for bone repair from various natural,[40] synthetic polymers,[29,51,52] and polymer blends[53,54] are often tested dry, which would make direct comparisons irrelevant. Moreover, the variations in processing techniques and pore sizes further complicate comparative analysis. Wet compressive properties were reported for silk scaffolds[17,55,56] and collagen/glycosaminoglycan[47] scaffolds which were mechanically inferior to the silk/silk composite scaffolds reported here in terms of both modulus (stiffness) and yield strength.
Specific Modulus and Specific Yield Strength
The bulk density of the scaffolds increased with a corresponding decrease in porosity due to the change in mass with the addition of silk particles. Theoretically, modulus and the yield strength of elastomeric open cell foams are proportional to the square of the relative density (density of scaffolds including the pores/density of material forming the scaffold) and also proportional to the stiffness (elastic modulus) of the material forming the scaffold.[47] In the present study, we have found that the relative density increased 2–3 times after 100–200% particle reinforcement. Hence, theoretically modulus and yield strength should have increased 4–9 times. However, with 200% reinforcement, both modulus and yield strength increased up to about 114 times. Specific modulus and yield strength (calculated by normalizing modulus and yield strength by the scaffold density) increased up to about 40-fold. Even for scaffolds with 50% silk particles with a corresponding increase in relative density of about 1.3 times, the increase in specific modulus was around 15.3 times. In an earlier study with silk scaffolds, the specific modulus increased by nearly 7.5 times when reinforced with 50% chitin whiskers[6] and such an increase was attributed to strong hydrogen bonds between filler and matrix. Similarly, about a two-fold increase in modulus and yield strength was reported in chitosan scaffolds reinforced with ceramic particles[24] and silk scaffolds reinforced with 70% nano-hydroxyapatite particles.[49] It is clear that a large increase in stiffness and strength of scaffolds in the present study was primarily due to the increase in stiffness of pore walls, by increasing the amount of stiffer silk particles, although an increase in relative density (change in pore wall dimensions) could also play a secondary role.
Effect of Pore Size
In the present study, mechanical property changes due to porogen size were not statistically significant for the scaffolds [Figure 8(b, c)] except that the 200% particle reinforced composite prepared from 850 to 1000 μm porogens had significantly higher modulus (p < 0.05) compared to scaffolds fabricated using smaller sized porogens for the same reinforcement mass. The yield strength was likewise higher, though not statistically significant (p > 0.05). Theoretically, increasing porogen size for increased pore size should have no significant effect on the overall stiffness of composites.[47] This is due to a confluence of opposing mechanisms related to the response of foam-like structures to mechanical deformation. Strength and stiffness properties of foams are directly proportional to strut surface area and inversely proportional to strut length. In processing scaffolds, increasing porogen size will increase wall length (i.e., strut length) and decrease the number of walls; this would decrease the global strength properties. However, this effect is counteracted by an increase in spaces between the large sized porogens, thus thicker walls (i.e., strut cross-sectional area) should theoretically increase global strength properties.[46] During HFIP-based composite processing, space between porogens was sufficient in scaffolds containing 850–1000 μm porogens to uniformly pack 200% silk particles. The penetration of the silk solution, as observed during the experiments, was better in such scaffolds compared to scaffolds having smaller porogens, resulting in more homogeneous composites. We observed that the penetration of the silk solution was difficult through the porogen particle mixture prepared with 210–300 μm porogens and 200% silk particles. These factors were probably responsible for improved processibility and thus enhanced mechanical properties in the 200% particle reinforced composites prepared from the large porogens. It has been observed experimentally in an earlier study also that an increase in pore size enhanced the modulus of both flexible and rigid foams.[57] The findings could further promote developing strategies to form functional bone tissue in vitro and in vivo, since previous data showed that using larger-sized pores enhanced bone development.[58]
Influence of Particle Type
100% reinforced composites were prepared using PCR-AM or PCR-AM-AJM with 500–600 μm porogens to compare particles from silk species and size. Table 3 shows that for such loading and porogen size, there was no significant difference (p < 0.05) in specific modulus between scaffolds made from BM-AM and PCR-AM, but BM-AM-formed scaffolds provided better specific yield strength (p < 0.05). This could be due to better interfacial strength between matrix protein and particles as both the components were prepared from the same silk species (B. mori). The amino acid composition and thus chemistry of P.c.ricini silk particles (PCR-AM) is different from the matrix protein B. mori.[59–61] Due to relatively poor mechanical properties of P.c. ricini particle reinforced scaffolds, most of the studies as discussed were confined to B. mori particle reinforcements only.
