

Abstract
Purpose.
Oxidative stress in the RPE is widely accepted as a contributing factor to AMD. We have previously shown that ribozyme-mediated reduction in the antioxidant enzyme manganese superoxide dismutase (MnSOD) leads to some of the features of geographic atrophy in mice. To develop a mouse model independent of viral injection, we used a conditional knockout of the Sod2 gene in the RPE to elevate mitochondrial oxidative stress in that cell layer.
Methods.
Experimental mice in which exon 3 of Sod2 was flanked by loxP sites were also transgenic for PVMD2-rtTA and tetO-PhCMV cre, so that cre recombinase was expressed only in the RPE. Pups of this genotype (Sod2flox/floxVMD2cre) were induced to express cre recombinase by feeding doxycycline-laced chow to nursing dams. Controls included mice of this genotype not treated with doxycycline and doxycycline-treated Sod2flox/flox mice lacking the cre transgene. Expression of cre in the RPE was verified by immunohistochemistry, and deletion of Sod2 exon 3 in the RPE was confirmed by PCR. Mice were followed up over a period of 9 months by spectral-domain optical coherence tomography (SD-OCT), digital fundus imaging, and full-field ERG. Following euthanasia, retinas were examined by light and electron microscopy or by immunohistochemistry. Contour length of rod outer segments and thickness of the RPE layer were measured by unbiased stereology.
Results.
Following doxycycline induction of cre, Sod2flox/flox cre mice demonstrated increased signs of oxidative stress in the RPE and accumulation of autofluorescent material by age 2 months. They showed a gradual decline in the ERG response and thinning of the outer nuclear layer (by SD-OCT), which were statistically significant by 6 months. In addition, OCT and electron microscopy revealed increased porosity of the choroid. At the same interval, hypopigmented foci appeared in fundus micrographs, and vascular abnormalities were detected by fluorescein angiography. By 9 months, the RPE layer in Sod2flox/flox cre mice was thicker than in nontransgenic littermates, and the rod outer segments were significantly longer over most of the retina, although localized atrophy of photoreceptors was also obvious in some eyes.
Conclusions.
Conditional tissue-specific reduction in MnSOD induced oxidative stress in mouse RPE, leading to RPE dysfunction, damage to the choroid, and death of photoreceptor cells. The RPE oxidative stress did not cause drusen-like deposits, but the model recapitulated certain key aspects of the pathology of dry AMD and may be useful in testing therapies.
Keywords: retinal degeneration, mouse model, reactive oxygen species, manganese superoxide dismutase, retinal pigment epithelium
The RPE-specific deletion of mitochondrial superoxide dismutase in mice leads to oxidative stress in the RPE, accumulation of autofluorescent material, a decline in the ERG response, and localized death of photoreceptors. This model may be useful in understanding the mechanism of geographic atrophy.
Introduction
Damage to tissue from reactive oxygen species and reactive nitrogen species, collectively termed oxidative stress, is implicated in degenerative diseases affecting the cardiovascular system1 and the central nervous system.2 In the eye, oxidative stress is a contributing factor to age-related conditions, including dry eye, cataract, glaucoma, and AMD.3–9 Evidence supporting the role of oxidative stress in AMD comes from both epidemiology and biochemistry. Smoking, a rich source of reactive oxygen species, is the major environmental risk factor for the development of AMD,10–13 and the Age-Related Eye Disease Study14 has indicated that the use of antioxidant vitamins plus zinc can retard the progression of intermediate-stage AMD to advanced stages. The oxidation of polyunsaturated fats in the retina leads to lipid peroxidation products such as carboxyethylpyrrole (CEP) and 4-hydroxy-2-nonenal (4-HNE), which can form adducts with proteins and accumulate in the outer retina and in drusen.15 The levels of antioxidant proteins such as catalase and superoxide dismutase are increased in RPE homogenates derived from late-stage AMD eyes, indicating a response to increased oxidative stress in these eyes.16
The RPE is thought to be a critical site of injury in AMD.9,17–19 The first clinical signs of early AMD are lipid-rich sub-RPE deposits known as drusen, which contain a variety of proteins, including vitronectin, components of the terminal complement cascade, and β-amyloid.20,21 Microscopic analysis of eyes donated by patients with AMD reveals lipid deposits within Bruch's membrane and apoptosis of RPE cells as features of the disease distinct from normal aging. The advanced form of dry AMD, which leads to vision loss, is termed geographic atrophy and is characterized by the breakdown of the RPE, choriocapillaris, and photoreceptors in regions of the retina, often where large drusen had been present.22 The phagocytosis of oxidized fatty acids from photoreceptor outer segments is certainly a contributing factor to the metabolic failure of RPE cells.23–25 In addition, the damage done by the free radical by-products of mitochondrial energy metabolism is implicated in age-related damage to the RPE.26 The mitochondrial electron transport chain generates superoxide radical through single-electron leak at respiratory complex I and III,27 but flavin-dependent enzymes in the mitochondrial matrix may be larger contributors of reactive oxygen species.28 Superoxide can directly damage mitochondrial DNA, proteins, and lipids and can also be converted to hydrogen peroxide by manganese superoxide dismutase (MnSOD encoded by Sod2) in the organelle. In primary RPE cells isolated from older donors, mitochondria are more elongated but less numerous than in eyes donated by younger people, and mitochondrial function is impaired as revealed by reduced membrane potential.29 Damage to mitochondria is qualitatively different in AMD than in normal aging as revealed by analysis of the mitochondrial proteome30 and the distribution of mitochondrial mutations in advanced stages of AMD.31 Sequence polymorphisms that comprise certain mitochondrial DNA haplogroups appear to confer increased or decreased risk of developing AMD.32
Animal models have been used to understand AMD pathogenesis and to develop treatments for this disease.33,34 Some of the most valuable of these models bear the least resemblance to physiology of AMD. For example, laser disruption of the RPE and Bruch's membrane has been used to model exudative AMD and is translatable to primate models. Laser injury served as the preclinical test system for the development of VEGF inhibitors now in widespread clinical application. Light injury in albino mice and rats is a useful model of photoreceptor/RPE injury and of oxidative stress in the retina.35 Mice, of course, lack a macula and a cone-rich central retina, but their genetic malleability has permitted the generation of models of retinal degeneration affecting the cellular immune response,36 the alternative complement system,37–39 and the inflammatory response to double-stranded RNA.40,41 Models of retinal degeneration that may have physiological relevance to AMD include mice bearing risk-associated variants of complement factor H,42 the ceruloplasmin/hephaestin–knockout model of iron overload,43 and ApoE4-knockin mice fed a high-fat/high-cholesterol diet.44 Mice immunized with CEP-modified albumin show severe damage to the RPE and Bruch's membrane, as well as death of photoreceptor cells.15 Deletion of the mitochondrial transcription factor Tfam in the RPE leads to dedifferentiation and atrophy of this cell layer and eventually death of the associated photoreceptors, a phenomenon that could be prevented by treatment with rapamycin.45 Hu and colleagues46 described a model based on the deletion of the aryl hydrocarbon receptor in which mice showed disruption of RPE tight junctions and accumulation of basal linear deposits and basal laminar deposits. These mice show choroidal thinning by age 11 months, and frank RPE atrophy is observed by 16 months.
Some mouse models focus on the impact of increased oxidative stress on the retina. Mice deficient in copper/zinc superoxide dismutase because of a homozygous deletion of the Sod1 gene develop thickened Bruch's membrane, drusenoid deposits, and in some cases choroidal neovascularization.47 Nrf2 is a transcription factor that regulates the response to environmental stress, and knockout mice develop age-dependent degeneration of the RPE, formation of drusen-like deposits, and choroidal neovascularization.48 We have used the tools of gene therapy in mice to test the hypothesis that mitochondrial oxidative stress contributes to AMD-like pathology. Adeno-associated virus (AAV) was used to deliver a ribozyme that cleaved the mRNA for MnSOD specifically in the RPE.49,50 We observed increased levels of nitrated and CEP-modified proteins in the RPE-choroid of mice, as well as a reduction in the ERG response and in the thickness of the outer nuclear layer (ONL) as revealed by morphometry and spectral-domain optical coherence tomography (SD-OCT), which also showed subretinal and sub-RPE deposits. Electron microscopic examination demonstrated vacuolization, thickening of the RPE and disorganization of Bruch's membrane, and shortening and disorganization of the photoreceptor outer and inner segments. The MnSOD depletion led to increased autofluorescence and elevated levels of bis-retinoid pigments. The Sod2-knockdown model has been useful in testing antioxidant pharmacological therapy to prevent retinal atrophy. Thampi et al.51 showed that systemic delivery of a 5-hydroxytryptamine 1a receptor agonist preserved ONL thickness and reduced lipofuscin accumulation in this model.
