

Abstract
Introduction
Tuberculosis (TB) remains the leading cause of death from a curable infectious disease; drug-resistant TB threatens to dismantle all prior gains in global control. Suboptimal circulating anti-TB drug concentrations can lead to lack of cure and acquired drug resistance.
Areas covered
This review will introduce pharmacokinetic parameters for key anti-TB drugs, as well as the indications and limitations of measuring these parameters in clinical practice. Current and novel methodologies for delivering anti-TB pharmacokinetic-pharmacodynamic data are highlighted and gaps in operational research described.
Expert opinion
Individual pharmacokinetic variability is commonplace, underappreciated and difficult to predict without therapeutic drug monitoring (TDM). Pharmacokinetic thresholds associated with poor TB treatment outcome in drug-susceptible TB have recently been described and may now guide the application of TDM, but require validation in a variety of settings and comorbidities. Dried blood spots for TDM and prepackaged multidrug plates for minimum inhibitory concentration testing will overcome barriers of accessibility and represent areas for innovation. Operationalizing pharmacokinetics has the potential to improve TB outcomes in the most difficult-to-treat forms of the disease such as multidrug resistance. Clinical studies in these areas are eagerly anticipated and we expect will better define the rational introduction of novel therapeutics.
Keywords: dried blood spot, minimum inhibitory concentration, multidrug-resistant tuberculosis, pharmacokinetics, therapeutic drug monitoring, tuberculosis
1. Introduction
In the past two decades, major progress has been made globally in reducing cases of tuberculosis (TB) and TB-related deaths, yet considerable work remains. TB is the leading cause of death from a curable infectious disease, and is second only to HIV/AIDS as a cause of death from any infectious disease. In 2011, there were an estimated 8.7 million new cases of TB and 1.4 million deaths from TB [1]. While cure rates can be high for drug-susceptible TB with appropriate multidrug therapy and when given in supervised treatment settings, success rates can drop as low as 65% in less-resourced areas [1,2].
Among the multitude of systemic contributors, treatment failure can result from drug resistance prior to initiation of therapy (primary drug resistance) and drug resistance developing while on therapy (acquired drug resistance). Novel and rapid drug-susceptibility testing has targeted primary drug resistance, but acquired resistance was thought almost exclusively to be the result of patient nonadherence or programmatic barriers to receipt of guideline-endorsed multidrug regimens. While implementation of directly observed therapy in certain TB endemic settings improved adherence with TB treatment, relapse and acquired drug resistance were not entirely eliminated [3,4]. Recently, however, suboptimal drug concentrations, due to individual pharmacokinetic variability and independent of nonadherence, have been associated with acquired drug resistance in in vitro models [5,6]. Even more definitively, they have been associated with poor outcome in prospective patient cohorts [5,7,8]. This concept cannot be overstated as the majority of all TB cases in endemic settings are treated with empiric weight-based regimens, often in fixed-dose combination and without the benefit of pharmacokinetic monitoring.
The consequences of treatment failure and amplified drug resistance can be devastating for both patient and public health. Quite simply, drug-resistant TB threatens to eliminate all gains made in global TB control [9]. Multidrug-resistant (MDR)-TB is now estimated in 3.7% of new cases and 20% of previously treated cases worldwide, and by definition, is resistant to the two most potent anti-TB medications, isoniazid and rifampin [1]. Furthermore, approximately 9% of all MDR-TB cases are categorized as extensively drug-resistant (XDR)-TB with additional resistance to a fluoroquinolone and an injectable agent (kanamycin, amikacin or capreomycin). The so-called ‘second-line’ drugs used in the treatment of drug-resistant TB are of greater cost, less potency and increased toxicity [10]. While treatment duration is typically 6 months for drug-susceptible TB, already a burdensome duration for many patients and TB programs, treatment of MDR-TB can last more than 20 months. As such, global success rates for treatment of MDR-TB and XDR-TB are estimated at 48 and 33%, respectively [1].
Such dismal outcomes represent the end result of a considerable personal and financial toll on patient, community and the governmental TB program. Where such estimates are calculable in the European Union in 2011, for instance, the economic loss for care of drug-resistant TB in disability-adjusted life years was 10 times greater than the treatment cost itself [11]. Consequently, optimization of existing drug regimens for TB would carry tremendous downstream benefit if even modestly associated with improved treatment outcome or shortened treatment duration. Therefore, the following review will provide an introduction to standard pharmacokinetic parameters for critical anti-TB drugs, the clinical application and limitations of therapeutic drug concentration monitoring for TB, and discussion of specific settings, patient populations and anatomic disease sites that may influence this application. Opportunities of operational research and technological innovation are highlighted.
2. Pharmacokinetic and pharmacodynamic principles in TB treatment
Incidentally, the first anti-TB drug, streptomycin, played a key role in the foundational pharmacokinetic–pharmacodynamic experiments of Dr. Harry Eagle, and was demonstrated to kill in a concentration-dependent manner compared to penicillin, which was time dependent in activity [12,13]. Today, the most common pharmacokinetic–pharmacodynamic measures used to describe antibacterial agents, including those with anti-TB activity, are the duration of time a drug concentration remains above the minimum inhibitory concentration (T > MIC), the ratio of the peak drug concentration relative to the MIC (Cmax/MIC) and the ratio of the area under the concentration–time curve at the end of the dosing interval relative to the MIC (AUC0 – 24/MIC) [14].
Unconventionally, however, TB drug-susceptibility testing in clinical laboratories differs from that of nearly any other infectious disease where MICs are determined by dilution schemes [15]. In contrast, most TB susceptibility testing is performed with a version of the proportion method where equal amounts of organism are inoculated on plates or in liquid media with and without a single ‘critical’ concentration of drug. Prior studies to determine the single critical concentration have used a midpoint concentration between the lowest MICs of known resistant strains and the highest MIC of known wild-type strains, where the clinical outcome is well described [16-18]. Yet this method does not allow for accurate individual pharmacokinetic–pharmacodynamic assessment. Commercially available MIC plates for Mycobacterium tuberculosis are now available and may ultimately facilitate pharmacokinetic–pharmacodynamic testing for TB patients outside the research setting (Figure 1) [19-21]. Nevertheless, MIC remains a static parameter and like all phenotypic susceptibility testing for TB measures only the dominant subpopulation of infecting M. tuberculosis cultured from the specimen.
Figure 1. TREK MYCOTB Sensititre plate of a multidrug-resistant isolate.
