

Abstract
A structural perspective of drug target and anti-target proteins, and their molecular interactions with biologically active molecules, largely advances many areas of drug discovery, including target validation, hit and lead finding and lead optimisation. In the absence of experimental 3D structures, protein structure prediction often offers a suitable alternative to facilitate structure-based studies. This review outlines recent methodical advances in homology modelling, with a focus on those techniques that necessitate consideration of ligand binding. In this context, model quality estimation deserves special attention because the accuracy and reliability of different structure prediction techniques vary considerably, and the quality of a model ultimately determines its usefulness for structure-based drug discovery. Examples of G-protein-coupled receptors and ADMET-related proteins were selected to illustrate recent progress and current limitations of protein structure prediction. Basic guidelines for good modelling practice are also provided.
Keywords: Protein structure modelling, structure prediction, structure-based drug discovery, virtual screening, model quality
Introduction
The goal of drug discovery is to contrive bioactive molecules that efficaciously modify a disease in a way that is beneficial to the patient, while keeping adverse effects such as toxic responses controllable. On the molecular level these requirements translate into a picture where a drug molecule binds to one or more target proteins that are implicated in the pathophysiology of a disease and act as, for example, inhibitors, agonists or modulators. At the same time, binding to proteins that have a negative impact on efficacy, or cause unwanted side effects, has to be avoided. In this sense, drug design is an enterprise that aims to engineer molecules with a controlled interaction profile against a multitude of different target and off-target proteins in an organism. During the initial target validation and hit finding phases of a drug discovery programme the focus is usually on the main target and then, as a programme progresses into lead optimisation, the attention shifts to the interplay of the drug candidate with an increasing number of proteins. Obviously, a full characterisation of these interactions down to the 3D structural details would constitute a profound structural perspective of the mode of action (MOA) of a drug molecule, and hence greatly facilitate drug design. Nowadays, a vast amount of experimental structural data, mainly generated by X-ray crystallography, is available [1]. Yet, the number of known protein sequences vastly exceeds the number of corresponding 3D structures. This so-called sequence– structure gap implies that for many important proteins there are no structures available. Fortunately, 3D protein structure prediction often offers an appropriate remedy in such situations [2,3]. In this review, we discuss the current status, applicability and limitations of protein models derived from protein structure prediction methods. We briefly introduce the prevailing prediction methods, with a focus on their relevance in drug discovery. Using selected examples, we also demonstrate typical applications at various stages of the drug discovery process.
Although it might appear trivial, it is worth emphasising one of the most important achievements of protein 3D structure modelling: the transformation, integration and contextualisation of heterogeneous information, such as mutation and SAR data, in a 3D model. Numerous visualisation tools have been developed for inspecting, analysing and annotating such models [4]. Visualisation of valid models is not merely a decorative offshoot of modelling, rather a focal point where disparate facets of research efforts can amalgamate and converge into a detailed view of the underlying mechanistic basis, which in turn can become the driving force for further advances. It should be kept in mind that, even at relatively low resolution, ‘any level of physical characterisation of a protein, as opposed to its absence, is valuable’ [5].
Protein and binding site flexibility
Proteins are intrinsically dynamic systems that can exhibit significant flexibility and structural plasticity, also in their drug binding sites. A single structural model embodies only a static snapshot, regardless of whether it is an experimental or a predicted structure, and can therefore not always capture all the relevant characteristics of a protein. In essence, there is no entity such as ‘the’ structure of a protein, and this principle also applies to experimental structures for which, in addition to the issues associated with protein flexibility, experimental conditions, structural errors [6] and crystal packing effects [7] must be taken into account. Consideration of target and binding site flexibility is of paramount importance in computer-aided drug design (CADD), and disregarding them can dramatically hamper its success. Consequently, appropriate treatment of protein flexibility has become a major effort [8,9]. The ligand-steered modelling approaches outlined below have emerged as a result of these challenges.
Methods for protein modelling
Computational methods for predicting 3D protein models are widely used in the pharmaceutical industry, and much effort has been invested in improving model accuracy, and in expanding the scope of these methods (Table 1). Methods are generally categorised into template-based (i.e. homology) modelling and de novo modelling [10,11]. Traditional homology modelling (or comparative modelling) is considered to be the most accurate of these methods, and is thus most commonly applied in drug discovery research [12]. Homology modelling is based on the fundamental observation that all members of a protein family persistently exhibit the same fold, characterised by a core structure that is robust against sequence modifications [13]. It relies on experimentally determined structures of homologous proteins (templates), and enables the generation of models starting from given protein sequences (targets). The most accurate models can be obtained from close homologue structures; however, even with low sequence similarity (~20%) suitable models can be obtained [14,15].
Table 1.
Frequently used servers and tools for protein structure homology modelling
| Resource | Refs and URL | |
|---|---|---|
| Protein Model Portal | [94] | http://www.proteinmodelportal.org |
| HHpred | [95] | http://toolkit.tuebingen.mpg.de/hhpred |
| ICM | [96] | http://www.molsoft.com/ |
| IntFOLD | [97] | http://www.reading.ac.uk/bioinf/IntFOLD/ |
| Modeller, ModWeb | [98] | http://salilab.org/modeller/ |
| Phyre2 | [99] | http://www.sbg.bio.ic.ac.uk/phyre2/ |
| Robetta | [100] | http://robetta.bakerlab.org/ |
| SWISS-MODEL | [101] | http://swissmodel.expasy.org/ |
A homology modelling pipeline generally comprises the following steps which can be repeated until a suitable model is obtained: (i) template selection for identifying the most suitable experimentally determined structures; (ii) target–template sequence alignment; (iii) 3D model structure building; (iv) model refinement; and (v) model quality estimation. Model refinement usually involves clash removal and geometrical regularisation of bond lengths and angles but can also involve additional more-sophisticated structural amendments. As a rule of thumb, most attention should be devoted to steps i, ii, iii and v, whereas global model refinement (iv) typically has a disappointing return on investment [16].