The AAV-knockdown–induced model of retinal atrophy has the advantage of being applicable to larger animals with cone-rich central retinas or even to primates with a macula. Furthermore, knockdown of MnSOD in one eye leaves a control eye in the same animal for comparison. The AAV-Sod2 ribozyme model has some disadvantages, however. For one, it is relatively aggressive: mice develop a statistically significant decrease in ERG amplitudes by 3 months after injection. In addition, subretinal injection leads to a variable extent of retinal transduction, and the retina does not reattach to the RPE in some eyes. Because germline disruption of Sod2 leads to neonatal fatality,52 we have turned to the cre/lox system to achieve RPE-specific deletion of Sod2. We crossed mice doubly transgenic for cre recombinase under the control of the tetracycline operator and for the tetracycline transactivator under the control of an RPE-specific promoter with mice in which exon 3 of the Sod2 gene was flanked with loxP sites. Following induction of cre in neonates, we monitored retinal degeneration by fundus imaging, ERG, and SD-OCT in the same cohort of mice for 9 months. We report age-dependent geographic atrophy that recapitulates some of the features of dry AMD and of our AAV-ribozyme–induced model.
Methods
Mouse Strains and Induction of Cre
To generate an RPE-specific MnSOD (Sod2)–deletion mouse, we used a tetracycline (tet)-on system for cre-mediated recombination. The inducible RPE-specific cre mice carried 3-kb human VMD2 gene promoter, which directs the expression of tetracycline-inducible transactivator gene rtTA.53 This gene was inserted at the same chromosomal locus as a transgene expressing cre from the tetO-PhCMV chimeric promoter. Mice with loxP sites flanking exon 3 of Sod2 (Sod2-floxed mice) were originally used to study the role of mitochondrial oxidative stress in the heart.54 Both parental strains were backcrossed a minimum of six generations to C57Bl/6J mice, and breeders were monitored for the absence of the rd1 and rd8 alleles. We bred tet-on VMD2-cre mice with Sod2-floxed mice to generate mice homozygous for the floxed Sod2 gene and hemizygous for the tetracycline transactivator/cre transgenes (Sod2 flox/flox VMD2-cre mice). Expression of the cre transgene was induced in neonatal mice by feeding nursing dams doxycycline-containing chow (200 mg/kg rodent diet [T-7012, 200 doxycycline]; Harlan Laboratories, Inc., Tampa, FL, USA) for 2 weeks immediately after birth of the litter.
To analyze induction of cre recombinase in the RPE, ROSA26-lacZ–reporter mice on a C57Bl/6J background (003474; The Jackson Laboratory, Bar Harbor, ME, USA) were bred with VMD2-cre mice to generate ROSA26-lacZ, VMD2-cre mice. In ROSA26-reporter mice, a loxP flanked cassette prevents the lacZ expression in the absence of cre. However, with the induction cre by doxycycline, ROSA26VMD2-cre mice expressed lacZ and produced Escherichia coli β-galactosidase. Genotyping was performed on tail-tip biopsy specimens using the primers and conditions described by Le and colleagues53 for the cre gene and those described by Strassburger and colleagues54 for the floxed Sod2 allele.
All the animal procedures were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. In addition, they were approved by the University of Florida Institutional Animal Care and Use Committee.
Electroretinography
For ERG, mice were dark-adapted overnight. They were anesthetized with ketamine-xylazine, and their eyes were dilated in dim red light with 2.5% phenylephrine solution. Small contact lenses with gold wire loops were placed on each cornea with a drop of 2.5% methylcellulose to maintain corneal hydration. A silver wire reference electrode was placed subcutaneously between the eyes, and a ground electrode was placed subcutaneously in a hind leg. Responses from both eyes were recorded simultaneously using a visual electrodiagnostic system (UTAS with a BigShot Ganzfeld dome; LKC, Gaithersburg, MD, USA). Scotopic ERGs, which primarily measure rod function, were elicited with 10-ms flashes of white light at 0 decibel (dB) (2.68 scotopic candelas per square meter [scot cd/m2]), −10 dB (0.18 scot cd/m2), and −20 dB (0.02 scot cd/m2) with appropriate delay between flashes. Five to ten scans were averaged at each light intensity. The a-wave amplitudes were measured from baseline to the peak in the cornea-negative direction, and b-wave amplitudes were measured from cornea-negative peak to major cornea-positive peak. Both a-wave and b-wave amplitudes were measured at 1, 2, 3, 6, and 9 months.
Fundus Imaging and Fluorescein Angiography
To image the retinas in living mice, we used a fundus imaging system (Micron III; Phoenix Research Labs, Pleasanton, CA, USA). Sodium fluorescein (0.02 mL 25% fluorescein; Akorn, Ann Arbor, MI, USA) was injected intraperitoneally for examination of the retinal circulation. We began taking images 1 minute after injection and continued at increasing intervals for 15 minutes. Changes in retinal integrity were evaluated by comparing the sequential photographs taken over time and between Sod2flox/floxVMD2-cre mice and control mice.
Spectral-Domain Optical Coherence Tomography
Spectral-domain optical coherence tomography used an ultrahigh-resolution instrument (Bioptigen, Research Triangle Park, NC, USA) at 1, 2, 3, 6, and 9 months to measure ONL thickness and subretinal morphology. Mice were anesthetized, and pupils were dilated with drops of 1% atropine sulfate and 2.5% phenylephrine. Three hundred linear B-scans were obtained, and 30 averaged images were captured to minimize the background noise and to achieve a better resolution. Four measurements at the same distance from the optical nerve head were recorded from each eye and were analyzed to determine the difference in ONL thickness. The results from each time point were averaged, and the means were compared between time points.
PCR Analysis
To verify the RPE-specific deletion of exon 3 of Sod2, genomic DNA was extracted from dissected RPE/choroid of Sod2flox/floxVMD2-cre mice with and without doxycycline treatment. For this purpose, we used a tissue PCR kit (REDExtract-N-AMP; Sigma-Aldrich Corp., St. Louis, MO, USA). Primers were located in intron 2 (GAGGGGCATCTAGTGGAGAA) and intron 3 (AACAATCGGGGCTAGTGAGA) of the Sod2 gene. A 0.4-kb PCR product represented the exon 3–deletion allele, while the wild-type allele presented as a 1.1-kb band only. The denaturation, annealing, and elongation temperatures for PCR reactions were 95°C, 55°C, and 68°C, respectively.
Immunoblots and ELISA
Dissected RPE/choroid was mechanically disrupted in ristocetin-induced platelet agglutination buffer with protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA) and 2 mM EDTA. Lysates were diluted in Laemmli sample buffer55 containing 100 μM dithiothreitol and boiled for 5 minutes. Equal amounts of protein were separated by SDS-PAGE and transferred into a polyvinylidene difluoride membrane using a transfer system (iBlot; Invitrogen, Grand Island, NY, USA). The membrane was blocked with a proprietary blocking buffer (Li-Cor Biosciences, Lincoln, NE, USA) for 1 hour at room temperature and incubated overnight with the designated primary antibody at 4°C. The immunoblot was scanned using an infrared imaging system (Odyssey CLx; Li-Cor Biosciences). Nitrotyrosine levels were quantified by competitive enzyme immunoassay using a nitrotyrosine assay kit with chemiluminescence detection (Millipore, Billerica, MA, USA) according to the instructions provided in the kit. The RPE/choroid was dissected from four Sod2flox/floxVMD2-cre (+dox) mice and four Sod2flox/floxVMD2-cre (−dox) mice at both 1 month and 2 months of age. The left and right RPE/choroid from each mouse were pooled and sonicated for 10 seconds in Tris-buffered saline supplied with the kit. Duplicates of each mouse sample and duplicates of each test standard were used in the assay.
