

Abstract
Background
Development of alcohol dependence, a chronic and relapsing disease, largely depends on the effects of alcohol on the brain reward systems. By elucidating the mechanisms involved in alcohol use disorder, novel treatment strategies may be developed. Ghrelin, the endogenous ligand for the growth hormone secretagogue receptor 1A, acts as an important regulator of energy balance. Recently ghrelin and its receptor were shown to mediate alcohol reward and to control alcohol consumption in rodents. However, the role of central versus peripheral ghrelin for alcohol reward needs to be elucidated.
Methods
Given that ghrelin mainly is produced by peripheral organs, the present study was designed to investigate the role of circulating endogenous ghelin for alcohol reward and for alcohol intake in rodents.
Results
We showed that the Spiegelmer NOX‐B11‐2, which binds and neutralizes acylated ghrelin in the periphery with high affinity and thus prevents its brain access, does not attenuate the alcohol‐induced locomotor activity, accumbal dopamine release and expression of conditioned place preference in mice. Moreover, NOX‐B11‐2 does not affect alcohol intake using the intermittent access 20% alcohol 2‐bottle‐choice drinking paradigm in rats, suggesting that circulating ghrelin does not regulate alcohol intake or the rewarding properties of alcohol. In the present study, we showed however, that NOX‐B11‐2 reduced food intake in rats supporting a role for circulating ghrelin as physiological regulators of food intake. Moreover, NOX‐B11‐2 did not affect the blood alcohol concentration in mice.
Conclusions
Collectively, the past and present studies suggest that central, rather than peripheral, ghrelin signaling may be a potential target for pharmacological treatment of alcohol dependence.
Keywords: Alcohol, Ghrelin, Spiegelmer, Reward, Food Intake
Development of alcohol dependence, a chronic and relapsing disease, largely depends on the effects of alcohol on the brain reward systems, specifically the cholinergic‐dopaminergic reward link (for review, see Larsson and Engel, 2004; Soderpalm et al., 2009; Volkow and Li, 2004). There is a comorbidity between alcohol dependence and compulsive over eating (for review, see Dickson et al., 2011), indicating that gut‐brain hormones, such as ghrelin, could be common biological mechanisms important for reward induced by food and alcohol.
Ghrelin, the endogenous ligand for the growth hormone secretagogue receptor 1A (GHS‐R1A), acts as an important regulator of energy balance (Kojima et al., 1999). Indeed, ghrelin induces adiposity and increases body weight in rats (Tschöp et al., 2000). Additionally, central as well as peripheral ghrelin administration increases food intake in rodents, an effect predominantly mediated via hypothalamic GHS‐R1A (Wren et al., 2000, 2001b). Human studies show that ghrelin increases the sensation of hunger and appetite. (Wren et al., 2001a). The plasma levels of ghrelin rise preprandially and fall postprandially in humans as well as in rodents, implying that ghrelin induces meal initiation (for review, see Egecioglu et al., 2011). The findings that GHS‐R1A are expressed in reward nodes associated with the cholinergic dopaminergic reward link including the ventral tegmental area, the nucleus accumbens, and the laterodorsal tegmental area (for review, see Dickson et al., 2011) imply that ghrelin may be involved in reward regulation. Initially, it was shown that ghrelin activates cholinergic‐dopaminergic reward link (Abizaid et al., 2006; Jerlhag et al., 2006a, 2007). This was corroborated by the finding showing that the rewarding properties of alcohol, as measured by locomotor stimulation, accumbal dopamine release, and conditioned place preference are attenuated in mice with suppressed GHS‐R1A and ghrelin signaling (Jerlhag et al., 2009, 2011) and that GHS‐R1A antagonists reduced the intake and the motivation to consume alcohol in rodents (Jerlhag et al., 2009; Kaur and Ryabinin, 2010; Landgren et al., 2012). Supportively, human genetic findings show that a single‐nucleotide polymorphism in the GHS‐R1A gene is associated with high alcohol consumption in humans (Landgren et al., 2008). The findings that this gut‐brain hormone is produced centrally (Cowley et al., 2003; Lu et al., 2002; Mondal et al., 2005) as well as in the gastrointestinal tract (Kojima et al., 1999) raises the need for investigations regarding the importance of central versus peripheral ghrelin for alcohol reward.
