

Abstract
Hypertension is generally attributed to perturbations of the vasculature, the kidney, and the central nervous system. During the past several years, it has become apparent that cells of the innate and adaptive immune system also contribute to this disease. Macrophages and T cells accumulate in the kidneys and vasculature of humans and experimental animals with hypertension, and likely contribute to end-organ damage. We have shown that mice lacking lymphocytes, such as recombinase-activating gene-deficient (RAG-1−/−) mice, have blunted hypertension in response to angiotensin II, increased salt levels, and norepinephrine. Adoptive transfer of T cells restores the blood pressure response to these stimuli. Others have shown that mice with severe combined immunodeficiency have blunted hypertension in response to angiotensin II. Deletion of the RAG gene in Dahl salt-sensitive rats reduces the hypertensive response to salt feeding. The central nervous system seems to orchestrate immune cell activation. We produced lesions of the anteroventral third ventricle and showed that these block T cell activation in response to angiotensin II. Likewise, we showed that genetic manipulation of reactive oxygen species in the subfornical organ modulates both hypertension and T cell activation. Current evidence indicates that production of cytokines including tumor necrosis factor alpha, interleukin 17, and interleukin 6 contribute to hypertension, likely by promoting vasoconstriction, production of reactive oxygen species, and sodium reabsorption in the kidney. We propose a working hypothesis linking the sympathetic nervous system, immune cells, the production of cytokines, and ultimately vascular and renal dysfunction, leading to augmentation of hypertension.
EARLY STUDIES IN IMMUNITY AND HYPERTENSION
The concept that immune cells contribute to hypertension is not new. In the 1960s, Grollman et al showed that immunosuppression blunts hypertension in a model of renal infarction and that transfer of lymphocytes from rats with renal infarction induces hypertension in previously non-hypertensive animals (1, 2). Later, Svenson showed that hypertension is not maintained in thymectomized or athymic nude mice with renal infarction (3). In the 1980s, Ba et al found that transplanting the thymus from a Wistar Kyoto rat to a spontaneously hypertensive rat (SHR) decreased blood pressure in the recipient SHR (4). Hypertension was reduced in the SHR by either anti-thymocyte serum or the immunosuppressive drug cyclophosphamide (5, 6). Whereas these early studies showed a role for the immune system in hypertension, an in-depth understanding of this phenomenon was limited by a lack of animal models, methodology, and understanding of immunology.
T CELLS AND HYPERTENSION
In 2007, we showed that T cells play a critical role in the development of hypertension (7). Mice lacking lack both T and B lymphocytes, that is, recombinase-activating gene-1(RAG-1−/−)–deficient mice, were found to have blunted hypertensive responses to angiotensin II and increased salt levels. These mice did not exhibit increased vascular superoxide production and endothelial dysfunction commonly observed in hypertensive animals. Hypertension and vascular dysfunction was restored when Rag-1−/− mice received adoptive transfer of T but not B cells. In these studies, we found that angiotensin II increased circulating CD69+, CCR5+, and CD44high T cells, markers of effector memory T cells. In addition, T cells with an effector phenotype accumulated in the perivascular adipose tissue of the aorta. In a subsequent study, Crowley et al showed that mice with severe combined immunodeficiency are protected against hypertension and exhibit reduced albuminuria and renal damage (8). Recently, Mattson et al deleted the RAG1 gene in the Dahl salt-sensitive rat using zinc finger nuclease technology and showed that this attenuates blood pressure, albuminuria, and kidney damage (9). Thus, T cells seem to play a role in the development of various forms of hypertension in different strains of mice and in rats.