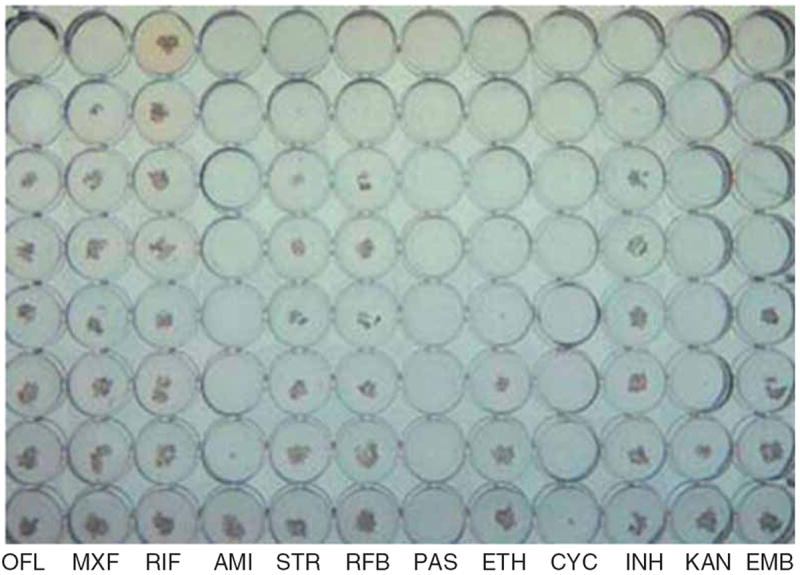
Each column contains prefilled wells of lyophilized drugs of increasing (from bottom to top) concentration. Shelf-life is 2 years at room temperature. Standardized inoculum of a patient’s Mycobacterium tuberculosis in liquid media is added to each well and can be read manually at set growth intervals thereby allowing MIC interpretation. Photo courtesy of Prof. Eric Houpt, University of Virginia.
AMI: Amikacin; CYC: Cycloserine; EMB: Ethambutol; ETH: Ethionamide; INH: Isoniazid; KAN: Kanamycin; MXF: Moxifloxacin; OFL: Ofloxacin; PAS: Para-aminosalicylic acid; RFB: Rifabutin; RIF: Rifampin; STR: Streptomycin.
Given that the clinical response to anti-TB treatment can take weeks to months to become apparent and adequate assessment of relapse-free cure may span years, early bactericidal studies in humans (measuring the decrement of bacteria growing in serially collected sputum from patients with pulmonary TB) as well as in vitro and animal models have played a growing role in determining anti-TB drug dosing and new drug development. Population-based statistical application, such as Monte Carlo simulation, has further allowed the integration of human pharmacokinetic data into models of large patient cohorts that otherwise could not be easily studied [22]. Additionally, novel multiple arm and multiple dosing strategies for TB clinical trials have been advocated for new drug assessment [23,24].
3. Anti-TB parameters
The bulk of pharmacokinetic–pharmacodynamic studies have focused on the first-line medications to treat drug-susceptible TB (Table 1) [7,24]. The majority of the best-studied agents, including rifampin, isoniazid, ethambutol and pyrazinamide, as well as the fluoroquinolones, have been shown to be concentration dependent in activity [25-29]. This effect was first seen in a guinea pig model for isoniazid in 1968, and was later confirmed in the modern pharmacokinetic–pharmacodynamic era in a murine model in 2004 and using the in vitro hollow fiber system (HFS) in 2007 [30-32]. The data for rifampin follow a similar trend, with guinea pig and murine models showing its concentration-dependent activity as early as 1969, and modern murine and HFS models confirming the link between its activity and the AUC/MIC ratio [33-36]. This is true for both bacterial killing [34] as well as prevention of resistance, which was calculated to occur at Cmax/MIC > 175 [35]. For rifampin in particular, the Cmax correlates well with AUC and may serve as a functional proxy [36]. Importantly, however, the standard rifampin dose for TB treatment, 10 mg/kg, was chosen in 1971 without a dose escalation study. Yet, a recent trial has found doses as high as 35 mg/kg as tolerable and bactericidal activity remains linear with dose escalation, and thus rifampin may be uniquely suitable for dose optimization to shorten total treatment duration [37]. Preclinical models of novel drug development seeking combination with rifampin must therefore view these more aggressive dosing strategies as presaging the norm.
Table 1.
Predictive pharmacokinetic parameters of currently recommended anti-tuberculosis drugs.
| Category | Route | Drug | Parameter best predictive of activity* | Predominant methods of evaluation | Threshold associated with poor outcome‡ |
|---|---|---|---|---|---|
| First line | Oral | Isoniazid | AUC/MIC > Cmax/MIC | Animal TB, HFS | AUC ≤ 52 mg·h/l |
| Rifamycins (rifampin, rifabutin) | AUC/MIC ~ Cmax/MIC | Animal TB, HFS | AUC ≤ 13 mg·h/l | ||
| Ethambutol | AUC/MIC | Animal TB, HFS, Human TB | N/A | ||
| Pyrazinamide | AUC/MIC | HFS, Human TB | AUC ≤ 363 mg·h/l | ||
| Second-line | Oral | Fluoroquinolones (ofloxacin, levofloxacin, moxifloxacin) | AUC/MIC > Cmax/MIC | Animal TB, HFS, human TB | N/A |
| Thioamides (ethionamide, prothionamide) | Unknown | N/A | |||
| Serine analogs (cycloserine, terizidone, thicetazone) | Unknown | N/A | |||
| Para-aminosalicylic acid | Unknown | N/A | |||
| Bedaquiline | T/MIC | Animal TB, human TB | N/A | ||
| Injectable | Aminoglycosides (streptomycin, amikacin, kanamycin) | AUC/MIC | Non-TB | N/A | |
| Capreomycin | Unknown | N/A | |||
| Third-line | Oral | Linezolid | T/MIC | Non-TB, human TB | N/A |
| Amoxicillin-clavulanate | T/MIC | Non-TB | N/A | ||
| Clarithromycin | T/MIC | Non-TB | N/A | ||
| Clofazimine | Unknown | N/A | |||
| Injectable | Carbapenems (Imipenem, meropenem) | T/MIC | Non-TB | N/A |
One recent study marks the first prospective effort to date that rigorously defines the pharmacokinetic thresholds associated with treatment failure in the first-line treatment of drug-susceptible TB [7]. A landmark investigation to disentangle the multiplicity of contributors to TB treatment failure, the study of 142 adults with TB from the Western Cape in South Africa found the top three independent predictors of poor long-term outcome to be AUC of pyrazinamide, rifampin and isoniazid in decreasing order of predictive magnitude. Secondary analysis allowed threshold values to be set whereby patients with an AUC of at least one drug below the threshold were over-whelmingly more likely to have a poor outcome (odds rate 14.14; 95% confidence interval 4.08 – 49.08) compared to patients with an AUC for each drug above the threshold (Table 1). Furthermore, the study clearly demonstrated that an individual’s pharmacokinetic variability was not able to be predicted by easily measurable factors, asserting the clinical relevance of individual drug concentration monitoring. It remains to be studied whether these findings are replicable in other settings or among patients with other comorbidities, and if similar pharmacokinetic thresholds can be established for key second-line drugs within the MDR-TB regimen.
Of the second-line TB agents, the fluoroquinolones have been subjected to rigorous testing, and like their first-line counterparts show concentration-dependent killing exemplified by AUC/MIC in murine models, HFS and human studies [38-42]. Though less so than rifampin, Cmax and AUC track reasonably well for the fluoroquinolones, covary over fixed doses and intervals, and Cmax alone has been used for clinical action regarding dose adjustment [38]. Notably, the fluoroquinolones have also exhibited considerable pharmacokinetic variability when tested in TB-infected human cohorts [43]. For instance, in a study of ofloxacin pharmacokinetics from South Africa, only 45% of patients reached target AUC/MIC, and for all those with an isolate harboring an increased MIC of 2.0 μg/ml, but still considered within the conventional susceptible range, 0% reached the target [44]. Given these observations and that fluoroquinolone anti-TB activity is not consistent across the class, the fluoroquinolones may be particularly applicable for pharmacokinetic monitoring and dose optimization in the treatment of MDR-TB [44,45]. Less is known about the pharmacokinetic–pharmacodynamic parameters of other second-line drugs in TB-specific treatment settings, and has instead been extrapolated from non-TB models [46]. Linezolid, and the recently FDA-approved diarylquinoline, bedaquiline, are exceptions, given their comparatively recent introduction [47,48]. In an early bactericidal study in human TB, linezolid activity was linear at AUC/MIC values up to 120, but the correlation was diminished once T > MIC reached 100% [49]. Both animal and human data on bedaquiline support time-dependent bactericidal activity with a rather delayed onset of action [50].