Table 3.
Specific compressive properties of silk scaffolds. Mechanical property values are average ± standard deviation; N = 5.
| Type of particles | Porogen size | Percentage of silk particles | Specific modulus | Specific yield strength |
|---|---|---|---|---|
|
|
|
|
||
| μm | kPa · g−1 · cm3 | kPa · g−1 · cm3 | ||
| No particles | 210–300 | 0 | 205.8 ± 93.7 | 19.5 ± 8.5 |
| BM-AM | 210–300 | 50 | 3150.6 ± 215.1 | 339.3 ± 56.1 |
| BM-AM | 210–300 | 100 | 3774.6 ± 808.2 | 685.3 ± 103.7 |
| BM-AM | 210–300 | 200 | 4616.4 ± 1033.5 | 648.8 ± 224.5 |
| No particles | 500–600 | 0 | 232.8 ± 96.3 | 24.6 ± 7.8 |
| BM-AM | 500–600 | 100 | 4193.5 ± 17.6 | 670.6 ± 149.7 |
| BM-AM | 500–600 | 200 | 5065.5 ± 650.5 | 663.6 ± 88.9 |
| No particles | 850–1000 | 0 | 189.2 ± 104.2 | 21 ± 6.2 |
| BM-AM | 850–1000 | 100 | 3735.2 ± 649.3 | 516 ± 59.2 |
| BM-AM | 850–1000 | 200 | 7722.4 ± 812.6 | 853.8 ± 179.4 |
| PCR-AM | 500–600 | 100 | 4055.8 ± 790.8 | 449.9 ± 49.2 |
| PCR-AM-AJM | 500–600 | 100 | 3725.4 ± 649.3 | 435.8 ± 163.9 |
The results in Table 3 do not indicate a noticeable difference between scaffolds prepared from PCR-AM and PCR-AM-AJM particles, though the finer and non-aggregated particles are expected to provide better composite mechanical properties as they have less interparticle contact points, from which cracks generate and propagate.[21] In our earlier study, PCR-AM-AJM particles showed poor bulk density despite their smaller size, which was attributed to the generation of static charges during particle preparation in the AJM.[33] A lack of uniform and compact packing in PCR-AM-AJM particles probably failed to further enhance the composite performance despite their submicron size and non-aggregated nature. Further studies are warranted to understand the effect of particle size on composite properties.
Enzymatic Degradation
Figure 9 shows the mass change of the HFIP-derived scaffolds prepared using 210–300 μm NaCl with and without added silk BM-AM particles over a degradation period of 8 d by protease IXV (1 mg · mL−1, 4.7 U · mg−1). The scaffolds in phosphate buffer without protease showed no degradation. The scaffolds prepared without silk particles rapidly degraded and the average mass remaining was only 29% after 8 d. The results in Figure 9 also show that the degradation was slower due to the reinforcing particles; scaffolds having 50% BM-AM and PCR-AM particles retained 76 and 74% mass, respectively after 8 d. Enzymatic degradation of aqueous-derived scaffolds under similar conditions revealed that the scaffold structure was completely lost within 2 d. Rapid loss of weight of aqueous scaffolds compared to HFIP scaffolds was also reported earlier.[8,16] Such differences in degradability can be attributed to difference in secondary structure between silk fibroins processed differently; the influence of secondary structure on silk degradability has been established.[15,62] The ability to control degradability by particle reinforcement along with excellent mechanical properties compared to most other biodegradable scaffolds offers important potential for these silk–silk macro-porous composites for skeletal tissue engineering. Apart from weight loss, additional studies on degradability of the particles will be needed to shed light on the degradation behavior of the composites.