THE IMMUNE SYSTEM AND PRE-ECLAMPSIA
Pre-eclampsia is characterized by the onset of hypertension during pregnancy accompanied by proteinuria. Pre-eclampsia is associated with the production of autoantibodies that stimulate the angiotensin II (AT1) receptor (10) and infusion of these antibodies can induce pre-eclampsia–like symptoms in pregnant mice (11). It has been established that a specific subset of B cells produces these antibodies (12). Depletion of B cells using the anti-CD20 antibody rituximab blunts the blood pressure response in the reduced uterine perfusion pressure (RUPP) rat model of pre-eclampsia (13). Adoptive transfer of CD4+ T cells from RUPP rats to normal pregnant rats increased blood pressure in the recipients (14). Treatment with either rituximab or an AT1 receptor antagonist blunts this response, in keeping with the helper function of these cells in stimulating B cell production of agonistic antibodies. Supporting the role of T cells in pre-eclampsia, mice deficient in the cytokines interleukin-4 (IL-4) or IL-10, which skew T cells to an anti-inflammatory phenotype, develop pre-eclampsia–like symptoms when pregnant (15, 16). The pro-inflammatory cytokine IL-17 (discussed below) mediates placental oxidative stress and increases in blood pressure in pregnant rats (17). Together, these observations support a role for the adaptive immune system in pre-eclampsia where T and B cells act in a synergistic manner.
ROLE OF THE CENTRAL NERVOUS SYSTEM IN IMMUNE MEDIATED HYPERTENSION
The blood vessels, kidney, and central nervous system (CNS) have been implicated in the genesis of hypertension. Immune cells likely link these systems. Secondary lymphoid tissues, including lymph nodes and the spleen, are richly innervated (18). Ganta et al showed that intracerebroventricular (ICV) angiotensin II increases splenic efferent nerve firing and splenic mRNA expression of multiple cytokines (19). The circumventricular organs of the brain, including the subfornical organ (SFO), the organum vasculosum of the lateral terminalis, the median eminence and the area postrema, have an incomplete blood-brain barrier and can therefore be influenced by circulating hormones such as angiotensin II.
Several studies from our group have implicated the CNS in immune cell activation in hypertension. We produced lesions in the anteroventral third cerebral ventricle (AV3V), which are known to prevent angiotensin II–induced hypertension (20, 21). We found that AV3V lesions protected against T cell activation and aortic infiltration of T cells in response to angiotensin II (21). Mice that were infused with norepinephrine became hypertensive and exhibited T cell activation and aortic infiltration even after AV3V lesions. This supports the concept that sympathetic drive, and its attendant release of norepinephrine, likely mediates T cell activation and hypertension. This study showed that angiotensin II–induced T cell activation is not due to direct actions of angiotensin II on T cells but rather that central signals are required for T cell activation.
Prior studies by Davisson et al have shown that superoxide in the SFO promotes hypertension. The SFO both sends and receives signals from other cardiovascular centers of the brain. Mice with loxP sites flanking the extracellular superoxide dismutase (ecSOD) coding region were used for ICV injections of an adenovirus encoding Cre recombinase, deleting ecSOD in the SFO. This increased reactive oxygen species (ROS) levels in the SFO, increased sympathetic outflow as indicated by the ratio of low-to-high frequency heart rate variability, and caused an elevated blood pressure (22). Deletion of ecSOD in the SFO also sensitized mice to a normally subpressor dose of angiotensin II (140 ng/kg/min). This central manipulation increased the presence of activated T cells in the circulation and markedly increased infiltration of leukocytes into the vasculature. In a separate investigation, when ecSOD was specifically deleted in vascular smooth muscle, despite increases in vascular reactive oxygen species, blood pressure and T cell responses were not altered compared to mice with intact vascular ecSOD (23).
The NADPH oxidases are major sources of superoxide in mammalian cells. The subunit p22phox mediates trafficking of the Nox catalytic subunits to the cell membrane, and is required for enzyme complex assembly and ultimately superoxide production. Complementing the studies that used deletion of ecSOD, our laboratory also deleted p22phox in the SFO in a similar manner (24). Deletion of p22phox in the SFO blunted the pressor response to angiotensin II and decreased sympathetic outflow as assessed by heart rate variability. In addition, p22phox deletion abolished angiotensin II–induced aortic T cell infiltration and activation of circulating T cells.
More recently, we performed studies showing that the T cells play a role in stress-induced hypertension (25). We exposed mice to 7 days of an experimental stress paradigm involving restraint and cage switching. This increased blood pressure, activation of circulating T cells, and aortic T cell infiltration. RAG-1−/− mice were protected from stress-induced hypertension and adoptive transfer of T cells restored the hypertensive response. These findings emphasize the crucial role of the CNS in orchestrating the T cell response leading to hypertension.