4. Therapeutic drug concentration monitoring in practice and emerging methods to optimize treatment
The clinical application of drug concentration monitoring and adjustment of drug dosages in individual patients, termed therapeutic drug monitoring (TDM), has been employed regularly by specialized TB treatment centers [51]. The Cmax is practically targeted for TDM for many of the key first-line drugs, and can be closely approximated by a 2- and 6-h post-dose blood draw. Many of the first-line drugs reach peak concentrations within 1 – 3 h of administration, yet the 6-h blood draw can help distinguish between delayed absorption and malabsorption among those patients with abnormal 2-h concentrations. Blood is centrifuged after it is drawn, and the serum is immediately frozen. Methods differ for each drug but commonly utilize high-performance liquid chromatography, gas chromatography or, more recently, tandem mass spectrometry for determining serum concentrations. Consequently, drug concentration analysis is usually performed by experienced laboratories, in relatively few settings worldwide. Even in settings with access to specialized laboratories, overall cost including serum transport and logistical coordination has limited more widespread uptake. Furthermore, concentrations below the expected range for key drugs in the anti-TB regimen have been found in patients responding well to treatment [10,52], and even more refined means of interpreting TDM results may evolve, such as measuring plasma protein binding of the drugs in question [53]. Thus, some large governmental programs have sought guidance for the strategic selection of patients that would benefit most from TDM.
The application of dried blood spot (DBS) analysis instead of serum analysis may ultimately overcome the obstacle of specimen storage and transport [54]. For DBS analysis, blood is obtained by single-use lancing devices and dropped directly on DBS paper, generally cellulose- or a cotton-based filter. The paper is left to dry at room temperature and then stored in a plastic bag with desiccant packages. The samples can be sent to outside laboratories using normal postal systems without the need for special mailing cartons or preservation of the cold chain. Once received by the laboratory, a disc of uniform size is punched from the paper and subjected to solvent extraction with subsequent drug quantification through standard means [55]. The lower blood volume required for DBS reduces its biohazardous risk compared to conventional serum sampling and makes it more applicable to pediatric populations. A recent study using second-line drugs for MDR-TB found little variation with ranges of hematocrit between 20 and 50% and acceptable correlation between whole blood and plasma values by calculable conversion factors from the geometric mean DBS/plasma concentration ratios [56]. The stability in DBS does vary from drug to drug, depending on temperature ranges [54]; hence, further study is needed to refine drug-specific protocols.
Other TB bioassays provide functional pharmacodynamic information without the use of chromatography or mass spectrometry and have utilized TB culture accessible in many mycobacteriology laboratories worldwide. Wallis et al. developed a method of performing culture of M. tuberculosis with a subject’s whole blood while on TB therapy to quantify the predominately intracellular bactericidal activity of a drug individually, or in a combination regimen [57]. Given the bactericidal activity observed for common drugs in whole blood culture closely approximated that observed in clinic practice, and total bactericidal activity correlated with two-month sputum culture conversion in subjects on a drug-susceptible TB regimen, whole blood culture has been proposed as a functional biomarker and continues to be used in new drug development [58-60]. Others have constrained similar analysis to plasma and employed the patient’s own M. tuberculosis isolate, finding significant correlation with drug concentrations for isoniazid and rifampin in a drug-susceptible TB regimen [61]. The plasma drug activity assay has also been applied to subjects with MDR-TB in Tanzania, and it was found that subjects with the worst plasma drug activity against their M. tuberculosis isolate had significantly lower kanamycin Cmax/MIC [43]. While lacking the precision of individual drug concentration and MIC information, plasma drug activity is being considered for use in stratifying MDR-TB patients for early hospital discharge and decentralized management. In addition to ensuring adequate drug exposure, methods are being designed to limit toxicity. Approaches include CY450 enzyme activity and hERG gene channel pathway assessment used in the evaluation of new anti-TB compounds [62].
5. Pharmacokinetics of TB drugs in special populations
5.1 HIV
Of the 8.8 million incident cases of TB in 2010, 1.1 million were infected with HIV and at risk for shortened survival and higher rates of TB recurrence compared to TB patients without HIV [63-65]. In a cohort of TB patients from Botswana, the majority HIV infected, those with low pyrazinamide peak concentrations were 3 times more likely to have poor TB treatment outcome even when adjusting for HIV infection [8]. Furthermore, low rifampin concentrations were so common, found in 84%, that differences in outcome may have been difficult to appreciate in dichotomous analysis. Additionally, the increased number of medications required for both diseases early in the course of TB therapy [66-69] may lead to drug toxicity or poor adherence that further limits treatment success. Independent of drug interactions, TB drugs may be improperly absorbed in HIV patients with various forms of enteropathy [70-72]. This may be particularly problematic for rifampin in which absorption is dependent upon both gut permeability and solubility, the latter affected by both pH and gut transit time [73]. Indeed, HIV patients with CD4 < 100 on intermittent (twice or thrice weekly regimens) in the clinical trial setting have shown increased rates of acquired resistance to the rifamycins, explained by poor exposure (as marked by AUC0 – 24) to the study drug of rifabutin, but also isoniazid, and for these reasons daily dosing is recommended [74,75].
HIV-related TB also presents the paradoxical problem that rifampin is a potent CYP450 inducer, limiting the role of key antiretrovirals such as protease inhibitors and some non-nucleoside reverse transcriptase inhibitors (NNRTIs) that are heavily metabolized through this system [76]. Unfortunately, HIV patients treated with rifamycin sparing anti-TB regimens have been shown to have worse outcomes [77]. Of the NNRTI class, efavirenz, at standard doses, has been shown to be effective in patients treated with rifampin without compromise of HIV virological suppression [78,79]. Yet while rifampin induces CYP2B6, isoniazid also inhibits a secondary pathway of CYP2A6-related metabolism and may actually increase efavirenz concentrations during TB treatment in those patients with a slow CYP2B6 metabolizer genotype, thereby risking efavirenz-related toxicity [80,81]. Rifabutin, a much less potent inducer, may be used in replacement of rifampin, but when employed in combination with protease inhibitor-based antitretroviral regimens plasma concentrations of rifabutin are increased due to protease inhibitor inhibition of CYP3A4 necessitating considerable dose reduction [82-84]. Thus, for these complex interactions and to balance efficacy and toxicity, HIV-infected TB patients represent an ideal population for operational research in TDM-directed management.