The present series of experiments was designed to investigate the role of circulating endogenous ghrelin, by using Spiegelmer NOX‐B11‐2, for alcohol‐induced reward, as measured by locomotor stimulation, accumbal dopamine release, and the expression of conditioned place preference in mice, as well as for alcohol intake in rats. NOX‐B11‐2 inhibits ghrelin's activation of GHS‐R1A‐expressing CHO cells by directly binding to the bioactive, acylated form of ghrelin (Shearman et al., 2006). NOX‐B11‐2 is expected to selectively neutralize ghrelin in the periphery since autoradiography studies of related compounds in rats and cynomolgus monkeys have shown that Spiegelmers do not enter the central nervous system. The results of the present experiments may be of clinical interest since prevention of ghrelin's brain access tentatively could be used as novel treatment of alcohol addiction.
Materials and Methods
Animals
Adult post pubertal age‐matched male NMRI mice (8 to 12 weeks old and 25 to 40 g body weight; B&K Universal AB, Sollentuna, Sweden) were used. We have extensive experience with NMRI mice, and they are considered to be a good model for general use and are extensively used in behavioral studies used in psychopharmacology research (Jerlhag et al., 2009). All mice were group‐housed and maintained at a 12/12 hour light/dark cycle. Adult post pubertal age‐matched male Rcc Han Wistar rats (Harlan, Horst, Netherlands) were used as they are known to voluntarily consume alcohol using the intermittent access model and thereby reach physiological relevant blood alcohol concentrations (Simms et al., 2008). They were housed individually in high Macrolon III cages covered with filter tops (Tecniplast, Varese, Italy) and maintained on a 12‐hour reversed light dark cycle.
All animals were maintained at 20°C with 50% humidity and tap water and food (Normal chow; Harlan Teklad, Norfolk, UK) were supplied ad libitum. These studies were carried out in strict accordance with the recommendations in the Swedish Animal Welfare Act, and all experiments were approved by the Swedish Ethical Committee on Animal Research in Gothenburg.
Drugs
For studies investigating alcohol‐induced locomotor stimulation, accumbal dopamine release, and conditioned place preference in mice, alcohol (VWR International AB, Stockholm, Sweden) was diluted in saline (0.9% NaCl) to 15% v/v for intraperitoneal (IP) injections and was administered 5 minutes prior to initiation of the experiments. A dose of 1.75 g/kg was used since it induces a locomotor stimulation, accumbal dopamine release, and conditioned place preference in NMRI mice (Jerlhag et al., 2006b, 2009; Larsson et al., 2004). For the intermittent access 20% alcohol 2‐bottle‐choice drinking paradigm alcohol was diluted in tap water to a final concentration of 20% v/v. Saccharin (Sigma‐Aldrich, Stockholm, Sweden) and quinine hemisulfate (Sigma‐Aldrich) was diluted in tap water to a final concentration of 0.1% and 0.02 mM, respectively. The Spiegelmer compound NOX‐B11‐2 (NOXXON Pharma AG, Berlin, Germany) was diluted in water and thereafter in saline (0.9% NaCl). NOX‐B11‐2 has in previous studies been found to bind to the biologically active ghrelin and thereby preventing the peptide penetrating into the brain (Shearman et al., 2006). A dose–response study was conducted in mice where the lowest dose (20 mg/kg, IP) with no effect per se was selected (Fig. 1). In rats, a dose of 20 mg/kg (IP) was administrated since previous studies show that a similar dose reduces ghrelin‐induced food intake (Kobelt et al., 2006; Shearman et al., 2006). NOX‐B11‐2 was always administered IP 60 minutes prior to drug challenge as this is the time frame required for NOX‐B11‐2 to bind to circulating ghrelin and prevents its access to the brain (Shearman et al., 2006). The selected dose did not affect the rodent's gross behavior. A balanced or within subject design was used for all drug challenges. All doses refer to the PEGylated compound as anhydrous free acid with a molecular weight of 53.4 g/mol. For IP injections, NOX‐B11‐2 was diluted in 10 ml/kg and in 2 ml/kg for mice and rats, respectively.