T CELL SUBTYPES, CYTOKINES, AND MECHANISMS OF ACTIVATION
The above studies show that T cells play a role in the development of hypertension; however, they do not define the subsets of T cells involved. CD4+ T cells have been generally classified as either Th1 or Th2, depending on their activation markers and cytokine production (26). Th17 cells are a newly characterized subset of T cells; these cells produce the cytokine IL-17 and play a role in numerous autoimmune diseases, obesity, and cardiovascular disease (27–29). To investigate the role of IL-17 in hypertension, our group studied IL-17a−/− mice. Angiotensin II caused an initial increase in blood pressure in these animals; however, the hypertension was not sustained after 1 week (30). The angiotensin II–induced aortic T cell infiltration observed in wild-type mice was absent in IL-17a−/− mice, as were increases in vascular oxidative stress and endothelial dysfunction. Recent reports have shown that direct infusion of IL-17a induces hypertension and endothelial dysfunction in mice (31) and that IL-17 causes oxidative stress in the placenta of pregnant rats, promoting hypertension (17).
In addition to IL-17a, other cytokines have been implicated in the pathogenesis of hypertension. Etanercept, the tumor necrosis factor-α (TNF-α) antagonist, is effective in preventing hypertension (7, 32, 33). IL-6–deficient mice are also protected in certain models of hypertension (34–36). Interferon-γ (IFN-γ) is upregulated in the kidneys of hypertensive mice (8) and deletion of the IFN-γ receptor prevents angiotensin II–induced end-organ damage (37). Together these observations suggest that hypertension is mediated by multiple pro-inflammatory T cell subsets. In accord with this concept, T regulatory cells, which act to restrain pro-inflammatory T cells, attenuate hypertension-induced end-organ damage in mice (38) and blunt hypertension in rats (39).
T CELL CO-STIMULATION AND HYPERTENSION
Classically, T cells require two signals for activation: 1) interaction of the T cell receptor with an antigen presented in the context of a major histocompatibility complex, and 2) stimulation of co-stimulatory molecules on the T cell by ligands on the antigen presenting cell. A major co-stimulatory molecule on T cells is CD28, which is bound by the B7 ligands CD80 and CD86 of the antigen-presenting cell. Ligation of the T cell receptor in the absence of co-stimulation leads to T cell apoptosis (40). The pharmacologic agent CTLA4-Ig inhibits co-stimulation by binding to B7 ligands on antigen presenting cells. We found that CTLA4-Ig treatment blunts blood pressure, T cell activation, and vascular infiltration in both angiotensin II- and deoxycorticosterone acetate salt-induced hypertension (41). CTLA4-Ig treatment also abolished T cell production of TNF-α and IFN-γ induced by angiotensin II. Similar results were observed in B7−/− mice, which lack B7 ligands. These observations suggest that T cell receptor ligation and co-stimulation are necessary for T cell activation in hypertension.
MONOCYTES AND HYPERTENSION
In addition to cells of the adaptive immune system, there are data that cells of the innate immune system, and in particular those of the monocyte/macrophage lineage, contribute to hypertension. Wenzel et al deleted LysM+ cells in mice that have the diphtheria toxin receptor driven by the LysM promoter (42). Treatment of these mice with diphtheria toxin depleted both neutrophils and monocytes, and completely eliminated the hypertensive response to angiotensin II. Likewise, this prevented the vascular dysfunction, hypertrophy, and production of ROS caused by hypertension. Repletion of monocytes, but not granulocytes, restored hypertension in these mice. Given that monocytes are capable of presenting antigen to T cells, it is interesting to speculate that one effect of monocyte depletion is prevention of T cell activation in this model.
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, it has been known for almost 50 years that immune cells play a role in hypertension. In the last several years, investigations from our group and others have further shown the importance of immune cells in hypertension. In light of the data discussed here, we have formulated a working hypothesis for the development of overt hypertension (Figure 1). We propose that central actions of hypertensive stimuli, such as angiotensin II or salt, increase sympathetic outflow. This leads to an initial elevation in blood pressure, consistent with clinical “prehypertension,” which results in protein modifications, possibly due to oxidative alterations. These neoantigens, which are no longer recognized as self, are processed and presented by dendritic cells, ultimately leading to T cell activation. Activated T cells infiltrate the kidney and vasculature and produce cytokines that promote renal sodium and water retention and in the vasculature, vasoconstriction, and remodeling. Together these alterations result in overt hypertension. This working hypothesis will almost certainly require refinement as new information becomes available.