5.2 Diabetes mellitus
Diabetes is a risk factor for TB infection and progression to active disease with the largest meta-analysis to date concluding that diabetics were 3.1 times more likely than non-diabetics to develop active TB [85-88]. Furthermore, diabetic patients have been shown to have higher rates of treatment failure and death [89-91]. Concentrations of isoniazid and rifampin below the expected range are commonly found in TB patients with diabetes, and in some studies diabetes has been an independently associated with decreased serum concentrations of rifampin [92,93]. In settings where TDM is routinely practiced, some TB programs have elected to check drug concentrations early in the course of therapy in all diabetics [94]. In one such initiative, 76% of all diabetics had peak concentrations of isoniazid, rifampin or both medications below the expected range that were then corrected with a single dose increase [94]. Other means of regimen intensification or extended duration of anti-TB treatment have yet to be prospectively studied in diabetic patients, and no consensus recommendations for treatment modification currently exist. Therefore, as the incidence of diabetic-related TB is anticipated to rise, we expect considerable new study in this subpopulation [95].
5.3 Pediatrics
Pediatric TB is estimated at 500,000 cases annually contributing to 64,000 deaths among children in 2011 [1]. Pediatric patients metabolize drugs differently than adults, most notably evidenced by the more rapid metabolism of isoniazid and the subsequently higher mg/kg dose requirement [96]. Host genetic predisposition for the rate of isoniazid metabolism can be predicted by polymorphisms in the N-acetyltransferase 2 gene responsible for acetylation, but can more functionally be measured by the serum isoniazid and acetyl-isoniazid fraction [96]. Additionally, lower anti-TB drug concentrations have been found in malnourished children compared to those without malnourishment. As in adults, children with TB and HIV are also at risk for suboptimal drug exposure [97,98]. We expect an expansion of pharmacokinetic data in pediatric TB patients given requests for this study from WHO and other international advocacy groups and that evaluation of novel anti-TB drugs in pediatric patients is now a prerequisite for approval in the US and Europe [99-101]. Innovation in this setting is not only expected to center on novel compounds but also on pediatric friendly preparations and optimal dose combinations.
5.4 Extrapulmonary TB
Despite mainly infecting the lung, TB can affect any organ of the body, and does so with increased frequency in pediatric and HIV-infected populations [102]. While treatment of most forms of extrapulmonary TB (EPTB) does not differ from pulmonary TB, meningitis and bone/joint disease may require a longer duration, and TB meningitis in particular carries considerable morbidity and mortality [103]. Given anatomic barriers to drug penetration, these forms of EPTB may be apt for study of dose optimization.
For instance, studies of cerebrospinal fluid concentrations of anti-TB drugs show good penetration of isoniazid, but relatively poor penetration of rifampin, which is larger, more protein bound, and susceptible to alteration in membrane drug transporters. Consequently, rifampin rarely exceeds the MIC at standard doses [104]. Despite this, higher-dose rifampin for TB meningitis has not been formally recommended. Yet a recent study of adults with TB meningitis in Indonesia found lower cerebrospinal fluid than plasma concentrations of rifampin and those treated with higher-dose intravenous rifampin during the first 2 weeks of therapy had significantly lower 6-month mortality (35 vs 65%) than patients treated with standard-dose rifampin [105]. No increase in adverse effects was seen with increased dosages of rifampin. Given the high early mortality of TB meningitis in resource-limited settings, these patients may represent a subpopulation for an empiric higher dose of rifampin, followed by the application of DBS for dose adjustment. Similar to the ongoing trials of higher-dose rifampin to shorten the treatment duration for pulmonary TB, we anticipate a growing body of studies to inform the optimal dosing of rifampin and the later-generation fluoroquinolones in the treatment of TB meningitis.
6. Conclusion
Individual pharmacokinetic variability is far more common than previously understood and contributes to poor TB outcomes independent of adherence. Yet such pharmacokinetic variability has proven difficult to predict without TDM. Application of pharmacokinetic data has thus far been limited to experienced treatment centers, but may become increasingly accessible by means of DBS. Incorporation of TDM with the MIC from an individual patient’s M. tuberculosis isolate may be particularly beneficial for second-line drugs used in the treatment of drug-resistant TB. These applications require operational research in patients at high risk of poor treatment outcome. Doing so will better inform new drug development because unlike many other infectious diseases, TB is treated not with a single drug, but rather a complex multidrug regimen.
7. Expert opinion
Initially, we anticipate considerable operational research to validate the pharmacokinetic thresholds associated with poor TB treatment outcome in drug-susceptible TB, and determination if similar thresholds can be established for key second-line drugs in the MDR-TB regimen. In TB endemic settings, the vast majority of TB treatment is delivered by governmental or non-governmental organizations with poorly funded mandates from local health ministries informed by international or national guidelines. Resource constraint in these settings does not favor individualized management. Yet if validation studies continue to demonstrate that pharmacokinetic variability contributes as significantly to TB outcome as has been found in the work of Pasipanodya et al. in South Africa [7], then resource-constrained organizations and guiding bodies will be compelled to redefine empiric dosing or critical concentrations for drug resistance, or rather adopt a degree of individualized management in the form of applied pharmacokinetics (Figure 2). Each approach carries consequence for innovation and novel drug development.
Figure 2. Depicted are scenarios for operational research that can be studied to optimize anti-TB regimens in resource-limited and TB endemic settings.
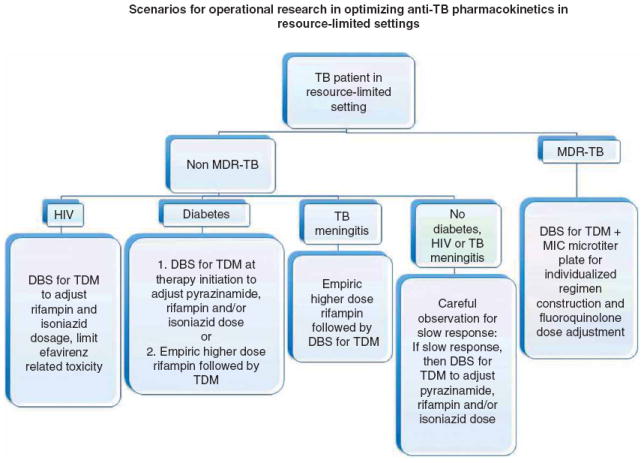
In these scenarios the term resource-limited setting assumes that therapeutic drug monitoring and minimum inhibitory concentration (MIC) may be available at a national or supranational reference laboratory and multidrug-resistant (MDR)-TB is diagnosed by rapid molecular test. Each subpopulation of MDR or non-MDR-TB patient may represent a separate interventional opportunity thereby necessitating their own control group.
DBS: Dried blood spot, a distinct delivery system with innovative opportunity; TB: Tuberculosis.
For instance, we may predict given the encouraging clinical trial results in higher-dose rifampin that empiric dose increase will eventually become standard practice for drug-susceptible pulmonary TB and TB meningitis, and may therefore lessen the need for individual TDM in that setting. TB programs may ultimately opt for individualized management in only specialized scenarios, limiting the market for innovative improvement in TDM delivery. Yet we agree with others that MDR-TB may represent an emerging niche [106,107]. Certainly TDM of plasma may not reflect drug concentrations at the site of infection, yet recent reports of correlative plasma concentrations of MDR-TB drugs with concentrations from pathologic specimens of affected lung are encouraging [106]. Furthermore, molecular diagnostics for M. tuberculosis drugresistance mutations will be more extensively informed by deeper sequencing methods and individual mutations may become increasingly precise in predicting quantitative change in MIC. In this scenario, we could anticipate TDM results for key MDR-TB drugs to be used in concert with rapid quantitative susceptibility in order to guide therapeutic decisions at the bedside. Regardless, rigorously performed studies of currently available second-line drug pharmacokinetics will better define the rational introduction of novel therapeutics.
highlights.