Figure 1.
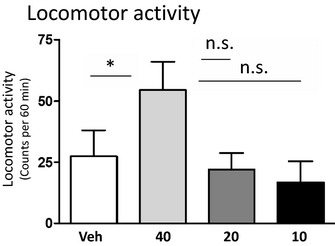
Dose–response effects of NOX‐B11‐2 on locomotor activity in mice. A dose–response study revealed that the highest NOX‐B11‐2 dose (40 mg/kg intraperitoneal [IP]) increased the locomotor activity compared to vehicle treatment. Neither of the lower doses of NOX‐B11‐2 (20 and 10 mg/kg IP) had any significant effect on locomotor activity compared with vehicle (n = 8 in each group; *p < 0.05 and n.s p > 0.05, 1‐way ANOVA followed by a Bonferroni post hoc test). All values represent mean ± SEM.
Locomotor Activity Experiments
Locomotor activity was recorded as described previously (Jerlhag et al., 2006a). In brief, locomotor activity was registered in sound‐attenuated, ventilated, and dim lit (45 lux) locomotor boxes. Photocell detection allowed a computer‐based system to register the activity of the mice. Locomotor activity was defined as the accumulated number of new photocell beams interrupted during a 60‐minute period.
The mice were allowed to habituate to the locomotor activity box 1 hour prior to drug administration. To identify a dose of NOX‐B11‐2 that did not affect the general gross or locomotor behavior, a dose–response study (NOX‐B11‐2 40, 20, 10 mg/kg or vehicle, n = 8 per treatment group) was conducted. Each mouse received 1 injection 60 minutes before the start of the test session. From this experiment the highest NOX‐B11‐2 dose with no effect per se (20 mg/kg, IP) was selected for further studies. Thereafter, the effects of NOX‐B11‐2 (20 mg/kg, IP) on alcohol‐induced (1.75 g/kg, IP) locomotor stimulation were investigated. NOX‐B11‐2 was administered 60 minutes prior to alcohol, and the activity registration started 5 minutes after the alcohol injection. Each mouse received 1 treatment combination (vehicle/vehicle, NOX‐B11‐2/vehicle, vehicle/alcohol, or NOX‐B11‐2/alcohol; n = 8 per treatment).
In Vivo Microdialysis and Dopamine Release Measurements
For measurements of extracellular dopamine levels, mice were implanted with a microdialysis probe positioned in the nucleus accumbens. The surgery was performed as described in detail previously (Jerlhag et al., 2006a). In brief, mice were anesthetized with isoflurane (Isoflurane Baxter; Univentor 400 Anaesthesia Unit, Univentor Ltd., Zejtun, Malta), placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) and kept on a heating pad to prevent hypothermia. The skull bone was exposed and 1 hole for the probe and 1 for the anchoring screw were drilled. The probe was randomly alternated to either the left or right side of the brain. The coordinates of 1.5 mm anterior to the bregma, ±0.7 lateral to the midline, and 4.7 mm below the surface of the brain surface was used (Franklin and Paxinos, 1996). All probes were surgically implanted 2 days prior to the experiment. After surgery the mice were kept in individual cages (Macrolon III).