Fig. 1.
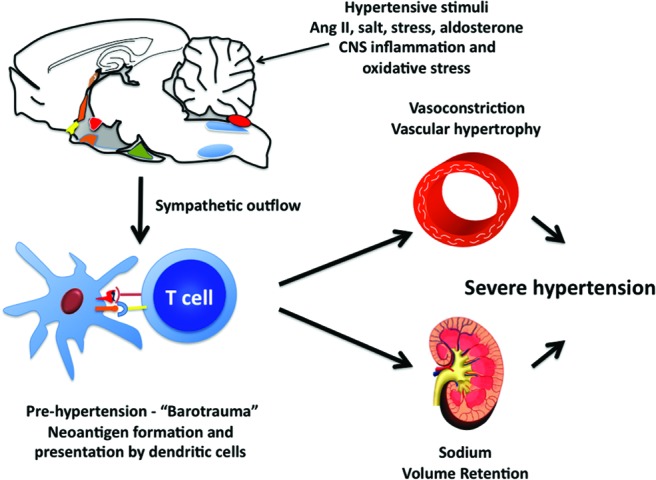
Working hypothesis describing the role of immune cells in hypertension.
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
del Rio, Emery: Great talk, David. I am glad to see that you now finally believe that inflammation is possible in all diseases. As I had a hypertensive-inducing breakfast this morning, I was thinking about this process and based on your hypothesis, do you think we could have a vaccine for hypertension?
Harrison, Nashville: I think it's possible. We would have to identify the specific peptides that are involved. I also think that we are learning more about how the dendritic cell is altered in this process, and it may be less specific than a vaccine would permit intervention for.
Reddy, Ann Arbor: So I have two related questions. What exactly is turning on the dendritic cells (DCs) to make the isoketals; any thoughts on that?
Harrison, Nashville: So can I answer that first? It might very well be sympathetic adrenergic activation. These cells, we've shown, have beta-2, alpha-1, and alpha-2 receptors, and we actually have some belief that these are beta-2 receptors that are important in responding. We've done renal denervation, which is a very popular thing, and that is what prevents this activation.
Reddy, Ann Arbor: The second question is — maybe you've done this as well — have you eliminated the DCs and tried to induce hypertension with DTR transgenics?
Harrison, Nashville: That has been done. A colleague — one of the fellows in my laboratory previously — has done that in his lab and that is dramatically effective. That was a paper in Circulation about 2 years ago.
Bryan, Columbia: One brief question going back to your starting point about the role of the sympathetic nervous system on T cell activation and cytokine production. As you know, the ongoing SIMPLICITY trial uses low-dose radiofrequency to selectively ablate sympathetic nerves in the renal arteries using catheterization. Is there any ongoing — or was there any background — work showing the effect of sympathetic nerve ablation in the adventitia of the arteries on T cell proliferation and cytokine production?
Harrison, Nashville: So we actually have a project in which we are doing that now. And we show that renal denervation dramatically reduces T cell activation and dendritic cell activation in the spleen. So when you denervate the kidney it has this striking effect in peripheral tissues, and we don't understand all of the mechanisms involved in that yet.
Bryan, Columbia: Thank you for that sneak preview.
Spivak, Baltimore: This is a great paper. I remember in medical school learning about essential hypertension, which meant hypertension we don't know anything about. And in hematology we have the same unfortunate misuse of the English language; essential thrombocytosis, which means that people have thrombocytosis and we used to not know what it was all about. I'd link essential hypertension and essential thrombocytosis together, because in 50% of the patients with essential thrombocytosis, they have over-activity constituently of JAK2, and LNK is the modifier molecule of JAK2. Some patients with myeloproliferative disorders actually have a mutation in LNK, which allows JAK2 to be overactive. So I am thinking not so much down at the dendritic part of your pathway, but down at the other end where you've released all these cytokines. There are a number of non-specific JAK inhibitors, particularly ones that hit both JAK1 and JAK2, and if you give these to patients with chronic myeloproliferative disorders who have tremendous cytokine storm — TNF alpha, interferon gamma, the whole gambit of these cytokine mediators — very rapidly, you turn off the signal. So to me, it seems that the LNK knockout model might allow you to have a therapeutic advantage and, perhaps, JAK inhibitors might be another way of attacking hypertension.