X-gal Staining and Immunohistochemistry
X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining was used to detect the presence of β-galactosidase activity by color change (from no color to blue) in ROSA26-reporter mice or ROSA26VMD2-cre mice. Briefly, freshly enucleated eyes were fixed in PBS with 0.2% glutaraldehyde, 2 mM magnesium chloride, and 5 mM EGTA at room temperature for 30 minutes. After that, the RPE was incubated in a wash buffer in PBS with 0.01% sodium deoxycholate, 0.02% octylphenoxypolyethoxyethanol (Nonidet P-40; Aldrich, Milwaukee, WI, USA), and 5 mM EGTA at room temperature for 30 minutes. Tissues were incubated overnight at 37°C with 1 mg/mL X-gal with 5 mM potassium ferricyanide and potassium ferrocyanide. To analyze the X-gal staining, pigmented RPE of C57Bl/6J mice was bleached with 0.25% potassium permanganate for 5 minutes and 1% oxalic acid for 20 minutes at room temperature. X-gal staining of ROSA26-reporter mice or ROSA26VMD2-cre mice was then visualized by bright-field microscopy.
Immunostaining of RPE flat mounts was performed to examine cre expression in doxycycline-induced and noninduced Sod2flox/flox VMD2-cre mice. Primary rabbit anti-cre polyclonal antibody (Novus Biologicals, Littleton, CO, USA) was applied to detect cre recombinase in Sod2flox/floxVMD2-cre mice. To examine the morphologic changes of Sod2flox/flox VMD2-cre mice and control mice, rabbit zona occludens protein 1 (ZO-1) polyclonal antibody (Invitrogen) was applied to RPE flat mount samples. The MnSOD reduction was evaluated by immunohistochemistry with MnSOD antibody (Millipore) using RPE flat mounts. On immunoblots, this antibody reacted with a single protein band that migrated with apparent molecular weight of 24 kD, as expected for MnSOD (Supplementary Fig. S1C). To study oxidative stress in the RPE, we used 8-hydroxydeoxyguanosine (8-OHdG) monoclonal antibody (Santa Cruz Biotechnology, Dallas, TX, USA) or 4-HNE polyclonal antibody (Alpha Diagnostic International, Inc., San Antonio, TX, USA) to stain RPE flat mounts. Species-specific, fluorescently tagged secondary antibodies were obtained from Invitrogen and used at 1:500 dilution in PBS.
Autofluorescence Analysis
Laser scanning confocal microscopy was used to detect autofluorescence in Sod2flox/flox VMD2-cre mice and control mice. Frozen serial sections (12 μm) were air-dried on slides for 30 minutes. Sections were washed in distilled water to eliminate the OCT cutting medium. Samples were again air-dried for 30 minutes and then mounted in Fluormount (Electron Microscopy Sciences, Fort Washington, PA, USA). Wavelength scans were performed using the 488-nm laser line of a laser scanning confocal microscope (TCS SP2 AOBS; Leica, Buffalo Grove, IL, USA).
Dihydroethidium Staining
To detect intracellular superoxide, we used dihydroethidium (DHE) as described by Komeima and colleagues.56 Superoxide oxidizes DHE to ethidium, which generates an emission signal at approximately 600 nm. Mice were given intraperitoneal injections (5 μg/mg in dimethyl sulfoxide) of freshly prepared DHE (hydroethidine; Invitrogen) and killed after 18 hours. Eyes were rapidly removed and frozen in OCT compound. Tissues were processed for cryomicroscopy and observed under a fluorescence microscope.
Histology
To process the tissue for plastic sectioning, we perfused mice with 2.5% glutaraldehyde and 2% paraformaldehyde in PBS. Eyes were removed freshly and fixed overnight in 4% paraformaldehyde and 2% glutaraldehyde at 4°C. After fixation, the tissue was washed with 0.1 M cacodylate buffer (pH 7.4) for 10 minutes and incubated in 1% osmium for 4 hours at 4°C. The tissue was incubated in 0.1 M cacodylate buffer overnight at 4°C. After that, the tissue was dehydrated in a graded series of ethanol concentrations and rotated in epoxy/propylene for embedding. For electron microscopy, sections of 80 to 100 nm were prepared.
Sections of 0.5 μm were made to measure the thickness of the RPE at each of 10 evenly spaced locations (400-μm distance, beginning from the optic nerve head) at five inferior loci and five superior loci along a vertical meridian. Data were collected from 10 readings at each locus, with three random samplings of the slides from each eye.
Statistical Analysis
The mean amplitudes of multiple groups were compared (i.e., changes in ERG amplitude over time). For statistical analysis, we used one-way ANOVA with Tukey post hoc test.
Results
Induction of Cre Recombinase
Cre recombinase was induced by feeding doxycycline-laced chow to nursing dams for 2 weeks after delivery of pups. The cre protein could be detected in RPE flat mounts in mice up to age 1 month (Figs. 1A, 1B), although by 2 months the protein was no longer detectable (data not shown). To verify the recombination activity of doxycycline-induced cre recombinase, we bred ROSA26-lacZ mice with VMD2-cre transgenic mice. In these mice, induction of cre removes a transcriptional terminator in front of the lacZ gene, leading to the expression of β-galactosidase in cells expressing cre. Cre-stimulated recombination was confirmed by the X-gal staining of RPE flat mounts of ROSA26-lacZ VMD2-cre mice 1 month after induction (Figs. 1D–F). To visualize X-gal staining, RPE pigmentation was partially bleached in the flat mount. The distribution of X-gal staining was widespread. In contrast, in nontransgenic littermates, also nursed by doxycycline-fed mothers, the lacZ gene was not induced, and we detected no X-gal staining (Figs. 1G–I). To confirm that the β-galactosidase was in the RPE, we also stained frozen sections of ROSA26-lacZ VMD2-cre eyes with antibody to this protein (Supplementary Figs. S1A, S1B). Immune reactivity was seen only in the RPE and only following induction of cre. The PCR analysis of genomic DNA indicated that doxycycline induction of cre led to deletion of exon 3 from both alleles of Sod2. Deoxyribonucleic acid extracted from the RPE of 1-month-old induced mice showed greater than 90% of the 0.4-kb product representative of the deletion allele, while DNA extracted from control tissue (no doxycycline) resulted primarily in the 1.1-kb band characteristic of wild-type Sod2, although there was some evidence of deletion even in the absence of doxycycline chow. No deletion occurred without the cre transgene even in doxycycline-fed mice (Fig. 1C).
Figure 1.

Expression of cre in the RPE leads to deletion of Sod2. (A, B) Immunostaining of cre recombinase in flat mounts of the RPE of Sod2flox/floxVMD2cre mice. In the absence of doxycycline treatment (A), cre recombinase was not detectable at age 1 month. However, after 2-week feeding of doxycycline via the mother's milk (B), cre recombinase accumulated in nuclei of the RPE from Sod2flox/floxVMD2cre mice. Cre has a eukaryotic nuclear localization signal.77 (C) The PCR analysis of the Sod2 gene using primers flanking exon 3. In the absence of the cre transgene (Sod2flfl), a 1.1-kb amplicon expected of intact Sod2 was observed in Sod2flox/flox mice54; less than 5% of the 0.4-kb–deleted allele was detected by densitometry of the image following this end point analysis. These are biological replicates from three individual mice. In three Sod2flox/floxVMD2cre mice, most of the product represented the intact gene in the absence of doxycycline treatment (−dox), and detection of the 0.4-kb band suggests leaky expression of cre in noninduced mice. Following doxycycline treatment of three mice (+dox), the 0.4-kb band representing the deleted allele was predominant. (D–E) Distribution of active cre in the RPE. Detection of β-galactosidase expression in doxycycline-treated Rosa26-lacZ VMD2-cre mice. Flat mounts of the RPE were briefly bleached, and the enzyme activity was detected by blue staining using X-gal. The original magnification in (D) was ×4, and the original magnification in (E) and (F) was ×10. (G–I) Rosa26-lacZ control mice without the cre transgene resulted in no enzymatic conversion of X-gal.