LSM: ligand-steered modelling
As mentioned above, appropriate modelling of the binding site and correct ligand placement are of the utmost importance in CADD. However, native protein ligands such as enzyme substrates or signalling molecules often exhibit only weak binding affinities and are therefore often lost during purification procedures. As a result, protein structures are often determined experimentally in the absence of ligands. Additionally, template selection procedures in traditional homology modelling are often based on sequence similarity as the only criterion, neglecting ligand information in the template structures. As a consequence of this, the resulting protein models often represent an unliganded state of the binding site.
Classically, docking approaches have been used to place the ligands into the binding sites of the final homology models as a post-processing step [17–19]. The shortcomings of this practice have been addressed by developing more ligand-aware approaches that treat ligands as an integral part of a model throughout the entire modelling process. Generally, two strategies can currently be distinguished. First, ligand-guided (or steered) receptor modelling (LSM) directly incorporates ligands in the modelling process for guiding the protein conformation sampling procedure. One pioneering approach is binding site remodelling, which uses restraints obtained from initially modelled complex structures to build a second refined model [20]. Such approaches often require expert knowledge and time-consuming manual intervention, and hence call for the development of fully automatic homology modelling pipelines. Dalton and Jackson [21] have developed and assessed two variants of LSM, both yielding significantly more accurate complex models than docking into static homology models, regardless of whether or not the ligand had been incorporated into the modelling process. The most successful variant utilises geometric hashing and shape-based superposition of the ligand to be built onto a known ligand in a template structure, prior to the modelling procedure. Generally, ligand-guided approaches can lead to highly accurate models but can be hindered by the fact that correct ligand placement is intrinsically linked to correct side-chain modelling, and even small inaccuracies can prevent the correct prediction of relevant interactions. The second approach, termed here: ligand-guided receptor selection, utilises a large number of homology models from which the model yielding the highest enrichment in docking calculations against known active and decoy compounds is determined [22]. Model generation usually encompasses extensive sampling of side chains in the binding cavity, but can also be extended to incorporate variations in the backbone conformation [23]. This method has recently been extended to a fully automated iterative sampling-selection procedure to generate an ensemble of optimised conformers [24]. This approach has the advantage that the models are optimised for a particular purpose; however, it is limited to cases where high-affinity ligands are known.
Model validation and quality estimation
Homology models are computationally derived approximations of a protein structure and can contain significant errors and inaccuracies. It should be noted that the quality required for a model depends largely on its intended use. For example, low-accuracy models can be completely sufficient for designing mutagenesis experiments, whereas structure-based virtual screening (SBVS) applications require greater accuracy [15], and for mechanistic studies the highest level of accuracy possible is essential [2,11]. Although the accuracy of a protein modelling method can be evaluated a posteriori based on experimental structures [14], the quality of an individual model can vary significantly and the a priori estimation of model quality it therefore of great importance. Common methods for estimating model quality use a combination of stereochemical plausibility checks, knowledge-based statistical potentials, physics-based energy functions or model consensus approaches [25–28]. Different scores have been developed for tasks ranging from ranking of an ensemble of models on a relative scale to the prediction of the absolute accuracy on a per residue basis.
Hit finding and virtual screening
Virtual screening (VS) has matured into an invaluable approach for identifying active compounds against drug targets by means of ‘smart’ computational approaches [29]. Basically, SBVS is the automated positioning (docking) of different 3D conformational models of compounds (poses) into a suitable binding site of a 3D protein structure. Subsequent post-processing of these poses aims to identify the compounds that are most likely to be active. See, for example, the reviews by Klebe [30], Waszkowycz [31] and Cheng et al. [32] for overviews. In the absence of appropriate experimental 3D structures, homology models can be used as an alternative. The usefulness of homology models in SBVS against many different targets has been demonstrated in various retrospective analyses [33–36]. A comprehensive survey of the scientific literature on prospective VS campaigns has also been published, analysing a total of 322 SBVS campaigns [37]. Out of these, homology models were successfully utilised in a total of 73 studies. Surprisingly, the potency of the hits identified using homology models was on average higher than for hits identified by docking into X-ray structures.
The selection of the most suitable model for docking from a pool of generated models remains a problem. Notably, there is only a weak correlation between global model quality parameters and docking success [15,34,38], indicating that the discrepancy between global and local structural accuracy cannot be adequately expressed by these measures. One fundamental reason is that protein flexibility, and in particular the adaptation of the ligand binding cavity to the bound ligand, can have a more significant impact on docking success than relatively small global modelling errors. Moreover, binding site residues generally exhibit a higher degree of conservation than the rest of a structure [39]. As a result, fairly accurate binding site models can be obtained from models with relatively poor overall quality [14,40].
There is a notion that models built on templates with sequence identity >50% are accurate enough for SBVS; however, this rule should be treated with caution. In a large-scale benchmarking study on 38 targets, Fan et al. [15] showed that the most enriching single models generally outperformed the apo X-ray structures, and even performed comparably to the holo X-ray structures. Docking into multiple models combined with consensus scoring further increased the enrichment rates, and was comparable to using the holo structure. This shows that the success of SBVS campaigns is intimately linked to adequate treatment of binding site flexibility. Approaches for handling protein flexibility in docking have been reviewed recently [41,42], and include docking to an ensemble of structures (ensemble docking) and ligand-guided homology modelling described above. Recently, basic guidelines for selecting the best binding site for docking from an ensemble of X-ray structures have been published [43], and it will be interesting to examine whether such rules could be conferred to homology models.