Harrison, Nashville: These animals, by the way, get profound hypertension. Even if you use normally sub-pressor doses of angiotensin II, they get blood pressures of 180. So they are activated. They are inflamed at baseline.
Spivak, Baltimore: So it might be that trying the JAK2 inhibitors in these animals may take the hypertension away.
Harrison, Nashville: Right
Czeisler, Boston: My question is also about essential hypertension. I wondered if you could comment on the recurrent hypoxia associated with obstructive sleep apnea as being potentially leading to increased inflammation and sympathetic activation which, at least in the meetings I go to, is thought to be ideologically responsible for about a third of patients with essential hypertension.
Harrison, Nashville: So sleep apnea is associated with profound increases in sympathetic outflow. The thing that's fascinating is that it occurs in bursts. So that you have, you know, these very high catecholamine levels during these episodes, and then presumably they resolve during the day. We actually have done some memory protocols. We give animals angiotensin II and wait a month, and we see, during that first exposure, the appearance of memory CD8 cells in the kidney. Then we wait a month, and then we use a very low dose of ang II — that sub-pressor dose — and you get an immediate increase in these memory T cells and severe hypertension if the animal has been exposed before, but not if they received a sham infusion before. So I think there is a memory component to hypertension and that, in fact, what people do when they have sleep apnea, is they challenge themselves. They get an immunization every night to activate their T cells and then ultimately this just sustains.
Luke, Cincinnati: It's extremely difficult to produce hypertension in a low-salt diet in any animal model. As far as I know, it's not been done.
Harrison, Nashville: I'm sorry; on what kind of diet?
Luke, Cincinnati: Low-salt diet.
Harrison, Nashville: Yes, exactly.
Luke, Cincinnati: So I think, in terms of population medicines, we are forgetting the role of salt. There is very good evidence that a high-salt diet — independent of hypertension — stimulates the production of reactive oxidative species and that sort of thing. I think that it may well be responsible for the sympathetic nervous system activity as well. So in terms of helping patients and population medicine for this really important disease, I think that we need to reiterate the goals of the World Health Organization, et cetera, that a low-salt diet would stop a lot of these inflammatory responses.
Harrison, Nashville: I couldn't agree more.
Tweardy, Houston: I enjoyed your talk. In terms of the T cells that are responsible for the hypertension downstream of the dendritic cells, you suggested CD8s were very important but the IL-17 production, do you think that's a T helper–17 outcome…
Harrison, Nashville: So we actually see both CD8 and CD4 cells produce IL-17. In fact, there is recent evidence that salt — you know, high sodium — will stimulate, through SGK1, T cell production of IL-17. We see that also in the CD8 population. So it's complex, and I'm not sure I'm willing to go into which cell type is absolutely most important.
Tweardy, Houston: There is a small group of patients with Job's syndrome that have defective T helper–17 production, and they get the syndrome that includes cold abscesses of the skin. I was wondering if you've looked to see whether those individuals have a decreased incidence of hypertension.
Harrison, Nashville: We've not done that. Those kinds of questions often come up. HIV patients tend to have low blood pressure until they are treated, and then they get hypertensive, but there could be lots of explanations for that.