Figure 9.
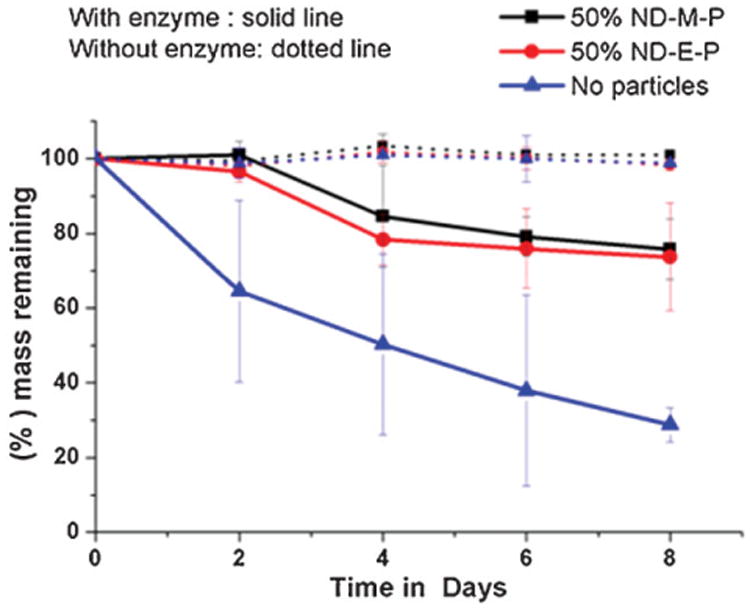
Mass remaining from enzymatic degradation of HFIP-derived scaffolds; N = 4.
Conclusion
New silk–silk composites were generated via the addition of silk particles. Up to 200% silk particles could be loaded into HFIP solutions of silk to prepare uniform and stiff scaffolds. The interfacial compatibility between the particles and the bulk phase was responsible for significant increase in mechanical properties. In addition, the partial solubility of mechanically fabricated silk particles in HFIP resulted in high interfacial cohesion between matrix and the particles and thus a substantial increase in mechanical properties. This increase was higher than expected from changes in dimensions of the pore walls. Specific modulus and yield strength increased nearly 40 times with the addition of 200% particle reinforcement prepared with large sized porogens (850–1000 μm), when compared to controls (without particle additions). The enzymatic degradation of the scaffolds was slowed by the presence of the added particles. Combined with the inherent biocompatibility of silk and ability to form large porous structures, the slowly degrading and mechanically stable silk–silk composites could be useful for bone and other skeletal tissue engineering applications.
Acknowledgments
The authors thank the Australian Research Council and National Institute of Health P41Tissue Engineering Resource Centre (EB002520) for supporting this work.
Contributor Information
Rangam Rajkhowa, Centre for Material and Fibre Innovation, Deakin University, Geelong, Victoria, Australia, Fax: (+61) 352272539; Department of Biomedical Engineering, Tufts University, 4 Colby St, Medford, Massachusetts 02155, USA.
Eun Seok Gil, Department of Biomedical Engineering, Tufts University, 4 Colby St, Medford, Massachusetts 02155, USA.
Jonathan Kluge, Department of Biomedical Engineering, Tufts University, 4 Colby St, Medford, Massachusetts 02155, USA.
Keiji Numata, Department of Biomedical Engineering, Tufts University, 4 Colby St, Medford, Massachusetts 02155, USA.
Lijing Wang, School of Fashion and Textiles, RMIT University, 25 Dawson Street, Brunswick, Vic. 3056, Australia.