Applications
Homology modelling has been applied to various areas of drug discovery including structure-related aspects of target validation such as site-directed mutagenesis and druggability assessment [44,45]. This review is confined to exemplifying recent progress in ligand-associated protein modelling, namely SBVS, and mapping of protein–ligand interactions and mutations. Many compelling studies involving diverse intricate proteins have been published recently, and cannot be appreciated here in detail but deserve attention. These include, for example, antibodies [46], gamma-aminobutyric acid (GABA)-A receptor [47], ion channels [48], cystic fibrosis transmembrane conductance regulator (CFTR) [49] and epigenetic targets such as DNA methyltransferases (DNMTs) [50], histone deacetylases (HDACs), sirtuins and HMTs [51].
G-protein-coupled receptors
G-protein-coupled receptors (GPCRs) represent the most prominent target class against which almost a third of all FDA-approved drugs are targeted [52]. Owing to the enormous difficulties of crystallising GCPRs, the era of GPCR structural biology began relatively late [53], with the first human GPCR structure, β2-adrenergic receptor (β2-AR), solved in 2007 [54]. At present, a total of 16 unique class A GPCR structures are available, and large efforts are being made to characterise more representative GPCR structures, including class B and C GPCRs [55]. Given that there are more than 800 GPCR genes in the human proteome, it is evident that there will be great demand for reliable structural models of GPCRs in forthcoming years.
GPCR structure prediction faces a number of challenges, inter alia the adoption of multiple conformations depending on their activation state [56] and interaction with different adaptor proteins that actuate multiple signalling pathways [57]. Whereas, for example, the differences between the orthosteric β2-AR binding pockets in its active and inactive state are small, the binding site volumes differ [58]. Related to this, subtle changes in chemical structure can turn agonists into antagonists, and vice versa [59]. By contrast, orthosteric GPCR binding sites represent deep cavities that are well buried between the transmembrane (TM) helices, and should therefore be particularly suitable for SBVS.
The usefulness of GPCR models in SBVS has been assessed in a number of publications: see, for example, a brief overview [58], and a description of an automated SBVS workflow [60]. Guidelines for building GPCR models to be used for docking have been worked out recently [61]. Although loop modelling, in spite of recent progress [62,63], is still a challenge, it has also been demonstrated that loop-less models can be successfully applied in SBVS [64]. In fact, carefully built models can even outperform X-ray crystal structures in docking performance, subject to selecting templates representing the correct activation state [65]. For a comprehensive discussion of retrospective and prospective SBVS campaigns with experimental and modelled GPCRs, including model building and VS approaches, incorporation of experimental data and results, we refer the reader to the extensive recent review by Kooistra et al. [66].
In a community-wide assessment it has been concluded that GPCR structures in complex with small molecules can be reliably predicted, and can ‘approach the level of accuracy observed in the experiment’, if templates exhibiting >35% sequence homology (in the TMD) are used [67]. Using this criterion, it is estimated that roughly 20% of nonolfactory class A GPCRs can be reliably modelled based on currently available structural data [55]. It is also likely that the recent discovery of a conserved network of noncovalent contacts between the TM helices [68] will aid the further development of GPCR homology models.
Whereas reasonable enrichments can be achieved with imprecise models in SBVS, the correct mapping of protein–ligand interactions in 3D models is a more ambitious task, because optimal accuracy is imperative. A recent example on the mapping of protein–ligand binding in H4R has conclusively established that this is feasible [69]. Homology models from various templates have been built, and a combination of docking and molecular dynamics (MD) has been employed. The determination of ligand-binding modes that could explain experimental data was complicated by a quasi-symmetric distribution of certain residues in the binding site, leading to different plausible configurations. Nevertheless, careful analysis of the ligand poses and pocket volumes in connection with the experimental data, consideration of ligand protonation states and, for one ligand class, quantum-mechanical evaluation of low-energy conformations [70] enabled the elucidation of binding models that could expound ligand-specific mutation effects and subtle experimental SAR data.
ADMET, cytochrome P450s
Managing the interplay of drug molecules with ADMET-relevant off-targets such as, for example, metabolic cytochrome P450 enzymes (CYPs), drug transporters or the hERG channel is paramount in lead-optimisation campaigns. The CYP gene family comprises 57 functional genes in man, and plays a major part in metabolising xenobiotics into more polar compounds with improved elimination [71]. In 2004, merely three CYP structures were available, which were intensively used for the homology modelling of various CYP subtypes [19]. Nowadays most CYP structures and many other ADMET-relevant ones have been solved, allowing structure-based methods in ADMET to be increasingly applied [72,73]. The impact of CYP homology models in ADMET prediction is therefore likely to attenuate; however, one should not conclude that an X-ray structure is necessarily the best possible representation of a particular structural state. A recent study for predicting the substrate site of metabolism (SOM) in CYP2D6 alludes to the drawbacks related to protein flexibility raised above. A model of CYP2D6 was generated based on published X-ray crystal structures of the substrate-bound CYP2C5 [74]. During the study, the structure of apo CYP2D6 also became available. Docking calculations were performed with the new structure and the homology model. Although the homology model was, overall, in good agreement with the CYP2D6 crystal structure, the model consistently outperformed the experimental structure; this has been attributed to structural differences in the substrate recognition sites (Figure 1).