The effect of systemic administration of NOX‐B11‐2 (20 mg/kg, IP) on alcohol‐induced (1.75 g/kg, IP) accumbal dopamine release was investigated using microdialysis in freely moving mice. On the day of the experiment, the probe was connected to a microperfusion pump (U‐864 Syringe Pump; AgnThós AB, Lidingö, Sweden) and perfused with Ringer solution at a rate of 1.5 μl/min. After 1 hour of habituation to the microdialysis set‐up, perfusion samples were collected every 20 minutes. The baseline dopamine level was defined as the average of 3 consecutive samples before the first alcohol or vehicle (saline, IP) challenge (Time 0). This initial alcohol‐challenge was given to establish that all mice included in the experiment would respond with an alcohol‐induced release of accumbal dopamine. The challenge‐induced increase in accumbal dopamine was calculated as the percent increase from baseline. Seven consecutive 20‐minute samples were collected after the initial challenge. At 140 minutes, the mice were injected with NOX‐B11‐2 (20 mg/kg, IP) or vehicle (second challenge), and 60 minutes later, vehicle or a second injection of alcohol (1.75 g/kg, IP) was administered (third challenge; 200 minutes) and followed by collection of four 20‐minute samples (experiment terminated at 280 min). Collectively, the following treatment groups (n = 8 in each group) was created: alcohol‐vehicle‐alcohol (Alc‐Veh‐Alc), alcohol‐NOX‐B11‐2‐alcohol (Alc‐NOX‐Alc), alcohol‐vehicle‐vehicle (Alc‐Veh‐Veh), and vehicle‐NOX‐B11‐2‐vehicle (Veh‐NOX‐Veh).
The dopamine levels in the dialysates were determined by high‐performance liquid chromatography with electrochemical detection as described previously (Jerlhag et al., 2006a).
After the microdialysis experiments were completed, the mice were decapitated. The brains were mounted on a vibroslice device (752M Vibroslice; Campden Instruments Ltd., Loughborough, UK) and cut in 50 μm sections. The location of the probe was determined by gross observation using light microscopy. The exact position of the probe was verified (Franklin and Paxinos, 1996), and only mice with correct placements were used in the statistical analysis.
Conditioned Place Preference
To evaluate the effects of NOX‐B11‐2 on the rewarding effects of alcohol, conditioned place preference tests were performed in mice as previously described (Jerlhag et al., 2009). In brief, a 2‐chambered conditioned place preference apparatus (45 lux) and distinct visual and tactile cues was used. The procedure consisted of preconditioning (day 1), conditioning (days 2 to 5), and postconditioning (day 6). At preconditioning, mice were injected IP with vehicle and were placed in the chamber with free access to both compartments during 20 minutes to determine the initial place preference. Conditioning (20 minutes per session) was done using a biased procedure in which alcohol (1.75 g/kg, IP) was paired with the least preferred compartment and vehicle with the preferred compartment. All mice received 1 alcohol and 1 vehicle injection every day and the injections were altered between morning and afternoon in a balanced design. At postconditioning, mice were injected with NOX‐B11‐2 (20 mg/kg, IP) or an equal volume of vehicle solution and, 60 minutes later, placed on the midline between the 2 compartments with free access to both compartments for 20 minutes (creating the following treatment groups; Alc‐Veh and Alc‐NOX). A control group of animals was subjected to the same procedure but received only vehicle injections throughout the conditioning (nonalcohol conditioned control group: creating the following treatment groups Veh‐Veh and Veh‐NOX). It should be noted that the present conditioned place preference design investigates the effect of NOX‐B11‐2 and vehicle on the expression, rather than acquisition, of conditioned place preference.
Conditioned place preference was calculated as the difference in % of total time spent in the drug‐paired (i.e., less preferred) compartment during the postconditioning and the preconditioning session.
Intermittent Access 20% Alcohol 2‐Bottle‐Choice Drinking Paradigm
The intermittent access 20% alcohol 2‐bottle‐choice drinking paradigm induces voluntary intake of high amounts of alcohol and pharmacological relevant blood alcohol concentrations (Carnicella et al., 2009; Simms et al., 2008). This drinking model has been extensively used as a preclinical screening tool for potential new treatments of alcohol dependence (Landgren et al., 2012; Steensland et al., 2007, 2012), and it seems to predict the efficacy of potential alcohol use disorder medications (e.g., varenicline) (McKee et al., 2009; Mitchell et al., 2012). In brief, the rats (n = 15) were given free access to 1 bottle of 20% alcohol and 1 bottle of water during three 24‐hour sessions per week (Mondays, Wednesdays, and Fridays), after the lights went out. The rats had unlimited access to 2 bottles of water between the alcohol access periods. Bottles were weighed manually at 1, 4, and 24 hours after the fluids were presented, and measurements were taken to the nearest 0.1 g. The body weight of each rat was measured daily prior to bottle presentation, to allow for calculating the grams of alcohol intake per kilogram of body weight (g/kg). The preference for alcohol over water (the ratio of alcohol to total fluid intake) was calculated at all time points. Administration of NOX‐B11‐2 began after the rats had voluntarily consumed high amounts of alcohol (3.4 ± 0.3 g/kg/24 h) for approximately 9 weeks. Each rat received NOX‐B11‐2 (20 mg/kg, IP) or an equal volume of vehicle solution the first day and vice versa the second treatment day (which corresponded to alcohol drinking days). Thus, each animal served as its own control.