Tuberculosis (TB) is responsible for approximately 1.4 million deaths per year, the leading cause of death from a curable infectious disease worldwide.
Individual pharmacokinetic variability is underappreciated and can lead to poor long-term treatment outcome including relapse and acquired drug resistance.
Threshold pharmacokinetic parameters associated with poor treatment outcome will guide further validation across diverse patient populations and comorbidities, and will be sought for second-line drugs used in the treatment of multidrug-resistant (MDR)-TB.
Therapeutic drug monitoring (TDM) is the most accurate means of assessing individual pharmacokinetic variability but is inaccessible in most TB endemic settings.
Optimizing treatment regimens based on individual pharmacokinetics in TB endemic settings may become more realizable with dried blood spot TDM and commercialized MIC plates – areas of innovative opportunity – but uptake will be tempered by programmatic unfamiliarity with individualized management.
A relative explosion of pharmacokinetic research is anticipated in special populations including MDR-TB and those at higher risk of poor treatment outcome and should inform new drug development and application.
This box summarizes key points contained in the article.
Footnotes
Declaration of interest
The authors have no competing interests to declare and have received no funding in preparation of the manuscript.
Bibliography
Papers of special note have been highlighted as either of interest
-
•
or of considerable interest
-
••
to readers.
- 1.World Health Organization. Global tuberculosis report 2012. 2012 Available from: http://www.who.int/tb/publications/global_report/en/index.html.
- 2.American Thoracic Society, Centers for Disease Control and Prevention, and Infectious Disease Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1203. [Google Scholar]
- 3.Weis SE, Slocum PC, Blais FX, et al. The effect of Directly Observed Therapy on the rates of drug resistance and relapse in tuberculosis. N Engl J Med. 1994;330:1179–84. doi: 10.1056/NEJM199404283301702. [DOI] [PubMed] [Google Scholar]
- 4.Burman WJ, Dalton CB, Cohn DL, et al. A cost-effectiveness analysis of directly observed therapy vs self-administered therapy for treatment of tuberculosis. Chest. 1997;112:63–70. doi: 10.1378/chest.112.1.63. [DOI] [PubMed] [Google Scholar]
- 5•.Srivastava S, Pasipanodya JG, Meek C, et al. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis. 2011;204:1951–9. doi: 10.1093/infdis/jir658. Preclinical model applying population modeling demonstrates acquired drug resistance possible from individual pharmacokinetic variability alone, even without nonadherence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner M, Burman W, Vernon A, et al. Tuberculosis Trials Consortium. Low isoniazid concentrations and outcome of tuberculosis treatment with once-weekly isoniazid and rifapentine. Am J Respir Crit Care Med. 2003;167(10):1341–7. doi: 10.1164/rccm.200208-951OC. [DOI] [PubMed] [Google Scholar]
- 7••.Pasipanodya J, McIlleron H, Burger A, et al. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis. 2013;208:1564–73. doi: 10.1093/infdis/jit352. Prospective cohort of tuberculosis (TB) patients demonstrates widespread individual pharmacokinetic variability independently predictive of poor TB outcomes, allowing threshold pharmacokinetic parameters for key first-line anti-TB drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chideya S, Winston CA, Peloquin CA, et al. Isoniazid, rifampin, ethambutol, and pyrazinamide pharmacokinetics and treatment outcomes among predominately HIV-infected cohort of adults with tuberculosis from Botswana. Clin Infect Dis. 2009;48:1685–94. doi: 10.1086/599040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falzon D, Jaramillo E, Wares F, et al. Universal access to care for multidrug-resistant tuberculosis: an analysis of surveillance data. Lancet Infect Dis. 2013;13:690–7. doi: 10.1016/S1473-3099(13)70130-0. [DOI] [PubMed] [Google Scholar]
- 10.Shenoi S, Heysell SK, Moll AP, Friedland G. Multidrug-resistant and extensively drug-resistant tuberculosis: consequences for the global HIV community. Curr Opin Infect Dis. 2009;22:11–17. doi: 10.1097/QCO.0b013e3283210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diel R, Vandeputte J, de Vries G, et al. Costs of tuberculosis disease in the EU – a systematic analysis and cost calculation. Euro Respir J. 2014;43(2):554–65. doi: 10.1183/09031936.00079413. [DOI] [PubMed] [Google Scholar]
- 12.Eagle H, Fleischman R, Musselman AD. The effective concentrations of penicillin in vitro and in vivo for streptococci, pneumococci, and Treponema pallidum. J Bacteriol. 1950;59:625–43. doi: 10.1128/jb.59.5.625-643.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eagle H, Fleischman R, Musselman AD. Effect of schedule of administration on the therapeutic efficacy of penicillin; importance of the aggregate time penicillin remains at effectively bactericidal levels. Am J Med. 1950;9:280–99. doi: 10.1016/0002-9343(50)90425-6. [DOI] [PubMed] [Google Scholar]
- 14.Ambrose PG, Bhavani SM, Rubino A, et al. Pharmacokinetics- Pharmacodynamics of Antimicrobial Therapy: it’s Not Just for Mice Anymore. Clin Infect Dis. 2007;44:79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- 15.NCCLS. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. 6. NCCLS; Wayne, PA, USA: 2003. [Google Scholar]
- 16.Kim SJ. Drug-susceptibility testing in tuberculosis: methods and reliability of results. Eur Respir J. 2005;25(3):564–9. doi: 10.1183/09031936.05.00111304. [DOI] [PubMed] [Google Scholar]
- 17.Heysell SK, Houpt ER. The future of molecular diagnostics for drug-resistant tuberculosis. Expert Rev Mol Diagn. 2012;12(4):395–405. doi: 10.1586/erm.12.25. [DOI] [PubMed] [Google Scholar]
- 18.Heifets LB, Cangelosi GA. Drug susceptibility testing of Mycobacterium: a neglected problem at the turn of the century. Int J Tuberc Lung Dis. 1999;3(7):564–81. [PubMed] [Google Scholar]