Figure 1.
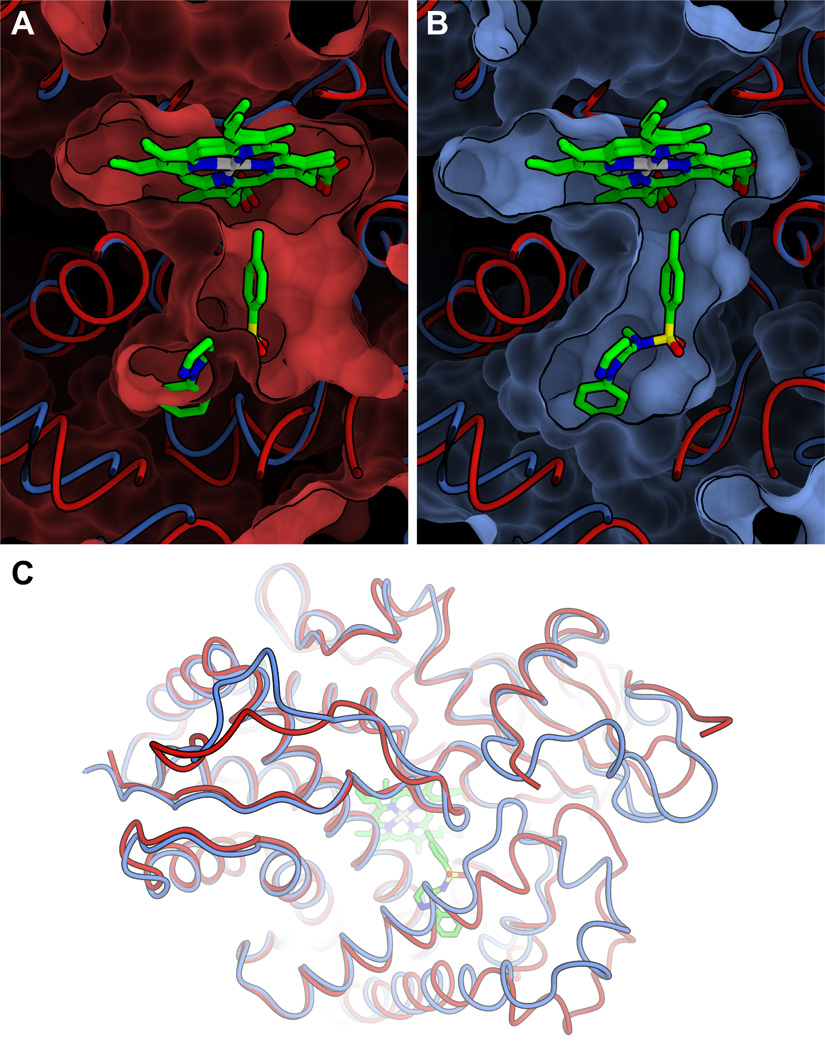
Selection of an appropriate structural template for model building is crucial for its successful application in structure-based virtual screening (SBVS), and often the most appropriate template is not necessarily the one with the highest sequence identity to the target protein. In a study by Unwalla et al. [74] docking calculations performed on the apo X-ray structure of cytochrome P450 (CYP)2D6 were consistently outperformed by a model of CYP2D6, generated based on the substrate-bound structure of CYP2C5. A superposition of the CYP2D6 apo structure (red) (PDB: 2F9Q) and the CYP2C5 holo structure (blue) (PDB: 1N6B) is shown here. Panels a and b display the binding site surfaces of apo CYP2D6 and holo CYP2C5, respectively. Panel a highlights the fact that the binding pocket observed in the apo structure is too small to accommodate the ligand. Panel c shows backbone structural variations close to the binding site. Graphics were produced with OpenStructure [105,106].
Several CYPs, including CYP2D6, appear in numerous polymorphic forms. These inter-individual variations result in large differences for drug clearance and clinical response of patients [10], delineating ADMET as a highly individual-specific phenomenon. Currently, more than 90 2D6 alleles are known, corresponding to roughly 30 non-synonymous protein-coding single nucleotide polymorphisms (nsSNPs). Amino acid residues corresponding to these SNPs are distributed across the whole CYP structure; however, distinct clusters in proximity to the haem cofactor or the substrate access channel could be found [10]. It is evident that structural modelling of these variants can facilitate the interpretation of SNP-specific genotype–phenotype relations. This approach can be seamlessly extended to examine variations across different species. Such ortholog models have, for example, been derived for Macaca CYPs [75] and various estrogen receptors [76], and can assist the selection of appropriate models for pharmacological studies. It can be hoped that, ultimately, and with the advance of NGS, such models will enable the prediction of individual and species-specific drug responses, thus truly promoting personalised medicine and reducing animal sacrifice.