Blood Alcohol Concentration
The effect of vehicle or NOX‐B11‐2 treatment on blood alcohol concentration in NMRI mice was investigated. The mice were injected with NOX‐B11‐2 (20 mg/kg, IP) or an equal volume of vehicle. Sixty minutes later, the mice were injected with alcohol (1.75 g/kg). Twenty minutes later, the mice were decapitated, and blood was collected in micro tubes (Vacuette; Greiner Bio‐one, Florence, Italy). The analysis of the blood alcohol concentration was outsourced to Sahlghrenska University Hospital (Gothenburg, Sweden; study agreement BML‐NEURO).
Saccharin and Quinine Consumption Paradigms
The mice were housed individually with continuous access to either tap water/saccharin solution (0.1%) or tap water/quinine solution (0.02 mM) for 5 days. On the sixth day, the mice were treated with NOX‐B11‐2 (20 mg/kg, IP) or an equal volume of vehicle. Bottles were weighed manually at 0 and 24 hours after the fluids were presented. The effect of NOX‐B11‐2 or vehicle treatment on the preference for saccharin or quinine was investigated. Food was freely supplied.
Statistical Analysis
Locomotor activity data were evaluated by a 1‐way analysis of variance (ANOVA) followed by Bonferroni post hoc tests. The microdialysis experiments were evaluated by a 2‐way ANOVA followed by Bonferroni post hoc test for comparisons between different treatments and specifically at given time points. The conditioned place preference data were evaluated by an unpaired t‐test. The data from intermittent access 20% alcohol 2‐bottle‐choice drinking paradigm were analyzed by a paired t‐test. Data from the blood alcohol concentration analysis as well as saccharin and quinine drinking experiments were analyzed with an unpaired t‐test. Data are presented as mean ± SEM. A probability value of p < 0.05 was considered as statistically significant.
Results
Dose–Response Effects of NOX‐B11‐2 on Locomotor Activity in Mice
A statistically significant overall effect of NOX‐B11‐2 treatment on locomotor activity was observed, F(3, 28) = 3.12, p = 0.042; n = 8 per group. Post hoc analysis revealed that the highest NOX‐B11‐2 dose (40 mg/kg) increased the locomotor activity compared to vehicle (p = 0.05) while the lower NOX‐B11‐2 doses (20 and 10 mg/kg) had no significant effect on locomotor activity compared to vehicle (p = 0.69 and p = 0.44, respectively) (Fig. 1).
Effects of NOX‐B11‐2 on Alcohol‐Induced Locomotor Stimulation, Accumbal Dopamine Release, and Conditioned Place Preference in Mice
An overall main effect of treatment was found on locomotor activity in mice following systemic administration of alcohol (1.75 g/kg) and NOX‐B11‐2 (20 mg/kg), F(3, 25) = 3.70, p = 0.025; n = 7 to 8 per group (Fig. 2A). As shown in Fig. 2A, post hoc analysis revealed that alcohol significantly increased the locomotor activity in both vehicle (p = 0.021; Veh‐Veh vs. Veh‐Alc) and NOX‐B11‐2 (p = 0.009; Veh‐Veh vs. NOX‐Alc) pretreated mice. Indeed, the magnitude of alcohol‐induced locomotor stimulation was not affected by pretreatment with a single injection of NOX‐B11‐2 as compared to vehicle pretreatment (p = 0.770; Veh‐Alc vs. NOX‐Alc). The selected dose of NOX‐B11‐2 had no significant effect on locomotor activity compared to vehicle treatment (p = 0.478; Veh‐Veh vs. NOX‐Veh).