- 19.Hall L, Jude KP, Clark SL, et al. Evaluation of the sensititre® MYCOTB plate for the susceptibility testing of mycobacterium tuberculosis complex against first and second line agents. J Clin Microbiol. 2012;50(11):3732–4. doi: 10.1128/JCM.02048-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mpagama S, Houpt ER, Stroup S, et al. Application of quantitative second-line drug susceptibility testing at a multidrug-resistant tuberculosis hospital in Tanzania. BMC Infect Dis. 2013;13:432. doi: 10.1186/1471-2334-13-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Lee JS, Armstrong DT, Ssengooba W, et al. The Sensititre MYCOTB MIC plate for susceptibility testing of Mycobacterium tuberculosis to 1st and 2nd line drugs. Antimicrob Agents Chemother. 2014;58(1):11–18. doi: 10.1128/AAC.01209-13. Comparison of over 200 Mycobacterium tuberculosis isolates by commercialized MIC plate to standard susceptibility testing, and demonstrating additional correlation of MIC to sequencing results of M. tuberculosis drug-resistance mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasipanodya J, Gumbo T. An Oracle: antituberculosis pharmacokinetic–pharmacodynamics, clinical correlation, and clinical trial simulations to predict the future. Antimicrob Agents Chemother. 2011;55(1):24–34. doi: 10.1128/AAC.00749-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bratton DJ, Phillips PP, Parmar MK, et al. A multi-arm multi-stage clinical trial design for binary outcomes with application to tuberculosis. BMC Med Res Methodol. 2013;13:139. doi: 10.1186/1471-2288-13-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burki T. PanACEA: a new approach to tuberculosis research. Lancet Infect Dis. 2012;12(3):184–5. doi: 10.1016/s1473-3099(12)70041-5. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava S, Musaka S, Sherman C, et al. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J Infect Dis. 2010;201:1225–31. doi: 10.1086/651377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gumbo T, Dona CS, Meek C, Leff R. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother. 2009;53:3197–204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anonymous. Ethambutol plus isoniazid for the treatment of pulmonary tuberculosis – a controlled trial of our regiments. Tubercle. 1981;62:13–29. doi: 10.1016/0041-3879(81)90031-3. [DOI] [PubMed] [Google Scholar]
- 28.British Medical Research Council. A controlled comparison of four regimens of streptomycin plus pyrazinamide in the retreatment of pulmonary tuberculosis. Tubercle. 1969;50:81–114. doi: 10.1016/0041-3879(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 29.Dickinson JM, Ellard GA, Mitchison DA. Suitability of isoniazid and ethambutol for intermittent administration in the treatment of tuberculosis. Tubercle. 1968;49:351–66. doi: 10.1016/s0041-3879(68)80016-9. [DOI] [PubMed] [Google Scholar]
- 30.Jayaram R, Shandil RK, Gaonkar S, et al. Isoniazid pharmacokinetics-pharmacodynamics in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2004;48:2951–7. doi: 10.1128/AAC.48.8.2951-2957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gumbo T, Louie A, Liu W, et al. Isoniazid bactericidal activity and resistance emergence: integrating pharmacodynamics and pharmacogenomics to predict efficacy in different ethnic populations. Antimicrob Agents Chemother. 2007;51:2329–36. doi: 10.1128/AAC.00185-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchison DA, Dickinson JM. Laboratory aspects of intermittent drug therapy. Postgrad Med J. 1971;47:737–41. doi: 10.1136/pgmj.47.553.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbist L. Rifampicin activity “in vitro” and in established tuberculosis in mice. Acta Tuberc Pneumol Belg. 1969;60:397–412. [PubMed] [Google Scholar]
- 34.Jayaram R, Gaonkar S, Kaur P, et al. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother. 2003;47:2118–24. doi: 10.1128/AAC.47.7.2118-2124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gumbo T, Louie A, Deziel MR, et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agnets Chemother. 2007;51:3781–8. doi: 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall R, Leff GRD, Gumbo T. Treatment of active pulmonary tuberculosis in adults: current standards and recent advances. Pharmacotherapy. 2009;29:1468–81. doi: 10.1592/phco.29.12.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boeree M. What Is the “Right” Dose of Rifampin?. 20th Conference on Retroviruses and Opportunistic Infections; 3 – 6 March 2013; Atlanta. Oral abstract and paper 148LB. [Google Scholar]
- 38.Shandil RK, Jayaram R, Kaur P, et al. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamics indices that best predict in vivo efficacy. Antimicrob Agents Chemother. 2007;51:576–82. doi: 10.1128/AAC.00414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gumbo T, Louie A, Dezeil MR, Drusano GL. Pharmacodynamic evidence that ciprofloxacin failure against tuberculosis is not due to poor microbial kill but to rapid emergence of resistance. Antimicrob Agents Chemother. 2005;49:3178–81. doi: 10.1128/AAC.49.8.3178-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gumbo T, Louie A, Deziel MR, et al. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamics infection model and mathematical modeling. J Infect Dis. 2004;190:1642–51. doi: 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 41.Peloquin CA, Hadad DJ, Molino LP, et al. Population pharmacokinetics of levofloxacin, gatifloxacin, and moxifloxacin in adults with pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52:852–7. doi: 10.1128/AAC.01036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.Mpagama SG, Ndusilo N, Stroup S, et al. Plasma drug activity in patients on treatment for multidrug-resistant tuberculosis. Antimicrob Agent Chemother. 2014;58(2):782–8. doi: 10.1128/AAC.01549-13. Field application of bioassay correlative with peak/MIC values of second-line drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chigutsa E, Meredith S, Wiesner L, et al. Population pharmacokinetics and pharmacodynamics of ofloxacin in south African patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2012;56(7):3857–63. doi: 10.1128/AAC.00048-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobson KR, Tierny B, Jeon CY, et al. Treatment outcomes among patients with extensively drug-resistant tuberculosis: systematic review and meta-analysis. Clin Infect Dis. 2010;51(1):6–14. doi: 10.1086/653115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Deun A, Maug AKJ, Salim MAH, et al. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med. 2010;182:684–92. doi: 10.1164/rccm.201001-0077OC. [DOI] [PubMed] [Google Scholar]
- 46.Mandell GL, Bennett JE, Dolin R. Mandell, douglas, and Bennett’s principles and practice of infectious diseases. 7. Vol. 1. Churchill Livingstone; Philadelphia: 2010. pp. 300–6. [Google Scholar]
- 47.Lee M, Lee J, Carroll MW, et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med. 2012;367:1508–18. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fox G, Menzies D. A review of the evidence for using bedaquiline (TMC207) to treat multi-drug resistant tuberculosis. Infect Dis Ther. 2013 doi: 10.1007/s40121-013-0009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dietze R, Hadad DJ, McGee B, et al. Early and extended bactericidal activity of linezolid in pulmonary tuberculosis. Am J Respir Crit Care Med. 2008;178(11):1180–5. doi: 10.1164/rccm.200806-892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rustomjee R, Diacon AH, Allen J, et al. Early bactericidal activity and pharmacokinetics of the diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrob Agents Chemother. 2008;52(8):2831. doi: 10.1128/AAC.01204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Peloquin CA. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs. 2002;62:2169–83. doi: 10.2165/00003495-200262150-00001. Comprehensive review of therapeutic drug monitoring for TB in clinical practice. [DOI] [PubMed] [Google Scholar]