Multidrug resistance protein 1
Multidrug resistance protein 1 (MDR1), also known as P-glycoprotein (P-gp) or ABCB1, is a membrane-bound ATP-driven efflux pump that belongs to the family of ABC transporters. MDR1 is a promiscuous transporter that extrudes numerous hydrophobic compounds and drugs as its substrates from cells. MDR1-mediated drug efflux thus impairs drug delivery, and hence plays a key part in ADMET [77]. In many cancer cell types, upregulation of MDR1 activity causes fast removal of drugs, triggering multidrug resistance [78]. Hence, the interplay of MDR1 with its substrates, modulators and inhibitors is crucial in different areas of drug discovery. Homology modelling of MDR1 had been hampered by the low sequence identity and even structural errors in the available templates [79,80]. Even with the murine structure [81] as a template with 87% homology to the human protein there are many obstacles, which can probably be attributed to the huge polyspecific ligand-binding site composed of several subsites, the low resolution of the template structures and the large dynamic rearrangements that occur during the transport cycle [82], see also the review by Ravna and Sylte [83] on homology modelling of transporters. Despite these complications, an intriguing study establishing detailed binding hypotheses for known MDR1 inhibitors (profanone derivatives) has been published recently [84]. To capture the different catalytic states of the transport cycle, 100 models were generated for each of the apo (open-inward) and the nucleotide-bound (open-outward) states; the templates used reflected the various states. The best models were selected using the MOE (Chemical Computing Group, Montreal, QC) geometry check functions and validated with different quality estimation methods. Exhaustive docking of five selected propafenone ligands was performed using the entire TM area as the binding site. Post-processing and analysis was accomplished using protein–ligand interaction fingerprints, common scaffold clustering and incorporation of SAR knowledge. This SAR-guided docking protocol led to the selection of binding hypotheses that convincingly concur with experimental data and previous pharmacophore studies. Other recent stimulating studies using MDR1 homology models include an explanation of enhanced substrate bioavailability [85] and modelling of catalytic transitions based on targeted MD [86].
hERG
The hERG K+ channel represents one of the primary ADMET-related anti-targets, because undesired binding of drugs can lead to life-threatening arrhythmias and sudden death [87]. In spite of major efforts hERG homology models have been notorious for being descriptive at best, and not predictive for applications in drug discovery [72]. Clearly, segments in the hERG sequence with very low sequence identity to all possible templates are a major impediment of hERG modelling. An analysis of seven published hERG models with inconsistent alignments in the S5 sectors emphasises the crucial importance of sound alignments [88]. The analysis enabled the selection of a model that exhibited good quality criteria, and conformed best to experimental data. It also stressed the potentially detrimental impact of alignment errors in the drug binding site, even in segments remote from drug interactions. Further progress in hERG channel modelling concerns the elucidation of channel blocker trapping. Open- and closed-state homology models in combination with docking of propafenone derivatives have been reported [89]. In another study, 12 blockers with known activity along with mutagenesis data were used for validating open- and closed-state models, also using docking [90]. It was concluded that the models were reliable enough for explaining inhibitor binding and for identifying strong binders; however, 3D pharmacophores and QSAR methods still remain more predictive than the 3D models.
Concluding remarks and future outlook
With the tremendous growth of available structural data one could naively assume that the need for protein structure prediction would decline. On the contrary, we anticipate the opposite. As discussed above, the availability of an experimental structure does not necessarily imply that this structure represents the best starting point for CADD studies. Although, in general, an experimental structure will undoubtedly be better than a predicted model, this should not be presupposed axiomatically. Because structures can vary depending on their functional state and form (apo vs holo form; agonist (active) vs antagonist (inactive) state), their applicability for CADD should always be appraised in their functional context.
Another aspect that should not be underestimated is the rapid availability of models, particularly in the light of advanced fully automated modelling pipelines. In lead optimisation, X-ray crystal structures of relevant lead compound complexes are undoubtedly always desirable; however, a good structural model can be instrumental and guide research a long time before the experimental structures become available.
Most importantly, the availability of new structures opens avenues for reliably modelling many more structures than previously possible, as shown for GPCRs and MDRs. Because it cannot be expected that experimental structures will ever be available for all structures of interest, it is probable that the impact of protein structure prediction will even increase for such target classes in drug discovery. As new experimental structures take over the arenas that have been dominated by predicted models, protein modelling will utilise those as seeds for pioneering new areas of structural relevance. The example CYP polymorphism in context with personalised medicine described above supports this optimism. Increasing reliability of fully automated modelling pipelines allows models to be generated on a proteomic scale, as for the structural characterisation of the entire human kinome [91]. Another enthralling area is the prediction of protein–protein complexes on an omics scale [5,92,93]. It remains to be seen whether or not this enterprise will become conducive to drug discovery and when, for example in the design of protein–protein interaction inhibitors.
To summarize, the recent literature endorses the supposition that accurate homology modelling suitable for many drug discovery applications can be achieved (Box 1). We trust that the co-action of newly solved X-ray crystal structures, constantly growing number of sequences, improved modelling techniques and quality assessment methods will furnish powerful homology modelling techniques to advance the complex structural challenges further in drug discovery.
Box 1. Tips to remember and questions to address when applying homology modelling and hints for best practice.
| Template selection |
|
| Sequence alignment |
|
| Modelling |
|
| Refinement |
|
| Validation |
|
| Application |
|
Highlights.