Deletion of Sod2 Induces Oxidative Stress in the RPE
Immunostaining of RPE flat mounts confirmed that the level of MnSOD was reduced in doxycycline-induced Sod2flox/floxVMD2-cre mice (Fig. 2B) compared with flat mount mice of the same genotype that were not treated with doxycycline (Fig. 2A), which had extensive MnSOD staining. The antibody used for immunohistochemistry reacts only with a band of the expected size of MnSOD in RPE protein extracts (Supplementary Fig. S1C). Although more than 90% of cells in doxycycline-induced mice were negative for MnSOD staining, Western blot of the dissected RPE/choroid showed only a 50% decrease in the level of total MnSOD, probably because of contaminating enzyme from the choroid (data not shown). To determine if Sod2 deletion in the RPE led to increased oxidative stress, we used an ELISA to detect nitrotyrosine, which results from the modification proteins by peroxynitrate (Fig. 2C). Although there was no significant difference between doxycycline-induced and noninduced eyes at 1 month, by age 2 months we measured an 18% increase in nitrotyrosine in RPE/choroid samples from doxycycline-induced Sod2flox/floxVMD2-cre animals compared with noninduced mice of the same age and genotype (n = 6; P < 0.05).
Figure 2.

Induction of cre leads to a reduction in MnSOD and an increase in oxidative stress in the RPE. (A) The MnSOD was detected by immunohistochemistry in RPE flat mounts prepared from 2-month-old Sod2flox/floxVMD2cre mice not treated with doxycycline as pups. Despite the specificity of the antibody (Supplementary Fig. S1), immune reactivity was detected in the cytoplasm but also surrounding the nuclei and along the surfaces of cells. (B) In mice of the same age and genotype, but induced with doxycycline as neonates, immune reactivity was dramatically reduced in greater than 90% of the RPE cells. (C) By ELISA, there was no significant difference in nitrotyrosine between doxycycline-induced mice and control mice at age 1 month, but by 2 months there was a significant elevation of nitrotyrosine in doxycycline-induced mice. n = 6; P < 0.05. (D–F) To examine oxidative damage to DNA, RPE flat mounts were decorated with primary antibodies to 8-OHdG and with antibodies to ZO-1 to visualize cell boundaries. There was a significant increase in 8-OHdG in Sod2flox/floxVMD2cre mice induced with doxycycline (E). The higher-magnification image (F) reveals that most of the immune reactivity was in the cytoplasm (red), although some oxidative damage could also be detected in nuclei (yellow).
Oxidative damage to DNA was revealed using an antibody that recognizes 8-OHdG, a product of oxidative injury to DNA, to decorate RPE flat mounts (Figs. 2D–F). The RPE from mice heterozygous for the floxed allele of Sod2 (Sod2flox/+) did not show immunoreactive 8-OHdG following doxycycline treatment (Fig. 2D), but even at age 6 weeks doxycycline treatment of Sod2flox/floxVMD2-cre mice led to extensive oxidative damage to DNA in the RPE (Fig. 2E). The 8-OHdG staining that occurred in Sod2flox/floxVMD2-cre mice appeared to be primarily in the cytoplasm, suggesting damage to mitochondrial DNA (Fig. 2F). At this same stage, increased oxidative stress resulted in an accumulation of 4-HNE–modified proteins as revealed by immunohistochemistry (Supplementary Figs. S1D, S1E), and increased accumulation of superoxide was detected in the RPE and photoreceptor layer by staining with DHE (Supplementary Figs. S1F, S1G). The 4-HNE–modified proteins are a feature of AMD eyes at all stages.57
Increased Autofluorescence in the RPE of Sod2-Deleted Mice
Fundus autofluorescence is increased in the aging retina, and alterations in autofluorescence are characteristic of AMD, although they do not necessarily associate with drusen.58,59 Fundus autofluorescence arises, for the most part, from lipofuscin in the RPE. Consequently, we measured fluorescence in frozen sections of the mouse eye using an excitation wavelength (488 nm) known to stimulate lipofuscin fluorescence. With laser scanning confocal microscopy, we examined autofluorescence in retinas excised from Sod2-deleted and control mice at age 4 months (Figs. 3A, 3B). In the nontransgenic mice (Sod2flox/flox), fluorescence in the choroid and RPE was at background levels (Fig. 3A), whereas in the doxycycline-induced Sod2flox/floxVMD2-cre mice the intensity of fluorescence was increased over 20-fold (Fig. 3B). There was also some increased fluorescence in the choroid in these animals. We are uncertain of the origin of this fluorescence. Autofluorescence in the RPE is attributable to increased accumulation of lipofuscin, and components of lipofuscin may activate the complement cascade.60 To determine if there was a complement response in mice deleted for Sod2 in the RPE, we dissected RPE/choroid from 4-month-old mice and monitored the levels of CD46, a membrane-bound regulator of the complement cascade and of complement factor 5. Both proteins are associated with drusen in AMD eyes,61,62 although CD46 has recently been shown to be depressed in the damaged RPE from a donor with early AMD.63 Immunohistochemistry revealed increased CD46 and complement factor 5 deposition in the RPE of Sod2flox/floxVMD2-cre mice treated with doxycycline relative to mice lacking the VMD2-regulated cre transgene (Supplementary Fig. S2).
Figure 3.

Deletion of Sod2 leads to increased autofluorescence in the RPE. (A, B) The RPE/choroid was dissected from 4-month-old doxycycline-treated Sod2flox/flox mice (no cre) (A) and from doxycycline-treated Sod2flox/floxVMD2cre mice (B), cryosectioned, and examined with a laser scanning confocal microscope (Leica TCS SP2 AOBS). Wavelength scans were performed at the 488-nm laser line (incident light), and fluorescence spectra were made from the regions of the image corresponding to the RPE and the choroid, as well as from a background region of the slide.
Diminished ERG Response and Thinning of the ONL Following Sod2 Deletion
Doxycycline-induced Sod2flox/floxVMD2-cre mice showed an age-related decline in the scotopic a-wave and b-wave amplitudes between age 1 and 9 months (Fig. 4A). The decrease in amplitudes was statistically significant (P < 0.05) by 6 months. The scotopic ERG response continued to decline; in five mice that survived to 12 months, the average a-wave amplitude was 82 mV (at 0 dB), and the average b-wave amplitude was 122 mV. Because these mice were not from the same cohort as those whose results are shown in Figure 4A, we did not include these results in the graph. Light-adapted (photopic) ERG recordings were made at age 2, 6, and 9 months and also revealed statistically significant depression in the b-wave amplitude by age 6 months. Representative wave forms are shown in Figure 4B and reflect the major loss in a-wave and b-wave response between 6 and 9 months. The rate of ERG decrease recorded in this model was slower and more modest than the rate we reported for MnSOD knockdown using an AAV-delivered Sod2 ribozyme. Using that approach, the ERG a-wave amplitude was reduced by 50% at 3 months after injection and declined to 29% by 6 months.50
Figure 4.

Gradual decline in ERG response following deletion of Sod2. (A) Dark-adapted ERG responses were measured in doxycycline-induced Sod2flox/floxVMD2cre mice at age 1, 2, 3, 6, and 9 months. The average a-wave amplitudes and the average b-wave amplitudes recorded at 0-dB (2.68 scot cd/m2) flash intensity are shown. The same cohort of mice was used for all data points. n = 20 for months 1 through 3; n = 16 for month 6; n = 12 for month 9. (B) Light-adapted ERG responses were recorded in doxycycline-induced Sod2flox/floxVMD2cre mice at age 2, 6, and 9 months. These were from a different group of mice than those used for scotopic ERG. n = 38 for 2 months; n = 30 for 6 months; n = 26 for 9 months. (C–G) Representative scotopic ERG waveforms of doxycycline-induced Sod2flox/floxVMD2cre mice at intensities of 0 dB (2.68 scot cd/m2), −10 dB (0.18 scot cd/m2), and −20 dB (0.02 scot cd/m2) at age 1, 2, 3, 6, and 9 months. *P = 0.05; **P < 0.01.