Xungai Wang, Email: xwang@deakin.edu.au, Centre for Material and Fibre Innovation, Deakin University, Geelong, Victoria, Australia, Fax: (þ61) 352272539.
David L. Kaplan, Department of Biomedical Engineering, Tufts University, 4 Colby St, Medford, Massachusetts 02155, USA
References
- 1.Vacanti JP, Langer R. Lancet. 1999;354:S32. doi: 10.1016/s0140-6736(99)90247-7. [DOI] [PubMed] [Google Scholar]
- 2.Rikli DA, Regazzoni P, Perren SM. Injury. 2002;33:2. doi: 10.1016/s0020-1383(02)00126-2. [DOI] [PubMed] [Google Scholar]
- 3.Rose FRAJ, Oreffo ROC. Biochem Biophys Res Commun. 2002;292:1. doi: 10.1006/bbrc.2002.6519. [DOI] [PubMed] [Google Scholar]
- 4.Fischbach C, Chen R, Matsumoto T, Tobias S, Brugge JS, Polverini DJM. Nat Methods. 2007;4:855. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 5.Meinel L, Betz O, Fajardo R, Hofmann S, Nazarian A, Cory E, Hilbe M, McCool J, Langer R, Vunjak-Novakovic G, Merkle HP, Rechenberg B, Kaplan DL, Kirker-Head C. Bone. 2006;39:922. doi: 10.1016/j.bone.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 6.Wongpanit P, Sanchavanakit N, Pavasant P, Bunaprasert T, Tabata Y, Rujiravanit R. Eur Polym J. 2007;43:4123. [Google Scholar]
- 7.Karageorgiou V, Kaplan D. Biomaterials. 2005;26:5474. doi: 10.1016/j.biomaterials.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Rudym DD, Walsh A, Abrahamsen L, Kim HJ, Kim HS, Kirker-Head C, Kaplan DL. Biomaterials. 2008;29:3415. doi: 10.1016/j.biomaterials.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vepari C, Kaplan DL. Prog Polym Sci. 2007;32:991. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandal BB, Kundu SC. Macromol Biosci. 2008;8:807. doi: 10.1002/mabi.200800113. [DOI] [PubMed] [Google Scholar]
- 11.Meinel L, Hofmann S, Karageorgiou V, Kirker-Head C, McCool J, Gronowicz G, Zichner L, Langer R, Vunjak-Novakovic G, Kaplan DL. Biomaterials. 2005;26:147. doi: 10.1016/j.biomaterials.2004.02.047. [DOI] [PubMed] [Google Scholar]
- 12.Dal Pra I, Freddi G, Minic J, Chiarini A, Armato U. Biomaterials. 2005;26:1987. doi: 10.1016/j.biomaterials.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 13.Arai T, Freddi G, Innocenti R, Tsukada M. J Appl Polym Sci. 2004;91:2383. [Google Scholar]
- 14.Horan RL, Antle K, Collette AL, Wang Y, Huang J, Moreau JE, Volloch V, Kaplan DL, Altman GH. Biomaterials. 2005;26:3385. doi: 10.1016/j.biomaterials.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Jin HJ, Park J, Karageorgiou V, Kim UJ, Valluzzi R, Cebe P, Kaplan DL. Adv Funct Mater. 2005;15:1241. [Google Scholar]
- 16.Kim UJ, Park J, Joo Kim H, Wada M, Kaplan DL. Biomaterials. 2005;26:2775. doi: 10.1016/j.biomaterials.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 17.Kim HJ, Kim UJ, Leisk GG, Bayan C, Georgakoudi I, Kaplan DL. Macromol Biosci. 2007;7:643. doi: 10.1002/mabi.200700030. [DOI] [PubMed] [Google Scholar]