The majority of proteins encoded in a genome are accessible by structure modelling
Modelling can provide accurate target predictions for structure-based drug design
Models can capture crucial receptor flexibility in a protein family beyond apo structures
Ligand steered modelling methods often outperform docking into static models
Automated modelling techniques have matured to provide accurate models
Acknowledgements
The authors would like to thank Karen J. Bergner for reviewing the manuscript and for help with the English.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berman HM, et al. The future of the protein data bank. Biopolymers. 2013;99:218–222. doi: 10.1002/bip.22132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwede T, et al. Outcome of a workshop on applications of protein models in biomedical research. Structure. 2009;17:151–159. doi: 10.1016/j.str.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwede T. Protein modeling: what happened to the "protein structure gap"? Structure. 2013;21:1531–1540. doi: 10.1016/j.str.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Donoghue SI, et al. Visualization of macromolecular structures. Nat. Methods. 2010;7(Suppl. 3):42–55. doi: 10.1038/nmeth.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vakser IA. Low-resolution structural modeling of protein interactome. Curr. Opin. Struct. Biol. 2013;23:198–205. doi: 10.1016/j.sbi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joosten RP, et al. Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank. Bioinformatics. 2011;27:3392–3398. doi: 10.1093/bioinformatics/btr590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergner A, et al. Use of Relibase for retrieving complex three-dimensional interaction patterns including crystallographic packing effects. Biopolymers. 2001;61:99–110. doi: 10.1002/1097-0282(2001/2002)61:2<99::AID-BIP10075>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Cozzini P, et al. Target flexibility: an emerging consideration in drug discovery and design. J. Med. Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durrant JD, McCammon JA. Computer-aided drug-discovery techniques that account for receptor flexibility. Curr. Opin. Pharmacol. 2010;10:770–774. doi: 10.1016/j.coph.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullins JG. Structural modelling pipelines in next generation sequencing projects. Adv. Protein Chem. Struct. Biol. 2012;89:117–167. doi: 10.1016/B978-0-12-394287-6.00005-7. [DOI] [PubMed] [Google Scholar]
- 11.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 12.Cavasotto CN, Phatak SS. Homology modeling in drug discovery: current trends and applications. Drug Discov. Today. 2009;14:676–683. doi: 10.1016/j.drudis.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariani V, et al. Assessment of template based protein structure predictions in CASP9. Proteins. 2011;79(Suppl. 10):37–58. doi: 10.1002/prot.23177. [DOI] [PubMed] [Google Scholar]
- 15.Fan H, et al. Molecular docking screens using comparative models of proteins. J. Chem. Inf. Model. 2009;49:2512–2527. doi: 10.1021/ci9003706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacCallum JL, et al. Assessment of protein structure refinement in CASP9. Proteins. 2011;79(Suppl. 10):74–90. doi: 10.1002/prot.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavasotto CN. Homology models in docking and high-throughput docking. Curr. Top. Med. Chem. 2011;11:1528–1534. doi: 10.2174/156802611795860951. [DOI] [PubMed] [Google Scholar]
- 18.Jacobson M, Sali A. Annual Reports in Medicinal Chemistry. Vol. 39. Academic Press; 2004. Comparative protein structure modeling and its applications to drug discovery; pp. 259–276. [Google Scholar]
- 19.Hillisch A, et al. Utility of homology models in the drug discovery process. Drug Discov. Today. 2004;9:659–669. doi: 10.1016/S1359-6446(04)03196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evers A, et al. Ligand-supported homology modelling of protein binding-sites using knowledge-based potentials. J. Mol. Biol. 2003;334:327–345. doi: 10.1016/j.jmb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 21.Dalton JA, Jackson RM. Homology-modelling protein-ligand interactions: allowing for ligand-induced conformational change. J. Mol. Biol. 2010;399:645–661. doi: 10.1016/j.jmb.2010.04.047. [DOI] [PubMed] [Google Scholar]
- 22.Cavasotto CN, et al. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 23.Katritch V, et al. Ligand-guided receptor optimization. Methods Mol .Biol. 2012;857:189–205. doi: 10.1007/978-1-61779-588-6_8. [DOI] [PubMed] [Google Scholar]
- 24.Rueda M, et al. ALiBERO: evolving a team of complementary pocket conformations rather than a single leader. J. Chem. Inf. Model. 2012;52:2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kryshtafovych A, et al. Assessment of the assessment: Evaluation of the model quality estimates in CASP10. Proteins. 2013 doi: 10.1002/prot.24347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benkert P, et al. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsson P, et al. Assessment of global and local model quality in CASP8 using Pcons and ProQ. Proteins. 2009;77(Suppl. 9):167–172. doi: 10.1002/prot.22476. [DOI] [PubMed] [Google Scholar]
- 28.McGuffin LJ, et al. The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res. 2013;41:W368–f372. doi: 10.1093/nar/gkt294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langer T, et al. Hit finding: towards 'smarter' approaches. Curr. Opin. Pharmacol. 2009;9:589–593. doi: 10.1016/j.coph.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Klebe G. Virtual ligand screening: strategies, perspectives and limitations. Drug Discov. Today. 2006;11:580–594. doi: 10.1016/j.drudis.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waszkowycz B. Structure-based approaches to drug design and virtual screening. Curr. Opin. Drug Discov. Devel. 2002;5:407–413. [PubMed] [Google Scholar]
- 32.Cheng T, et al. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012;14:133–141. doi: 10.1208/s12248-012-9322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oshiro C, et al. Performance of 3D-database molecular docking studies into homology models. J. Med. Chem. 2004;47:764–767. doi: 10.1021/jm0300781. [DOI] [PubMed] [Google Scholar]