To confirm that the attenuated ERG response was caused by doxycycline-induced deletion of Sod2, we examined Sod2flox/floxVMD2-cre mice that were not exposed to doxycycline as neonates. Such mice showed scotopic ERG responses similar to those of age-matched C57Bl/6J (wild-type) mice at age 9 months, whereas doxycycline induction led to more than 50% decrease in both a-wave (at 0 dB) and b-wave (at −20, −10, and 0 dB) in this group of mice (Supplementary Figs. S3A, S3B).
The reduction in ERG response was associated with the loss of photoreceptor cells as demonstrated by SD-OCT (Figs. 5A–E). It should be noted that using this approach we measured the thickness of the ONL only in the central 30% of the retina surrounding the optic nerve head. There was no significant change in ONL thickness in the first 3 months, but the average ONL thickness in the posterior retina was reduced by 15% by 6 months and by 24% by 9 months (Fig. 5F). We also noted distension and increased porosity in the choroid layer by age 6 months (Figs. 5D, 5E, white arrows). These findings were confirmed by electron microscopy: in 9-month-old doxycycline-treated Sod2flox/floxVMD2-cre mice, the choroid was less compact and more porous than in mice of the same genotype not treated with the inducer (Figs. 5G, 5H). Thinning of the ONL in these mice was dependent on the induction of cre by doxycycline: at 9 months, Sod2flox/floxVMD2-cre mice not exposed to doxycycline maintained ONL thickness similar to that of treated mice at 1 to 3 months (Supplementary Figs. S3C, S3D).
Figure 5.

Deletion of Sod2 in the RPE leads to thinning of the ONL and to choroidal abnormalities. (A–E) Representative SD-OCT images were captured to monitor the posterior retina in doxycycline-induced Sod2flox/floxVMD2cre mice at age 1, 2, 3, 6, and 9 months. White arrows indicate increased porosity of the choroid at 6 and 9 months. (F) Averaged ONL thickness measured between the outer plexiform layer and the external limiting membrane was measured at four standard locations relative to the optic nerve head at time points of 1, 2, 3, 6, and 9 months to demonstrate progressive degeneration of photoreceptor layers. These were the same mice and numbers of mice at each interval used for scotopic ERG (Fig. 4). **P < 0.01. (G) Electron micrographs reveal widened blood vessels and distended layers of the choroid in 9-month-old Sod2flox/floxVMD2cre mice treated with doxycycline (H) compared with untreated controls (scale bars: 10 μm).
Fundus Abnormalities and Vascular Leakage in Sod2-Deleted Eyes
Retinal atrophy developed slowly during aging of Sod2-deleted mice, which showed loss of pigmentation and abnormal vessels but no signs of choroidal neovascularization. During the first 3 months of life, the fundus of doxycycline-induced Sod2flox/floxVMD2-cre mice appeared unchanged (Fig. 6). This result differs from our AAV-Sod2 ribozyme–knockdown model, which caused obvious hypopigmentation as early as 1 month after injection.50 By the 6-month time point, discrete refractive foci were observed in the fundus images, suggesting localized retinal degeneration. Fluorescein angiography exhibited abnormalities in retinal blood vessels starting at approximately the same time (Fig. 7). In mice, the outer plexus forms approximately 1 week after birth.64 In several of the retinas, time-lapse imaging recorded what appeared to be active leaks (Figs. 7B–F) corresponding to a region of atrophy in the visible light image of the same retina (Fig. 7A). In contrast, at the 3-month time point no leakage or vascular abnormality was observed by fluorescein angiography in any of the mice (Supplementary Fig. S5).
Figure 6.

Deletion of Sod2 in the RPE leads to localized atrophy of the retina. (A–F) Fundus images were captured at age 1, 2, 3, 6, 9, and 10 months in doxycycline-induced Sod2flox/floxVMD2cre mice using a Micron III digital fundus microscope. By 6 months, hypopigmented areas (blotches) and white spots reflected thinning of the retina.
Figure 7.

Vascular leakage following Sod2 deletion. (A) A small atrophic region was apparent in a 6-month-old Sod2flox/floxVMD2cre mouse in fundus imaging. (B–F) Fluorescein angiography (FA) was performed to detect fluorescein leakage. The FA images were captured at 1, 3, 5, 10, and 15 minutes after fluorescein injection. Leaking fluorescein came from the same area of atrophic region from the fundus image; gradual enlargement of green fluorescein started at 3 minutes and reached maximum size at 15 minutes. In this and other retinas from doxycycline-induced Sod2flox/floxVMD2cre mice, we noted twisted major blood vessels, vascular beading, and an increase in branching and dilation of deeper blood vessels. Such changes were not observed at earlier time points (Supplementary Fig. S5).
RPE Dystrophy in Older Mice
To measure the thickness of the RPE, doxycycline-treated Sod2flox/floxVMD2-cre mice were killed at 1, 4, and 9 months and perfused with fixative, and their eyes were embedded for plastic sectioning. Using 0.5-μm sections imaged at ×40, we measured the RPE thickness for the whole retina up to 2000 nm from the center of the optic nerve head and averaged the readings for inferior, superior, and overall RPE (Fig. 8). The thickness of the RPE did not change significantly between age 1 month and 4 months but dramatically increased by 9 months, at which point it was almost 30% thicker than at age 1 month (Fig. 8, left). This was not simply a function of age, because 9-month-old mice of the same genotype that were not induced with doxycycline showed no hypertrophy of the RPE. The “spidergraph” of RPE thickness demonstrated thickening mainly in the superior region, which was over 40% thicker than the RPE from 1-month-old mice. While the RPE was intact in most of the eyes we examined by microscopy, in approximately 10% of the eyes of Sod2flox/floxVMD2-cre mice examined 6 or 9 months after doxycycline induction, large regions of the RPE appeared to be missing, and in these areas there was no intact ONL or photoreceptor layer (Supplementary Fig. S6). Such degeneration usually occurred only in induced mice and only at the 6-month time point or later. These eyes could not be used for morphometry.
Figure 8.
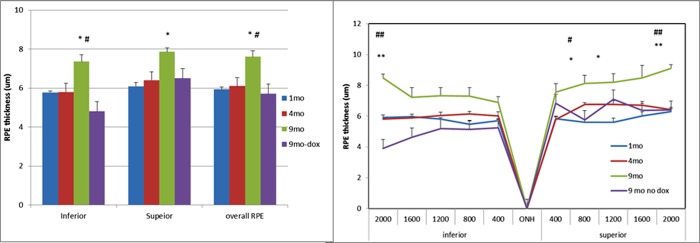
Increased thickness of the RPE in doxycycline-induced Sod2flox/floxVMD2cre mice. Left: Light micrographs of the retina and RPE were prepared from glutaraldehyde/paraformaldehyde–fixed tissue, and the RPE thickness was measured at 10 evenly spaced locations measuring from the center of the optic nerve head. The RPE thickness was averaged for the inferior hemisphere, the superior hemisphere, and the overall RPE. For each sample, four mice were used. *P < 0.05 (9-month time point compared with 1-month and 4-months); #P < 0.05 (9-month time point with doxycycline compared with 9 months without doxycycline). Right: The average RPE thickness at each measured point and age is displayed as a “spidergram.” *P < 0.05 (9-month time point compared with 1 month and 4 months); **P < 0.01 (9-month time point relative to 1-month time point); #P < 0.05 (9 months with doxycycline compared with 9 months without doxycycline).