- 18.Marolt D, Augst A, Freed LE, Vepari C, Fajardo R, Patel N, Gray M, Farley M, Kaplan D, Vunjak-Novakovic G. Biomaterials. 2006;27:6138. doi: 10.1016/j.biomaterials.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 19.Kim HJ, Kim UJ, Kim HS, Li C, Wada M, Leisk GG, Kaplan DL. Bone. 2008;42:1226. doi: 10.1016/j.bone.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchsa S, Jianga X, Schmidtb H, Dohlea E, Ghanaatia S, Ortha C, Hofmannc A, Mottad A, Migliaresid C, Kirkpatricka CJ. Biomaterials. 2009;30:1329. [Google Scholar]
- 21.Wang M. Biomaterials. 2003;24:2133. doi: 10.1016/s0142-9612(03)00037-1. [DOI] [PubMed] [Google Scholar]
- 22.Wang M. Am J Biochem Biotechnol. 2006;2:80. [Google Scholar]
- 23.Li J, Mak AFT. Key Eng Mater. 2007;334:1237. [Google Scholar]
- 24.Yong Zhang MZ. J Biomed Mater Res. 2001;55:304. doi: 10.1002/1097-4636(20010605)55:3<304::aid-jbm1018>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 25.Kim SS, Sun Park M, Jeon O, Yong Choi C, Kim BS. Biomaterials. 2006;27:1399. doi: 10.1016/j.biomaterials.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 26.Jung Y, Kim SS, Kim YH, Kim SH, Kim BS, Kim S, Choi CY, Kim SH. Biomaterials. 2005;26:6314. doi: 10.1016/j.biomaterials.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Minamiguchi S, Takechi M, Yuasa T, Momota Y, Tatehara S, Takano H, Miyamoto Y, Satomura K, Nagayama M. J Mater Sci: Mater Med. 2008;19:1165. doi: 10.1007/s10856-007-0162-x. [DOI] [PubMed] [Google Scholar]
- 28.Lv Q, Hu K, Feng Q, Cui F, Cao C. Compos Sci Technol. 2007;67:3023. [Google Scholar]
- 29.Olah L, Filipczak K, Jaegermann Z, Czigany T, Borbas L, Sosnowski S, Ulanski P, Rosiak JM. Polym Adv Technol. 2006;17:889. [Google Scholar]
- 30.Liao SS, Cui FZ, Zhang W, Feng QL. J Biomed Mater Res, Part B. 2004;69B:158. doi: 10.1002/jbm.b.20035. [DOI] [PubMed] [Google Scholar]
- 31.Yong Zhang MZ. J Biomed Mater Res. 2002;61:1. doi: 10.1002/jbm.10176. [DOI] [PubMed] [Google Scholar]
- 32.Moutos FT, Farshid G. Biotechnology. 2008;45:501. [Google Scholar]
- 33.Rajkhowa R, Wang L, Kanwar J, Wang X. Powder Technol. 2009;191:155. [Google Scholar]
- 34.Rajkhowa R, Wang L, Wang X. Powder Technol. 2008;185:87. [Google Scholar]
- 35.Wang L, Nemoto R, Senna M. J Eur Ceram Soc. 2004;24:2707. [Google Scholar]
- 36.Wang L, Li C. Carbohydr Polym. 2007;68:740. [Google Scholar]
- 37.Nazarov R, Jin HJ, Kaplan DL. Biomacromolecules. 2004;5:718. doi: 10.1021/bm034327e. [DOI] [PubMed] [Google Scholar]
- 38.Meinel L, Hofmann S, Karageorgiou V, Zichner L, Langer R, Kaplan D, Vunjak-Novakovic G. Biotechnol Bioeng. 2004;88:379. doi: 10.1002/bit.20252. [DOI] [PubMed] [Google Scholar]
- 39.Rajkhowa R, Gupta VB, Kothari VK. J Appl Polym Sci. 2000;77:2418. [Google Scholar]
- 40.Kim SE, Cho YW, Kang EJ, Kwon IC, Bae LE, Kim JH, Hessson C, Jeong SY. Fibers Polym. 2001;2:64. [Google Scholar]