Figure 2.
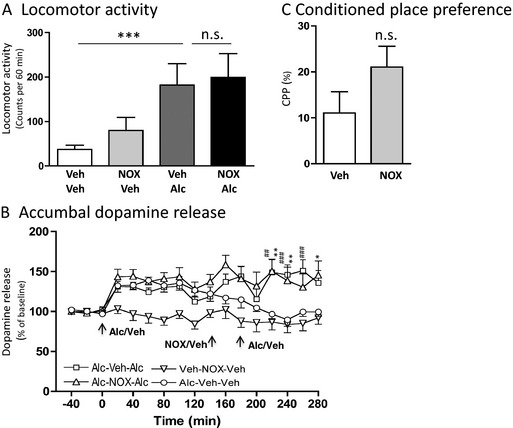
NOX‐B11‐2 does not affect the alcohol‐induced locomotor stimulation, accumbal dopamine release, and conditioned place preference in mice. (A) Alcohol‐induced (1.75 g/kg intraperitoneal [IP]) locomotor stimulation was not affected by a single injection of NOX‐B11‐2 (NOX, 20 mg/kg IP) (n = 7 to 8 in each group; ***p < 0.001 and n.s p > 0.05, 1‐way ANOVA followed by a Bonferroni post hoc test). (B) The alcohol‐induced (1.75 g/kg IP) increase in accumbal dopamine release was not effected in NOX‐B11‐2‐ (NOX, 20 mg/kg IP) compared with vehicle‐pretreated mice (n = 7 to 8 in each group). Arrows represent time points of injection of alcohol, NOX‐B11‐2 (NOX), or vehicle. Initial injections of alcohol increased dopamine release in all groups (Alc‐Veh‐Alc, Alc‐NOX‐Alc, and for Alc‐Veh‐Veh) in comparison to the group initially receiving vehicle treatment (Veh‐NOX‐Veh). Pretreatment with NOX‐B11‐2 (NOX) prior to the second injection of alcohol did not affect the alcohol‐induced increase in dopamine release (Alc‐Veh‐Alc vs. Alc‐NOX‐Alc), thus there was a significant difference in response between Alc‐Veh‐Veh and Alc‐NOX‐Alc (##p < 0.01 and ###p < 0.001, 2‐way ANOVA followed by a Bonferroni post hoc test), as well as between Alc‐Veh‐Veh and Alc‐Veh‐Alc (*p < 0.05 and **p < 0.01, 2‐way ANOVA followed by a Bonferroni post hoc test). (C) The alcohol‐induced (1.75 g/kg IP) conditioned place preference (CPP) was not attenuated by an acute single IP injection of NOX‐B11‐2 (NOX, 20 mg/kg IP) in mice (n = 7 to 8 in each group, n.s. p > 0.05, unpaired t‐test). All values represent mean ± SEM.
Accumbal microdialysis measurements of dopamine in mice revealed an overall main effect of treatment, F(3, 16) = 4.13, p < 0.0001, n = 7 to 8 in each group, time, F(16, 357) = 60.49, p < 0.0001, and a significant interaction of treatment × time, F(16, 357) = 2.01, p = 0.0002 (Fig. 2B). In the first part of the experiment, the responsiveness to alcohol (1.75 g/kg) per se was investigated. Initial injections of alcohol caused a significant increase in accumbal dopamine release compared to vehicle treatment at time point 80 (p < 0.05) and 180 minutes (p < 0.001) for the future Alc‐Veh‐Alc group, and at time points 60 to 80 (p < 0.05) and 120 minutes (p < 0.05) for the future Alc‐Veh‐Veh and at time points 20 (p < 0.05), 40 (p < 0.01), 60 (p < 0.05), 80 to 100 (p < 0.01), 120 (p < 0.05), 160 (p < 0.001), and 180 (p < 0.01) minutes for the future Alc‐NOX‐Alc group (Fig. 2B). In the subsequent part of the experiment, administration of NOX‐B11‐2 (20 mg/kg, IP at 140 minutes) 60 minutes prior to the second alcohol injection (1.75 g/kg, IP at 200 minutes) (Alc‐NOX‐Alc), did not affect the alcohol‐induced accumbal dopamine release compared to vehicle pretreatment (Alc‐Veh‐Alc) at any time point (220 to 280 minutes) (p > 0.05), Fig. 2B. In these treatment groups (Alc‐NOX‐Alc and Alc‐Veh‐Alc), there was a significant increase in accumbal dopamine releases compared to vehicle treatment (Alc‐Veh‐Veh) at time point 220 to 260 minutes (p < 0.01) for the Alc‐Veh‐Alc group, and at time points 220 to 240 (p < 0.01) and 280 minutes (p < 0.05) minutes for the Alc‐NOX‐Alc group, Fig. 2B. When administered alone, NOX‐B11‐2 had no significant effect on accumbal dopamine release compared to vehicle treatment (p > 0.05 at time points 200 to 280 minutes; Veh‐NOX‐Veh vs. Alc‐Veh‐Veh).