Ultrastructural Abnormalities in the Sod2-Deleted Mice
Even at age 4 months, mice deleted for Sod2 in the RPE exhibited enlargement and disorganization of Bruch's membrane and distention of the basal infoldings of the RPE (Fig. 9). Although the length of rod outer segments was not reduced, broken tips of the photoreceptor outer segments were apparent near the apical surface of the RPE (Figs. 9A, 9B), suggesting a problem in phagocytosis. By 9 months, the length of rod outer segments was increased in many areas of the retina (Fig. 9C; Supplementary Fig. S7), with gaps between disc membranes. At this time point, the mitochondria of the RPE were fragmented and localized near the basal surface of the RPE (Fig. 9D). Moreover, in some regions of the retinas we examined, photoreceptor inner and outer segments were severely shortened, and these regions corresponded to regions in which the RPE was highly vacuolated and apical microvilli were absent (Figs. 9E, 9F). At the same age in noninduced Sod2flox/floxVMD2-cre mice, the photoreceptors, RPE, and Bruch's membrane appeared normal (Figs. 9F, 9G). Note the apical microvilli, elongated mitochondria, compact basal infoldings, and ordered fibers in Bruch's membrane. In retinas from doxycycline-treated transgenic animals, we frequently observed deposits along the basal surface of the RPE, resembling basal laminar deposits seen in eyes donated by patients with AMD65 but lacking widely spaced fibrous inclusions (Supplementary Fig. S7A). At late stages (>6 months), the RPE of doxycycline-treated Sod2flox/floxVMD2-cre mice showed an increase in lipofuscin-containing vesicles (asterisk) and what appeared to be immature melanosomes (arrowhead) (Supplementary Fig. S7A), which are not typically seen in adult humans66 but have been reported in 1-year-old mice.67 We did not detect choroidal neovascularization in any of our light or electron micrographs, although we found evidence of a subretinal hemorrhage in one doxycycline-induced Sod2flox/floxVMD2-cre animal (Supplementary Fig. S7B).
Figure 9.

Ultrastructural evidence of the RPE and photoreceptor atrophy. Electron micrographs were prepared at low (top) and high (bottom) magnification (scale bars: 2 μm) from eyes of 4-month-old and 9-month-old Sod2flox/floxVMD2cre mice with or without doxycycline induction. (A, B) Even at 4 months, the photoreceptor outer segments appeared disordered in doxycycline-induced mice, and basal infoldings of the RPE were disordered. (C, D) By 9 months, disc membranes were more widely spaced, and fragmented mitochondria had accumulated along the basal surface of the RPE. (E, F), In some areas of the retina, membrane-filled vacuoles were apparent in the RPE, and inner and outer segments of the photoreceptors were truncated and disordered. (G, H) In Sod2flox/floxVMD2cre mice of the same age but not treated with doxycycline, Bruch's membrane retained its normal laminar appearance, basal infoldings were compact, elongated mitochondria and apical microvilli were visible, and photoreceptor disc membranes maintained their tight, parallel packing.
Timing of Pathologic Features
The timing of the salient features of this mouse model is summarized in the Table. The time at which features of the model were first detected is indicated. Note, however, that we did not measure every attribute at each time point and in every animal, so that features such as lengthening of outer segments or complement deposition might have been detected earlier if we had looked.
Table.
Timing of the Appearance of Pathologic Features in the Mouse Model
|
Feature |
1 Month |
2 Months |
4 Months |
6 Months |
9 Months |
| Cre expression | ∗ | ||||
| Sod2 deletion | ∗ | ||||
| Oxidative stress | † | ∗ | |||
| Increased lipofuscin | ∗ | ||||
| Decreased ERG | ∗ | ||||
| Fundus abnormalities | ∗ | ||||
| Vascular leakage | ∗ | ||||
| Longer OS | ∗ | ||||
| Distension of Bruch's membrane | † | ||||
| RPE hypertrophy | ∗ | ||||
| Spongy choroid | ∗ | ||||
| RPE atrophy | † | ||||
| Complement factor 5 deposition | † |
OS, outer segments.
* The time at which a specific feature of the model was first detected or was first statistically significant.
† A feature that was detected by some criteria but not others (oxidative stress) or that was detected only in a subset of mice (e.g., RPE atrophy).
Discussion
The mitochondrial theory of aging posits that reactive oxygen species that are a by-product of oxidative phosphorylation cause cumulative damage to lipids, proteins, and DNA, particularly mitochondrial DNA, and lead to age-related impairment of health.68 While recent evidence challenges the importance of somatic mitochondrial mutations, nutrient-sensing pathways signaling through peroxisome proliferator-activated receptor gamma coactivator 1-alpha that promote longevity are likely to function through regulation of mitochondrial energy metabolism.69 We initiated these experiments to test the hypothesis that oxidative stress associated with mitochondrial metabolism contributes to the RPE dysfunction and leads to geographic atrophy. We previously reported a mouse model of RPE oxidative stress based on an AAV-delivered ribozyme specific for Sod2 mRNA.49,50 The eyes treated with the Sod2 ribozyme showed significant accumulation of oxidative modified proteins and lipids and of the bis-retinoid compounds A2E and iso-A2E. These compounds are major fluorescent constituents of lipofuscin and are formed from nonenzymatic reactions of retinaldehyde in the photoreceptors. We speculated that their accumulation in the RPE of these mice reflects defects in phagocytic processing. Treated eyes exhibited a reduction in dark-adapted ERG a-wave and b-wave responses as early as 3 months. However, there was no further change in the ERG response after the 6-month time point, and there was no change in the ONL thickness between 7 months and 2 years, indicating that most of the damage had occurred early. This model proved useful in testing a pharmacological intervention to prevent photoreceptor cell death: the 5-hydroxytryptamine 1a receptor agonist 8-hydroxy-2-(di-n-propylamino)tetralin blocked retinal degeneration in this model when given systemically for 4 months.51
In the transgenic model presented herein, Sod2 was deleted specifically in the RPE using the cre-lox system following doxycycline induction of cre recombinase. The loxP sites flanked exon 3 of Sod2, which encodes the manganese-binding site of the enzyme.54 The PVMD2-rtTA; tetO-PCMV-cre transgenic mouse line was described by Le et al.,53 who reported no loss of the RPE or photoreceptors and no decrease in ERG amplitudes for 10 months following the induction of RPE-specific cre. Indeed, we induced expression of cre during the first 2 weeks of life but were unable to detect the cre protein 2 months later, and, like Le and colleagues,53 we detected no impact on ERG in mice induced for cre expression but not containing the floxed allele of Sod2 (data not shown). In order to obtain a uniform genetic background, we bred both lines with C57Bl/6J mice for at least six generations before beginning these experiments, monitoring our breeders for the rd1 and rd8 mutations.
Compared with our earlier AAV-Sod2 ribozyme model,49,50 doxycycline-induced Sod2flox/floxVMD2-cre provided a more uniform phenotype because of the variability of subretinal injections of the virus vector. As expected from the work by Le and colleagues,53 we obtained efficient, RPE-specific deletion of the floxed gene (Fig. 1), leading to a significant decrease in MnSOD and an increase in oxidative stress in the RPE as measured by 8-OH-dG, nitrotyrosine (Fig. 2), and 4-HNE (Supplementary Fig. S1). Similar to aged human retinas, oxidative stress led to an increase in autofluorescent material with the spectral properties of lipofuscin (Fig. 3). As we had anticipated, deletion of Sod2 in the RPE led to a slow, progressive reduction in photoreceptor function based on the decreasing scotopic and photopic ERG responses (Fig. 4) and to a thinning of the ONL (Fig. 5), reflecting death of photoreceptor cell bodies. Oxidative stress in the RPE also caused distension and increased porosity in the choroid, apparent in SD-OCT images and in micrographs by 6 months (Fig. 5). McLeod and colleagues70 have noted a close relationship between RPE injury and choriocapillaris damage in the eyes of patients with AMD. Fundus photography and microscopic analysis indicated RPE hypertrophy and localized atrophy of photoreceptor cells (Fig. 6); however, where photoreceptors survived, their outer segments were elongated (Supplementary Fig. S4), and disc membranes were more widely spaced than normal (Figs. 9C, 9D). In some eyes, electron microscopy revealed atrophy of the RPE associated with loss of photoreceptor cells or even the entire neural retina in specific regions (Supplementary Fig. S6). We also detected basal deposits within the RPE and Bruch's membrane reminiscent of those seen in eyes donated by patients with AMD (Supplementary Fig. S7). Unlike most patients with AMD but similar to patients with polypoidal choroidal vasculopathy, we noted localized vascular hemorrhaging in some of our late-stage Sod2-deleted mice (Fig. 7). We also observed dilation of the deeper vessels in retinas of most Sod2-deleted eyes.