REFERENCES
- 1.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–64. [PubMed] [Google Scholar]
- 2.White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron. 1964;1:93–102. doi: 10.1159/000179322. [DOI] [PubMed] [Google Scholar]
- 3.Svendsen UG. The role of thymus for the development and prognosis of hypertension and hypertensive vascular disease in mice following renal infarction. Acta Pathol Microbiol Scand A. 1976;84:235–43. doi: 10.1111/j.1699-0463.1976.tb00094.x. [DOI] [PubMed] [Google Scholar]
- 4.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol. 1982;128:1211–6. [PubMed] [Google Scholar]
- 5.Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun. 1981;99:600–7. doi: 10.1016/0006-291x(81)91787-3. [DOI] [PubMed] [Google Scholar]
- 6.Dzielak DJ. Immune mechanisms in experimental and essential hypertension. Am J Physiol. 1991;260:R459–67. doi: 10.1152/ajpregu.1991.260.3.R459. [DOI] [PubMed] [Google Scholar]
- 7.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II–dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–97. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–14. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallukat G, Homuth V, Fischer T, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou CC, Zhang Y, Irani RA, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–62. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen F, Wallukat G, Herse F, et al. CD19+CD5+ cells as indicators of preeclampsia. Hypertension. 2012;59:861–8. doi: 10.1161/HYPERTENSIONAHA.111.188276. [DOI] [PubMed] [Google Scholar]
- 13.LaMarca B, Wallace K, Herse F, et al. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension. 2011;57:865–71. doi: 10.1161/HYPERTENSIONAHA.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novotny SR, Wallace K, Heath J, et al. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R1197–201. doi: 10.1152/ajpregu.00623.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee P, Chiasson VL, Kopriva SE, et al. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension. 2011;58:489–96. doi: 10.1161/HYPERTENSIONAHA.111.172114. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee P, Kopriva SE, Chiasson VL, et al. Interleukin-4 deficiency induces mild preeclampsia in mice. J Hypertens. 2013;31:1414–1423. doi: 10.1097/HJH.0b013e328360ae6c. [DOI] [PubMed] [Google Scholar]
- 17.Dhillion P, Wallace K, Herse F, et al. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R353–8. doi: 10.1152/ajpregu.00051.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Felten DL, Livnat S, Felten SY, Carlson SL, Bellinger DL, Yeh P. Sympathetic innervation of lymph nodes in mice. Brain Res Bull. 1984;13:693–9. doi: 10.1016/0361-9230(84)90230-2. [DOI] [PubMed] [Google Scholar]
- 19.Ganta CK, Lu N, Helwig BG, et al. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–91. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- 20.Brody M, Fink G, Buggy J, et al. Critical role of the anteroventral third ventricle (AV3V) region in development and maintenance of experimental hypertension. Perspect Nephrol Hypertens. 1979;6:76–84. [Google Scholar]
- 21.Marvar PJ, Thabet SR, Guzik TJ, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–70. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lob HE, Marvar PJ, Guzik TJ, et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:276–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension. 2011;58:232–9. doi: 10.1161/HYPERTENSIONAHA.111.172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension. 2013;61:382–7. doi: 10.1161/HYPERTENSIONAHA.111.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marvar PJ, Vinh A, Thabet S, et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry. 2012;71:774–82. doi: 10.1016/j.biopsych.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57. [PubMed] [Google Scholar]
- 27.Eid RE, Rao DA, Zhou J, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. 2009;119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winer S, Paltser G, Chan Y, et al. Obesity predisposes to Th17 bias. Eur J Immunol. 2009;39:2629–35. doi: 10.1002/eji.200838893. [DOI] [PubMed] [Google Scholar]
- 30.Madhur MS, Lob HE, McCann LA, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330:219–28. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- 33.Venegas-Pont M, Manigrasso MB, Grifoni SC, et al. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56:643–9. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56:879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee DL, Sturgis LC, Labazi H, et al. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–40. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 36.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–81. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 37.Marko L, Kvakan H, Park JK, et al. Interferon-gamma signaling inhibition ameliorates angiotensin II–induced cardiac damage. Hypertension. 2012;60:1430–6. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 38.Kvakan H, Kleinewietfeld M, Qadri F, et al. Regulatory T cells ameliorate angiotensin II–induced cardiac damage. Circulation. 2009;119:2904–12. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 39.Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H938–44. doi: 10.1152/ajpheart.00707.2009. [DOI] [PubMed] [Google Scholar]
- 40.Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest. 2002;109:295–9. doi: 10.1172/JCI14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vinh A, Chen W, Blinder Y, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wenzel P, Knorr M, Kossmann S, et al. Lysozyme M–positive monocytes mediate angiotensin II–induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–81. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]