- 34.Kairys V, et al. Screening drug-like compounds by docking to homology models: a systematic study. J. Chem. Inf. Model. 2006;46:365–379. doi: 10.1021/ci050238c. [DOI] [PubMed] [Google Scholar]
- 35.Fernandes MX, et al. Comparing ligand interactions with multiple receptors via serial docking. J. Chem. Inf. Comput. Sci. 2004;44:1961–1970. doi: 10.1021/ci049803m. [DOI] [PubMed] [Google Scholar]
- 36.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 37.Ripphausen P, et al. Quo vadis, virtual screening? A comprehensive survey of prospective applications. J. Med. Chem. 2010;53:8461–8467. doi: 10.1021/jm101020z. [DOI] [PubMed] [Google Scholar]
- 38.Ferrara P, Jacoby E. Evaluation of the utility of homology models in high throughput docking. J. Mol. Model. 2007;13:897–905. doi: 10.1007/s00894-007-0207-6. [DOI] [PubMed] [Google Scholar]
- 39.Novoa EM, et al. Ensemble docking from homology models. J. Chem. Theory Comput. 2010;6:2547–2557. doi: 10.1021/ct100246y. [DOI] [PubMed] [Google Scholar]
- 40.Thorsteinsdottir HB, et al. How inaccuracies in protein structure models affect estimates of protein-ligand interactions: computational analysis of HIV-I protease inhibitor binding. Proteins. 2006;65:407–423. doi: 10.1002/prot.21096. [DOI] [PubMed] [Google Scholar]
- 41.B-Rao C, et al. Managing protein flexibility in docking and its applications. Drug Discov. Today. 2009;14:394–400. doi: 10.1016/j.drudis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Beier C, Zacharias M. Tackling the challenges posed by target flexibility in drug design. Expert Opin. Drug Discov. 2010;5:347–359. doi: 10.1517/17460441003713462. [DOI] [PubMed] [Google Scholar]
- 43.Ben Nasr N, et al. Multiple structures for virtual ligand screening: defining binding site properties-based criteria to optimize the selection of the query. J. Chem. Inf. Model. 2013;53:293–311. doi: 10.1021/ci3004557. [DOI] [PubMed] [Google Scholar]
- 44.Fauman EB, et al. Structure-based druggability assessment--identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 2011;15:463–468. doi: 10.1016/j.cbpa.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 45.Nisius B, et al. Structure-based computational analysis of protein binding sites for function and druggability prediction. J. Biotechnol. 2012;159:123–134. doi: 10.1016/j.jbiotec.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Kuroda D, et al. Computer-aided antibody design. Protein Eng. Des. Sel. 2012;25:507–521. doi: 10.1093/protein/gzs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergmann R, et al. A unified model of the GABA(A) receptor comprising agonist and benzodiazepine binding sites. PLoS One. 2013;8:e52323. doi: 10.1371/journal.pone.0052323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maffeo C, et al. Modeling and simulation of ion channels. Chem. Rev. 2012;112:6250–6284. doi: 10.1021/cr3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalton J, et al. New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J. Chem. Inf. Model. 2012;52:1842–1853. doi: 10.1021/ci2005884. [DOI] [PubMed] [Google Scholar]
- 50.Medina-Franco JL, Caulfield T. Advances in the computational development of DNA methyltransferase inhibitors. Drug Discov. Today. 2011;16:418–425. doi: 10.1016/j.drudis.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Heinke R, et al. Computer- and structure-based lead design for epigenetic targets. Bioorg. Med. Chem. 2011;19:3605–3615. doi: 10.1016/j.bmc.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 52.Overington JP, et al. How many drug targets are there? Nat. Rev. Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 53.Granier S, Kobilka B. A new era of GPCR structural and chemical biology. Nat. Chem. Biol. 2012;8:670–673. doi: 10.1038/nchembio.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rasmussen SG, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 55.Stevens RC, et al. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov. 2013;12:25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abrol R, et al. Characterizing and predicting the functional and conformational diversity of seven-transmembrane proteins. Methods. 2011;55:405–414. doi: 10.1016/j.ymeth.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajagopal S, et al. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujioka M, Omori N. Subtleties in GPCR drug discovery: a medicinal chemistry perspective. Drug Discov. Today. 2012;17:1133–1138. doi: 10.1016/j.drudis.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 60.Tautermann CS. Target based virtual screening by docking into automatically generated GPCR models. Methods Mol. Biol. 2012;914:255–270. doi: 10.1007/978-1-62703-023-6_15. [DOI] [PubMed] [Google Scholar]
- 61.Beuming T, Sherman W. Current assessment of docking into GPCR crystal structures and homology models: successes, challenges, and guidelines. J. Chem. Inf. Model. 2012;52:3263–3277. doi: 10.1021/ci300411b. [DOI] [PubMed] [Google Scholar]
- 62.Nikiforovich GV, et al. Modeling the possible conformations of the extracellular loops in G-protein-coupled receptors. Proteins. 2010;78:271–285. doi: 10.1002/prot.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldfeld DA, et al. Loop prediction for a GPCR homology model: algorithms and results. Proteins. 2013;81:214–228. doi: 10.1002/prot.24178. [DOI] [PubMed] [Google Scholar]
- 64.de Graaf C, et al. Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins. 2008;71:599–620. doi: 10.1002/prot.21724. [DOI] [PubMed] [Google Scholar]
- 65.Tang H, et al. Do crystal structures obviate the need for theoretical models of GPCRs for structure-based virtual screening? Proteins. 2012;80:1503–1521. doi: 10.1002/prot.24035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kooistra AJ, et al. From heptahelical bundle to hits from the Haystack: structure-based virtual screening for GPCR ligands. Methods Enzymol. 2013;522:279–336. doi: 10.1016/B978-0-12-407865-9.00015-7. [DOI] [PubMed] [Google Scholar]
- 67.Kufareva I, et al. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 69.Schultes S, et al. Mapping histamine H4 receptor-ligand binding modes. Med. Chem. Commun. 2013;4:193–204. [Google Scholar]