- 41.Lv Q, Cao C, Zhang Y, Man X, Zhu H. J Mater Sci: Mater Med. 2004;15:1193. doi: 10.1007/s10856-004-5918-y. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Kim HS, Matsumoto A, Chin I, Jin H, Kaplan DL. Aust J Chem. 2005;58:716. [Google Scholar]
- 43.Slotta UK, Rammense S, Gorb S, Scheibel T. Angew Chem, Int Ed. 2008;47:4592. doi: 10.1002/anie.200800683. [DOI] [PubMed] [Google Scholar]
- 44.Tretinnikov ON, Tamada Y. Langmuir. 2001;17:7406. [Google Scholar]
- 45.Chang G, Kim HJ, Kaplan D, Vunjak-Novakovic V, Kandel RA. Eur Spine J. 2007;16:1848. doi: 10.1007/s00586-007-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibson LJ. J Biomech. 2005;38:377. doi: 10.1016/j.jbiomech.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 47.Harley BA, Leung JH, Silva ECCM, Gibson LJ. Acta Biomater. 2007;3:463. doi: 10.1016/j.actbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 48.Nukavarapu SP, Kumbar SG, Brown JL, Krogman NR, Weikel AL, Hindenlang MD, Nair LS, Allcock HR, Laurencin CT. Biomacromolecules. 2008;9:1818. doi: 10.1021/bm800031t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu L, Liu J, Wang M, Min s, Cai Y, Zhu L, Yao J. J Biomater Sci, Polym Ed. 2008;19:325. doi: 10.1163/156856208783721010. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan D, Adams WW, Farmer B, Vinev C. ACS Symposium Series. American Chemical Society; Washington DC: 1994. Silk Polymers: Materials Science and Biotechnology. [Google Scholar]
- 51.Harris LD, Kim BS, Mooney DJ. J Biomed Mater Res. 1998;42:396. doi: 10.1002/(sici)1097-4636(19981205)42:3<396::aid-jbm7>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 52.Hou Q, Grijpma DW, Feijen J. J Biomed Mater Res, Part B: Appl Biomater. 2003;67B:732. doi: 10.1002/jbm.b.10066. [DOI] [PubMed] [Google Scholar]
- 53.Lv Q, Hu K, Feng Q, Cui F. J Appl Polym Sci. 2008;109:1577. [Google Scholar]
- 54.She Z, Zhang B, Jin C, Feng Q, Xu Y. Polym Degrad Stab. 2008;93:1316. [Google Scholar]
- 55.Tamada Y. Biomacromolecules. 2005;6:3100. doi: 10.1021/bm050431f. [DOI] [PubMed] [Google Scholar]
- 56.Gellynck K, Verdonk PCM, Nimmen EV, Almqvist KF, Gheysens T, Schokens G, Langenhove LV, Kiekens P, Mertens J, Verbruggen G. J Mater Sci: Mater Med. 2008;19:3399. doi: 10.1007/s10856-008-3474-6. [DOI] [PubMed] [Google Scholar]
- 57.Shutov FA. Adv Polym Sci. 1983;51:155. [Google Scholar]
- 58.Kim HJ, Kim UJ, Vunjak-Novakovic G, Min BH, Kaplan DL. Biomaterials. 2005;26:4442. doi: 10.1016/j.biomaterials.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Bhat NV, Nadiger GS. J Appl Polym Sci. 1980;25:921. [Google Scholar]
- 60.Asakura T, Kashiba H, Yoshimizu H. Macromolecules. 1988;21:644. [Google Scholar]
- 61.Lefevre T, Rousseau ME, Pezolet M. Biophys J. 2007;92:2885. doi: 10.1529/biophysj.106.100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Min BM, Jeong L, Lee KY, Ho Park W. Macromol Biosci. 2006;6:285. doi: 10.1002/mabi.200500246. [DOI] [PubMed] [Google Scholar]