What surprised us about the Sod2flox/floxVMD2-cre model was how slow it was. Based on fluorescence microscopy, we detected a substantial reduction in MnSOD in almost every cell in the RPE (Fig. 2), whereas with the ribozyme model we observed a 60% reduction in MnSOD in RPE cells in culture.49 Nevertheless, the ribozyme model led to a statistically significant reduction in a-wave and b-wave amplitudes in 3 months, which is half the time needed to detect significant ERG decrease in this Sod2-deletion model. While there are several plausible explanations for the difference in the rate of retinal degeneration between the two models, the largest difference is the subretinal injection required in the AAV-ribozyme model. We controlled for the impact of injection by injecting control eyes with AAV-green fluorescent protein or AAV expressing an inactive ribozyme, and we did not detect significant loss of photoreceptors or a decline in ERG amplitudes in those eyes. However, retinal detachment is associated with an increase in the release of proinflammatory cytokines such as monocyte chemotactic protein 1, TNF-α, and IL-1β.71 Because oxidative stress is thought to elicit inflammation in AMD,15,72,73 it is likely that the combination of RPE oxidative stress (caused by Sod2 knockdown) and inflammation (caused by subretinal injection) made the AAV-ribozyme model more severe than the induction of RPE oxidative stress without physical injury to the retina. This hypothesis could be tested by subretinal injection of sterile saline in one eye of a doxycycline-induced Sod2flox/floxVMD2-cre mouse or by inducing inflammation with endotoxin. While speeding up the degeneration might be useful for drug testing, doing so by injection would reintroduce the variability we sought to avoid.
Backcrossing the parental lines to C57Bl/6 might have also affected the rate of retinal degeneration in this model. C57Bl/6 mice carry a hypomorphic allele (Met450) of RPE65, in which Rpe65 protein levels are reduced and regeneration of the 11-cis chromophore is slowed.74,75 We note, however, that C57Bl/6 mice aged 5 to 6 months accumulated A2E and iso-A2E to the same level as DBA1/J mice aged 4 to 5 months (Leu450) following treatment with the AAV-Sod2 ribozyme.49
As mentioned in the introduction, there are a variety of mouse models that attempt to recapitulate the pathology of AMD. Some, like our model, do so by increasing oxidative stress in the retina and RPE, as in the iron overload model by Hadziahmetovic and colleagues43 or the Sod1-knockout mice.47 Other mouse models use different strategies based on our understanding of factors that contribute to AMD. These include the mouse lines expressing chimeric variants of complement factor H42 and the ApoE4-knockin mouse fed a high-fat/high-cholesterol diet44,76 and the aryl hydrocarbon receptor–knockout mice.46 Some of these models exhibit features similar to those we report herein, including accumul ation of RPE autofluorescence, RPE hypertrophy or atrophy, basal deposits in the RPE, and death of photoreceptor cells. In several of the models, additional features of AMD such as drusen-like deposits and choroidal neovascularization are also observed. We have not detected these in the RPE-specific Sod2-knockout mice. We perhaps did not wait long enough. Our experimental design calling for a 9-month time course was based on our expectation that deletion of Sod2 at the DNA level would produce a more dramatic phenotype than knockdown of Sod2 mRNA. Because the ERG decline appears to continue past 9 months, it is likely that a more extreme phenotype would be observed at later times. One difference between our model and many of the other mouse models of AMD is that the genetic defect in our mice is restricted to the RPE and will not lead to oxidative stress outside the retina, RPE, and choroid. This was intentional: we wanted to test the specific contribution of mitochondrial oxidative stress in the RPE to AMD-like pathology. To the extent that AMD reflects a systemic proinflammatory environment, our approach could not recapitulate this aspect of the disease.
What we have obtained is a progressive model of localized atrophy of the retina associated with damage to the underlying RPE and Bruch's membrane. Because these effects are not associated with drusen and because we cannot examine the macula-specific impacts of RPE oxidative stress, this is not a precise model of dry AMD. Nevertheless, these mice should allow us to test gene-based and drug-based therapies to prevent retinal degeneration associated with RPE damage and to look for additional contributing factors (second hits) that lead to a more extreme phenotype.
Acknowledgments
The authors thank Karin Scharffetter-Kochanek, University of Ulm, Ulm, Germany, for providing the mouse line containing the floxed allele of Sod2.
Supported by Grants R01EY020825 and P30-EY021721 from the National Eye Institute, Grant 1KG08 from the James and Esther King Biomedical Research Foundation, the Macula Vision Research Foundation, and the Shaler Richardson professorship endowment.
Disclosure: H. Mao, None; S.J. Seo, None; M.R. Biswal, None; H. Li, None; M. Conners, None; A. Nandyala, None; K. Jones, None; Y.-Z. Le, None; A.S. Lewin, None
References
- 1. Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005; 115: 500–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995; 38: 357–366 [DOI] [PubMed] [Google Scholar]
- 3. Uchino Y, Kawakita T, Miyazawa M, et al. Oxidative stress induced inflammation initiates functional decline of tear production. PLoS One. 2012; 7: 45805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brennan LA, Kantorow M. Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res. 2009; 88: 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tezel G, Yang X, Luo C, et al. Oxidative stress and the regulation of complement activation in human glaucoma. Invest Ophthalmol Vis Sci. 2010; 51: 5071–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sacca SC, Pascotto A, Camicione P, Capris P, Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch Ophthalmol. 2005; 123: 458–463 [DOI] [PubMed] [Google Scholar]
- 7. Dong A, Xie B, Shen J, et al. Oxidative stress promotes ocular neovascularization. J Cell Physiol. 2009; 219: 544–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barnett BP, Handa JT. Retinal microenvironment imbalance in dry age-related macular degeneration: a mini-review. Gerontology. 2013; 59: 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004; 122: 598–614 [DOI] [PubMed] [Google Scholar]
- 10. Cano M, Thimmalappula R, Fujihara M, et al. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and age-related macular degeneration. Vision Res. 2010; 50: 652–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seddon JM, Reynolds R, Maller J, Fagerness JA, Daly MJ, Rosner B. Prediction model for prevalence and incidence of advanced age-related macular degeneration based on genetic, demographic, and environmental variables. Invest Ophthalmol Vis Sci. 2009; 50: 2044–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tan JS, Mitchell P, Kifley A, Flood V, Smith W, Wang JJ. Smoking and the long-term incidence of age-related macular degeneration: the Blue Mountains Eye Study. Arch Ophthalmol. 2007; 125: 1089–1095 [DOI] [PubMed] [Google Scholar]
- 13. Tomany SC, Wang JJ, van Leeuwen R, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology. 2004; 111: 1280–1287 [DOI] [PubMed] [Google Scholar]
- 14. Age-Related Eye Disease Study Research Group. The relationship of dietary carotenoid and vitamin A, E, and C intake with age-related macular degeneration in a case-control study: AREDS report No. 22. Arch Ophthalmol. 2007; 125: 1225–1232 [DOI] [PubMed] [Google Scholar]
- 15. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage–induced inflammation initiates age-related macular degeneration. Nat Med. 2008; 14: 194–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007; 143: 607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai J, Nelson KC, Wu M, Sternberg P Jr, Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000; 19: 205–221 [DOI] [PubMed] [Google Scholar]
- 18. Hageman GS, Luthert PJ. Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE–Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001; 20: 705–732 [DOI] [PubMed] [Google Scholar]
- 19. Liang FQ, Godley BF. Oxidative stress–induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003; 76: 397–403 [DOI] [PubMed] [Google Scholar]
- 20. Crabb JW, Miyagi M, Gu X, et al. From the cover: drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682–14687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang L, Clark ME, Crossman DK, et al. Abundant lipid and protein components of drusen. PLoS One. [ serial online]. 2010; 5: e10329 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2859054/. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holz FG, Strauss EC, Schmitz-Valckenberg S, van Lookeren Campagne M. Geographic atrophy: clinical features and potential therapeutic approaches. Ophthalmology. 2014; 121: 1079–1091 [DOI] [PubMed] [Google Scholar]