- 70.Schultes S, et al. Mapping histamine H4 receptor-ligand binding modes. ChemMedChem. 2013;8:49–53. doi: 10.1002/cmdc.201200412. [DOI] [PubMed] [Google Scholar]
- 71.Dong D, et al. Substrate selectivity of drug-metabolizing cytochrome P450s predicted from crystal structures and in silico modeling. Drug Metab. Rev. 2012;44:192–208. doi: 10.3109/03602532.2011.645580. [DOI] [PubMed] [Google Scholar]
- 72.Stoll F, et al. Utility of protein structures in overcoming ADMET-related issues of drug-like compounds. Drug Discov. Today. 2011;16:530–538. doi: 10.1016/j.drudis.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 73.Moroy G, et al. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today. 2012;17:44–55. doi: 10.1016/j.drudis.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 74.Unwalla RJ, et al. Using a homology model of cytochrome P450 2D6 to predict substrate site of metabolism. J. Comput. Aided Mol. Des. 2010;24:237–256. doi: 10.1007/s10822-010-9336-6. [DOI] [PubMed] [Google Scholar]
- 75.Rua F, et al. Toward reduction in animal sacrifice for drugs: molecular modeling of Macaca fascicularis P450 2C20 for virtual screening of Homo sapiens P450 2C8 substrates. Biotechnol. Appl. Biochem. 2012;59:479–489. doi: 10.1002/bab.1051. [DOI] [PubMed] [Google Scholar]
- 76.Toschi L, et al. Protein-structure-based prediction of animal model suitability for pharmacodynamic studies of subtype-selective estrogens. ChemMedChem. 2006;1:1237–1248. doi: 10.1002/cmdc.200600183. [DOI] [PubMed] [Google Scholar]
- 77.Szakacs G, et al. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox) Drug Discov. Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 78.Gottesman MM, et al. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 79.Demel MA, et al. Predicting ligand interactions with ABC transporters in ADME. Chem. Biodivers. 2009;6:1960–1969. doi: 10.1002/cbdv.200900138. [DOI] [PubMed] [Google Scholar]
- 80.Ecker GF, et al. Computational models for prediction of interactions with ABC-transporters. Drug Discov. Today. 2008;13:311–317. doi: 10.1016/j.drudis.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 81.Aller SG, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen L, et al. Computational models for predicting substrates or inhibitors of P-glycoprotein. Drug Discov. Today. 2012;17:343–351. doi: 10.1016/j.drudis.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 83.Ravna AW, Sylte I. Homology modeling of transporter proteins (carriers and ion channels) Methods Mol. Biol. 2012;857:281–299. doi: 10.1007/978-1-61779-588-6_12. [DOI] [PubMed] [Google Scholar]
- 84.Klepsch F, et al. Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comput. Biol. 2011;7:e1002036. doi: 10.1371/journal.pcbi.1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singh DV, et al. A plausible explanation for enhanced bioavailability of P-gp substrates in presence of piperine: simulation for next generation of P-gp inhibitors. J. Mol. Model. 2013;19:227–238. doi: 10.1007/s00894-012-1535-8. [DOI] [PubMed] [Google Scholar]
- 86.Wise JG. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry. 2012;51:5125–5141. doi: 10.1021/bi300299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 88.Stary A, et al. Toward a consensus model of the HERG potassium channel. ChemMedChem. 2010;5:455–467. doi: 10.1002/cmdc.200900461. [DOI] [PubMed] [Google Scholar]
- 89.Thai KM, et al. The hERG potassium channel and drug trapping: insight from docking studies with propafenone derivatives. ChemMedChem. 2010;5:436–442. doi: 10.1002/cmdc.200900374. [DOI] [PubMed] [Google Scholar]
- 90.Du-Cuny L, et al. A critical assessment of combined ligand- and structure-based approaches to HERG channel blocker modeling. J. Chem. Inf. Model. 2011;51:2948–2960. doi: 10.1021/ci200271d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brylinski M, Skolnick J. Comprehensive structural and functional characterization of the human kinome by protein structure modeling and ligand virtual screening. J. Chem. Inf. Model. 2010;50:1839–1854. doi: 10.1021/ci100235n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang QC, et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mosca R, et al. Interactome3D: adding structural details to protein networks. Nat. Methods. 2013;10:47–53. doi: 10.1038/nmeth.2289. [DOI] [PubMed] [Google Scholar]
- 94.Arnold K, et al. The protein model portal. J. Struct. Funct. Genomics. 2009;10:1–8. doi: 10.1007/s10969-008-9048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hildebrand A, et al. Fast and accurate automatic structure prediction with HHpred. Proteins. 2009;77(Suppl. 9):128–132. doi: 10.1002/prot.22499. [DOI] [PubMed] [Google Scholar]
- 96.Abagyan RA, et al. ICM - a new method for protein modeling and design. Applications to docking and structure prediction from the distorted native conformation. J. Comp. Chem. 1994;15:488–506. [Google Scholar]
- 97.McGuffin LJ, Roche DB. Automated tertiary structure prediction with accurate local model quality assessment using the IntFOLD-TS method. Proteins. 2011;79(Suppl. 10):137–146. doi: 10.1002/prot.23120. [DOI] [PubMed] [Google Scholar]
- 98.Eswar N, et al. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 99.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 100.Raman S, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl. 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bordoli L, et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 102.Guex N, et al. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis. 2009;30(Suppl. 1):162–173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 103.Kleywegt GJ, et al. The Uppsala Electron-Density Server. Acta CrystallogrDBiol. Crystallogr. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 104.Emsley P, et al. Features and development of Coot. Acta CrystallogrDBiol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Biasini M, et al. OpenStructure: a flexible software framework for computational structural biology. Bioinformatics. 2010;26:2626–2628. doi: 10.1093/bioinformatics/btq481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Biasini M, et al. OpenStructure: an integrated software framework for computational structural biology. Acta CrystallogrDBiol. Crystallogr. 2013;69:701–709. doi: 10.1107/S0907444913007051. [DOI] [PMC free article] [PubMed] [Google Scholar]