- 52.Chang KC, Leung CC, Yew WW, et al. Peak plasma rifampicin level in tuberculosis patients with slow culture conversion. Eur J Clin Microbiol Infect Dis. 2008;27:467–72. doi: 10.1007/s10096-007-0454-6. [DOI] [PubMed] [Google Scholar]
- 53.Rowland M. Plasma protein binding and therapeutic drug monitoring. Ther Drug Monit. 1980;2(1):29–37. doi: 10.1097/00007691-198001000-00005. Review. [DOI] [PubMed] [Google Scholar]
- 54.Edelbroek PM, van der Heijden J, Stolk LM. Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther Drug Monit. 2009;31(3):327–36. doi: 10.1097/FTD.0b013e31819e91ce. [DOI] [PubMed] [Google Scholar]
- 55.Harmelink IM, Alffenaar JW, Wessels AM, et al. A rapid and simple liquid chromatography-tandem mass spectrom- etry method for the determination of linezolid in human serum. EJHP Sci. 2008;14:3–7. [Google Scholar]
- 56••.Vu DH, Blhuis MS, Koster RA, et al. Dried blood spot analysis for therapeutic drug monitoring of linezolid in patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother. 2012;56(11):5758–63. doi: 10.1128/AAC.01054-12. Innovative use of dried blood spot analysis in emerging niche of multidrug-resistant-TB and second-line or third-line drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallis RS, Palaci M, Vinhas S, et al. A whole blood bactericidal assay for tuberculosis. J Infect Dis. 2001;183:1300–3. doi: 10.1086/319679. [DOI] [PubMed] [Google Scholar]
- 58.Wallis RS, Vinhas SA, Johnson JL, et al. Whole blood bactericidal activity during treatment of pulmonary tuberculosis. J Infect Dis. 2003;187:270–8. doi: 10.1086/346053. [DOI] [PubMed] [Google Scholar]
- 59.Wallis RS, Kim P, Cole S, et al. Tuberculosis biomarkers discovery: developments, needs, and challenges. Lancet Infect Dis. 2013;13:362–72. doi: 10.1016/S1473-3099(13)70034-3. [DOI] [PubMed] [Google Scholar]
- 60••.Wallis RS, Jakubiec WM, Kumar V, et al. Pharmacokinetics and whole-blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU-100480 in healthy volunteers. J Infect Dis. 2010;202(5):745–51. doi: 10.1086/655471. Intracellular model of anti-TB drug activity applied for new drug development. [DOI] [PubMed] [Google Scholar]
- 61.Heysell SK, Mtabho C, Mpagama SG, et al. A plasma drug activity assay for treatment optimization in tuberculosis patients. Antimicrob Agents Chemother. 2011;55(12):5819–25. doi: 10.1128/AAC.05561-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rao SP, et al. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med. 2013;5:214. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 63.Wallis RS. Immune activation, allergic drug toxicity and mortality in HIV-positive tuberculosis. Tuber Lung Dis. 1996;77:516–23. doi: 10.1016/s0962-8479(96)90049-0. [DOI] [PubMed] [Google Scholar]
- 64.Hawken M, Nunn P, Gathua S, et al. Increased recurrence of tuberculosis in HIV-1-infected patients in Kenya. Lancet. 1993;342:332–8. doi: 10.1016/0140-6736(93)91474-z. [DOI] [PubMed] [Google Scholar]
- 65.Perriens JH, Colebunders RL, Karahunga C, et al. Increased mortality and tuberculosis treatment failure rate among human immunodeficiency virus (HIV) seropositive compared with HIV seronegative patients with pulmonary tuberculosis in Kinshasa, Zaire. Am Rev Respir Dis. 1991;122:750–5. doi: 10.1164/ajrccm/144.4.750. [DOI] [PubMed] [Google Scholar]
- 66.Abdool Karim SS, Naidoo K, Grobler A, et al. Timing of initiation of antiretroviral drugs during tuberculosis therapy. N Eng J Med. 2010;362(8):697–706. doi: 10.1056/NEJMoa0905848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abdool Karim SS, Naidoo K, Grobler A. Integration of antiretroviral therapy with tuberculosis treatment. N Engl J Med. 2011;365(16):1492–501. doi: 10.1056/NEJMoa1014181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blanc FX, Sok T, Laureillard D, et al. Earlier versus later start of antiretroviral therapy in HIV-infected adults with tuberculosis. N Engl J Med. 2011;365(16):1471–81. doi: 10.1056/NEJMoa1013911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Havlir DV, Kendall MA, Kumwenda J, et al. Timing of antiretroviral therapy for HIV-1 infection and tuberculosis. N Engl J Med. 2011;352(16):1482–91. doi: 10.1056/NEJMoa1013607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kotler DP, Gaetz HP, Lange M, et al. Enteropathy associated with the acquired immunodeficiency syndrome. Ann Intern Med. 1984;101:421–8. doi: 10.7326/0003-4819-101-4-421. [DOI] [PubMed] [Google Scholar]
- 71.Gillin JS, Shike M, Alcock N, et al. Malabsorption and mucosal abnormalities of the small intestine in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;102:619–22. doi: 10.7326/0003-4819-102-5-619. [DOI] [PubMed] [Google Scholar]
- 72.Bartlett JG, Belitsos PC, Sears CL. AIDS enteropathy. Clin Infect Dis. 1992;15:726–35. doi: 10.1093/clind/15.4.726. [DOI] [PubMed] [Google Scholar]
- 73.Ashokraj Y, Kaur KJ, Singh I, et al. In vivo dissolution: predominant factor affecting the bioavailability of rifampicin in its solid oral dosage forms. Clin Res Reg Aff. 2008;25(1):1–12. [Google Scholar]
- 74.Burman W, Benator D, Vernon A, et al. Acquired rifamycin resistance with twice-weekly treatment of hiv-related tuberculosis. Am J Respir Crit Care Med. 2006;173(3):350–6. doi: 10.1164/rccm.200503-417OC. [DOI] [PubMed] [Google Scholar]
- 75.Weiner M, Benator D, Burman W, et al. Association between acquired rifamycin resistance and the pharmacokinetics of rifabutin and isoniazid among patients with HIV and tuberculosis. Clin Infect Dis. 2005;40(10):1481–91. doi: 10.1086/429321. [DOI] [PubMed] [Google Scholar]
- 76.Centers for Disease Control and Prevention (CDC) Updated guidelines for the use of rifabutin or rifampin for the treatment and prevention of tuberculosis among HIV-infected patients taking protease inhibitors or nonnucleoside reverse transcriptase inhibitors. MMWR Morb Mortal Wkly Rep. 2000;49(9):185–9. [PubMed] [Google Scholar]
- 77.Okwera A, Whalen C, Byekwaso F, et al. Randomized trial of thiacetazone and rifampicin-containing regimens for pulmonary tuberculosis in HIV infected Ugandans. Makere University-Case Western Reserve University Research Collaboration. Lancet. 1994;344:1323–8. doi: 10.1016/s0140-6736(94)90693-9. [DOI] [PubMed] [Google Scholar]
- 78.Boulle A, van Custsem G, Cohen K, et al. Outcomes of nevirapine- and efavirenz-based antiretroviral therapy when coadministered with rifampicin-based antitubercular therapy. JAMA. 2008;300(5):530–9. doi: 10.1001/jama.300.5.530. [DOI] [PubMed] [Google Scholar]
- 79.Manosuthi W, Sungkanuparph S, Tantanathip P, et al. A randomized trial comparing plasma drug concentrations and efficacies between 2 nonnucleoside reverse-transcriptase inhibitor-based regimens in HIV-infected patients receiving rifampicin: the N2R study. Clin Infect Dis. 2009;8(12):1752–9. doi: 10.1086/599114. [DOI] [PubMed] [Google Scholar]