- 23. Kaarniranta K, Sinha D, Blasiak J, et al. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. 2013; 9: 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004; 18: 562–564 [DOI] [PubMed] [Google Scholar]
- 25. Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2002; 99: 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med. 2012; 33: 399–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010; 45: 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem. 2014; 289: 8312–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He Y, Ge J, Burke JM, Myers RL, Dong ZZ, Tombran-Tink J. Mitochondria impairment correlates with increased sensitivity of aging RPE cells to oxidative stress. J Ocul Biol Dis Inf. 2010; 3: 92–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008; 49: 2848–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010; 51: 5470–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kenney MC, Hertzog D, Chak G, et al. Mitochondrial DNA haplogroups confer differences in risk for age-related macular degeneration: a case control study. BMC Med Genet. [ serial online]. 2013; 14: 4 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3566905/. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rakoczy PE, Yu MJ, Nusinowitz S, Chang B, Heckenlively JR. Mouse models of age-related macular degeneration. Exp Eye Res. 2006; 82: 741–752 [DOI] [PubMed] [Google Scholar]
- 34. Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. 2012; 33: 487–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunter JJ, Morgan JI, Merigan WH, Sliney DH, Sparrow JR, Williams DR. The susceptibility of the retina to photochemical damage from visible light. Prog Retin Eye Res. 2012; 31: 28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Garland DL, Fernandez-Godino R, Kaur I, et al. Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum Mol Genet. 2014; 23: 52–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Radu RA, Hu J, Yuan Q, et al. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011; 286: 18593–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rohrer B, Long Q, Coughlin B, et al. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 3056–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011; 471: 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008; 452: 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ufret-Vincenty RL, Aredo B, Liu X, et al. Transgenic mice expressing variants of complement factor H develop AMD-like retinal findings. Invest Ophthalmol Vis Sci. 2010; 51: 5878–5887 [DOI] [PubMed] [Google Scholar]
- 43. Hadziahmetovic M, Dentchev T, Song Y, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008; 49: 2728–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malek G, Johnson LV, Mace BE, et al. Apolipoprotein E allele–dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci U S A. 2005; 102: 11900–11905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao C, Yasumura D, Li X, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011; 121: 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu P, Herrmann R, Bednar A, et al. Aryl hydrocarbon receptor deficiency causes dysregulated cellular matrix metabolism and age-related macular degeneration–like pathology. Proc Natl Acad Sci U S A. 2013; 110: E4069–E4078 Available at: http://www.pnas.org/content/110/43/E4069.long. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Imamura Y, Noda S, Hashizume K, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2006; 103: 11282–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao Z, Chen Y, Wang J, et al. Age-related retinopathy in NRF2-deficient mice. PLoS One. [ serial online]. 2011; 6: e19456 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3084871/. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Justilien V, Pang JJ, Renganathan K, et al. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci. 2007; 48: 4407–4420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seo SJ, Krebs MP, Mao H, Jones K, Conners M, Lewin AS. Pathological consequences of long-term mitochondrial oxidative stress in the mouse retinal pigment epithelium. Exp Eye Res. 2012; 101: 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thampi P, Rao HV, Mitter SK, et al. The 5HT(1a) receptor agonist 8-Oh DPAT induces protection from lipofuscin accumulation and oxidative stress in the retinal pigment epithelium. PLoS One. [ serial online]. 2012; 7: e34468 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3317995/. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li Y, Huang TT, Carlson EJ, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995; 11: 376–381 [DOI] [PubMed] [Google Scholar]
- 53. Le YZ, Zheng W, Rao PC, et al. Inducible expression of cre recombinase in the retinal pigmented epithelium. Invest Ophthalmol Vis Sci. 2008; 49: 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Strassburger M, Bloch W, Sulyok S, et al. Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med. 2005; 38: 1458–1470 [DOI] [PubMed] [Google Scholar]
- 55. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 56. Komeima K, Usui S, Shen J, Rogers BS, Campochiaro PA. Blockade of neuronal nitric oxide synthase reduces cone cell death in a model of retinitis pigmentosa. Free Radic Biol Med. 2008; 45: 905–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA. Age-related macular degeneration and retinal protein modification by 4-hydroxy-2-nonenal. Invest Ophthalmol Vis Sci. 2007; 48: 3469–3479 [DOI] [PubMed] [Google Scholar]
- 58. Lois N, Owens SL, Coco R, Hopkins J, Fitzke FW, Bird AC. Fundus autofluorescence in patients with age-related macular degeneration and high risk of visual loss. Am J Ophthalmol. 2002; 133: 341–349 [DOI] [PubMed] [Google Scholar]
- 59. Bindewald A, Bird AC, Dandekar SS, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005; 46: 3309–3314 [DOI] [PubMed] [Google Scholar]
- 60. Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009; 50: 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. 2001; 15: 390–395 [DOI] [PubMed] [Google Scholar]
- 62. Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001; 73: 887–896 [DOI] [PubMed] [Google Scholar]
- 63. Ebrahimi KB, Fijalkowski N, Cano M, Handa JT. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J Pathol. 2013; 229: 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007; 10: 77–88 [DOI] [PubMed] [Google Scholar]
- 65. Curcio CA, Presley JB, Millican CL, Medeiros NE. Basal deposits and drusen in eyes with age-related maculopathy: evidence for solid lipid particles. Exp Eye Res. 2005; 80: 761–775 [DOI] [PubMed] [Google Scholar]
- 66. Boulton M, Docchio F, Dayhaw-Barker P, Ramponi R, Cubeddu R. Age-related changes in the morphology, absorption and fluorescence of melanosomes and lipofuscin granules of the retinal pigment epithelium. Vision Res. 1990; 30: 1291–1303 [DOI] [PubMed] [Google Scholar]
- 67. Lindquist NG, Larsson BS, Stjernschantz J, Sjöquist B. Age-related melanogenesis in the eye of mice, studied by microautoradiography of 3H-methimazole, a specific marker of melanin synthesis. Exp Eye Res. 1998; 67: 259–264 [DOI] [PubMed] [Google Scholar]
- 68. Miquel J, Economos AC, Fleming J, Johnson J. Mitochondrial role in cell aging. Exp Gerontol. 1980; 15: 575–591 [DOI] [PubMed] [Google Scholar]
- 69. Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013; 123: 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McLeod DS, Grebe R, Bhutto I, Merges C, Baba T, Lutty GA. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 4982–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nakazawa T, Matsubara A, Noda K, et al. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006; 12: 867–878 [PubMed] [Google Scholar]
- 72. Kanda A, Abecasis G, Swaroop A. Inflammation in the pathogenesis of age-related macular degeneration. Br J Ophthalmol. 2008; 92: 448–450 [DOI] [PubMed] [Google Scholar]
- 73. Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells: implications for age-related macular degeneration (AMD). Immunol Lett. 2012; 147: 29–33 [DOI] [PubMed] [Google Scholar]
- 74. Danciger M, Matthes MT, Yasamura D, et al. A QTL on distal chromosome 3 that influences the severity of light-induced damage to mouse photoreceptors. Mamm Genome. 2000; 11: 422–427 [DOI] [PubMed] [Google Scholar]
- 75. Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of Rpe65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nat Genet. 2000; 25: 63–66 [DOI] [PubMed] [Google Scholar]
- 76. Ding JD, Johnson LV, Herrmann R, et al. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2011; 108: E279–E287 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3136266/. Accessed June 25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Le Y, Gagneten S, Tombaccini D, Bethke B, Sauer B. Nuclear targeting determinants of the phage P1 cre DNA recombinase. Nucleic Acids Res. 1999; 27: 4703–4709 [DOI] [PMC free article] [PubMed] [Google Scholar]