- 80.Ramachandran G, Ramesh K, Hemanth Kumar AK, et al. Association of high T allele frequency of CYP2B6 G516T polymorphism among ethnic south Indian HIV-infected patients with elevated plasma efavirenz and nevirapine. J Antimicrob Chemother. 2009;63:841–84. doi: 10.1093/jac/dkp033. [DOI] [PubMed] [Google Scholar]
- 81•.McIlleron HM, Schomaker M, Ren Y, et al. Effects of rifampin-based antituberculosis therapy on plasma efavirenz oncentrations in children vary by CYP2B6 genotype. AIDS. 2013;27:1933–40. doi: 10.1097/qad.0b013e328360dbb4. Illustrative study of contribution of pharmacogenomics in concurrent HIV/TB treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lan NT, Thu NT, Varrail-Tran A, et al. Randomised pharmacokinetic trial of rifabutin with lopinavir/ritonavirantiretroviral therapy in patients with HIV-associated tuberculosis in Vietnam. PLoS One. 2014 doi: 10.1371/journal.pone.0084866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loeliger A, Suthar AB, Ripin D, et al. Protease inhibitor-containing antiretroviral treatment and tuberculosis: can rifabutin fill the breach? Int J Tuberc Lung Dis. 2012;16:6–15. doi: 10.5588/ijtld.10.0626. [DOI] [PubMed] [Google Scholar]
- 84.Baciewicz AM, Chrisman CR, Finch CK, Self TH. Update on rifampin and rifabutin drug interactions. Am J Med Sci. 2008;335:126–36. doi: 10.1097/MAJ.0b013e31814a586a. [DOI] [PubMed] [Google Scholar]
- 85.Shetty N, Shemko M, Vaz M, D’Souza G. An epidemiological evaluation of risk factors for tuberculosis in South India: a matched case control study. Int J Tuberc Lung Dis. 2006;10:80–6. [PubMed] [Google Scholar]
- 86.Coker R, McKee M, Atun R, et al. Risk factors for pulmonary tuberculosis in Russia: case-control study. BMJ. 2006;332:85–7. doi: 10.1136/bmj.38684.687940.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stevenson CR, Forouhi NG, Roglic G, et al. Diabetes and tuberculosis: the impact of the diabetes epidemic on tuberculosis incidence. BMC Public Health. 2007;7:234. doi: 10.1186/1471-2458-7-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jeon CY, Murray MB. Diabetes mellitus increases the risk of active tuberculosis: a systematic review of 13 observational studies. PLoS Med. 2008;5:e152. doi: 10.1371/journal.pmed.0050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mboussa J, Monabeka H, Kombo M, et al. Course of pulmonary tuberculosis in diabetics. Rev Pneumol Clin. 2003;59:39–44. [PubMed] [Google Scholar]
- 90.Oursler KK, Moore RD, Bishai WR, et al. Survival of patients with pulmonary tuberculosis: clinical and molecular epidemiologic factors. Clin Infect Dis. 2002;34:752–9. doi: 10.1086/338784. [DOI] [PubMed] [Google Scholar]
- 91•.Alisjahbana B, Sahiratmadja E, Nelwan EJ, et al. The effect of type 2 diabetes mellitus on the presentation and treatment response of pulmonary tuberculosis. Clin Infect Dis. 2007;45:428–35. doi: 10.1086/519841. Example of growing body of literature associating diabetes with poor TB treatment outcome. [DOI] [PubMed] [Google Scholar]
- 92.Nijland HM, Ruslami R, Stalenhoef JE, et al. Exposure to rifampicin is strongly reduced in patients with tuberculosis and type 2 diabetes. Clin Infect Dis. 2006;43:848–54. doi: 10.1086/507543. [DOI] [PubMed] [Google Scholar]
- 93.Heysell SK, Moore JL, Keller SJ, Houpt ER. Therapeutic drug monitoring for slow response to tuberculosis treatment in a state control program, Virginia, USA. Emerg Infect Dis. 2010;16(101):1546–53. doi: 10.3201/eid1610.100374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heysell SK, Moore JL, Staley D, et al. Early therapeutic drug monitoring for isoniazid and rifampin among diabetics with newly diagnosed tuberculosis in Virginia, U.S.A. Tuberc Res Treat. 2013 doi: 10.1155/2013/129723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95•.Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis. 2009;9:737–46. doi: 10.1016/S1473-3099(09)70282-8. Authoritative review on the gaps in knowledge in our understanding of the interaction of diabetes and TB with course charted for further research priorities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McIlleron H, Willemse M, Werely CJ, et al. Isoniazid plasma concentrations in a cohort of South African children with tuberculosis: implications for international pediatric dosing guidelines. Clin Infect Dis. 2009;48:1547–53. doi: 10.1086/598192. [DOI] [PubMed] [Google Scholar]
- 97.Ramachandran G, Hemanth Kumar AK, Bhavani PK, et al. Age, nutritional status and INH acetylator status affect pharmacokinetics of anti-tuberculosis drugs in children. Int J Tuberc Lung Dis. 2013;17(6):800–6. doi: 10.5588/ijtld.12.0628. [DOI] [PubMed] [Google Scholar]
- 98.Schaaf HS, Willemse M, Cilliers K, et al. Rifampin pharmacokinetics in children, with and without human immunodeficiency virus infection, hospitalized for the management of severe forms of tuberculosis. BMC Med. 2009;7:19. doi: 10.1186/1741-7015-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seddon JA, Hesseling AC, Schaaf HS. Retooling existing tuberculosis drugs for children. Clin Infect Dis. 2013;56(1):167–8. doi: 10.1093/cid/cis819. [DOI] [PubMed] [Google Scholar]
- 100.United States Congress. Food and drug administration amendments act of 2007. HR3580; 110th congress; 2007. [Google Scholar]
- 101.European Medicines Agency. The EU paediatric regulation. 2007 Available from: http://www.emea.europa.eu/htms/human/paediatrics/regulation.htm.
- 102.Golden MP, Vikram HR. Extrapulmonary tuberculosis: an overview. Am Fam Physician. 2005;72(9):1761–8. [PubMed] [Google Scholar]
- 103.World Health Organization. Guidelines for treatment of tuberculosis. 4. World Health Organization; Geneva: 2010. [Google Scholar]
- 104.Donald PR. Cerebrospinal fluid concentrations of antituberculosis agents in adults and children. Tuberculosis. 2010;90(5):279–92. doi: 10.1016/j.tube.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 105•.Ruslami R, Ganiem AR, Dian S, et al. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open-label, randomised controlled phase 2 trial. Lancet Infect Dis. 2013;13(1):27–35. doi: 10.1016/S1473-3099(12)70264-5. Mortality benefit demonstrated in TB meningitis by understanding pharmacokinetics and optimized dosing. [DOI] [PubMed] [Google Scholar]
- 106.Akkerman OW, van Altena R, Klinkenberg T, et al. Drug concentration in lung tissue in multidrug-resistant tuberculosis. Eur Respir J. 2013;42:1750–2. doi: 10.1183/09031936.00047413. [DOI] [PubMed] [Google Scholar]
- 107.Srivastava S, Peloquin CA, Sotgiu G, Migliori GB. Therapeutic drug management: is it the future of multidrug-resistant tuberculosis treatment? Eur Respir J. 2013;42:1449–53. doi: 10.1183/09031936.00073213. [DOI] [PubMed] [Google Scholar]