In the conditioned place preference experiments, NOX‐B11‐2 (NOX, 20 mg/kg, IP, n = 7 to 8) had no effect on the alcohol‐induced (1.75 kg/kg) (Veh, IP, n = 7 to 8) expression of conditioned place preference (p < 0.146) (Fig. 2C). In the second series of conditioned place preference experiments, conducted in vehicle‐conditioned mice, NOX‐B11‐2 (NOX, 20 mg/kg, IP, n = 7 to 8) per se did not affect conditioned place preference compared to vehicle injections (Veh‐Veh: 1.0 ± 7.8%; Veh‐NOX: 1.6 ± 6.2%, p = 0.959, n = 7 to 8 in each group).
Effects of NOX‐B11‐2‐B11‐2 on Voluntary Alcohol Intake Using the Intermittent Access Model in Rats
The effect of NOX‐B11‐2 (NOX, 20 mg/kg, IP) or an equal volume of vehicle on voluntary alcohol intake was evaluated in rats that had consumed high amounts of alcohol (3.4 ± 0.3 g/kg/24 h, n = 15) for 9 weeks prior to the test. There was no effect of NOX‐B11‐2 treatment on alcohol intake (g/kg) at any time points analyzed compared to vehicle treatment (1 hour: p = 0.544; 4 hours: p = 0.757; 24 hours: p = 0.768) (Fig. 3A–C). Furthermore, NOX‐B11‐2 treatment had no effect on water intake at any time point (1 hour: p = 0.728; 4 hours: p = 0.892; 24 hours: p = 0.856) (Fig. 3D–F). There was no effect of NOX‐B11‐2 treatment on alcohol preference at any time point (1 hour: p = 0.600; 4 hours: p = 0.907; 24 hours: p = 0.941) (1 hour: 55.5 ± 5.4 and 51.9 ± 5.5%; 4 hours: 47.9 ± 3.6 and 47.4 ± 3.0%; 24 hours: 42.5 ± 2.9 and 42.7 ± 2.4% for vehicle and NOX‐B11‐2, respectively). There was no effect on the total fluid intake following NOX‐B11‐2 treatment at any time point (1 hour: p = 0.857; 4 hours: p = 0.752; 24 hours: p = 0.657) (1 hour: 4.4 ± 0.4 and 4.5 ± 0.3 g; 4 hours: 9.8 ± 0.4 and 9.7 ± 0.6 g; 24 hours: 24.4 ± 1.0 and 24.0 ± 1.1 g for vehicle and NOX‐B11‐2, respectively). However, NOX‐B11‐2 treatment significantly reduced the food intake at the 1‐hour time point (p = 0.013, Fig. 3G), but not at 4‐ or 24‐hours time points compared to vehicle treatment (4 hours: p = 0.079; 24 hours: p = 0.284) (Fig. 3H,I). There was no effect on the body weight following NOX‐B11‐2 treatment (p = 0.656) (439 ± 9 and 439 ± 10 g for vehicle, and NOX‐B11‐2, respectively).
Figure 3.
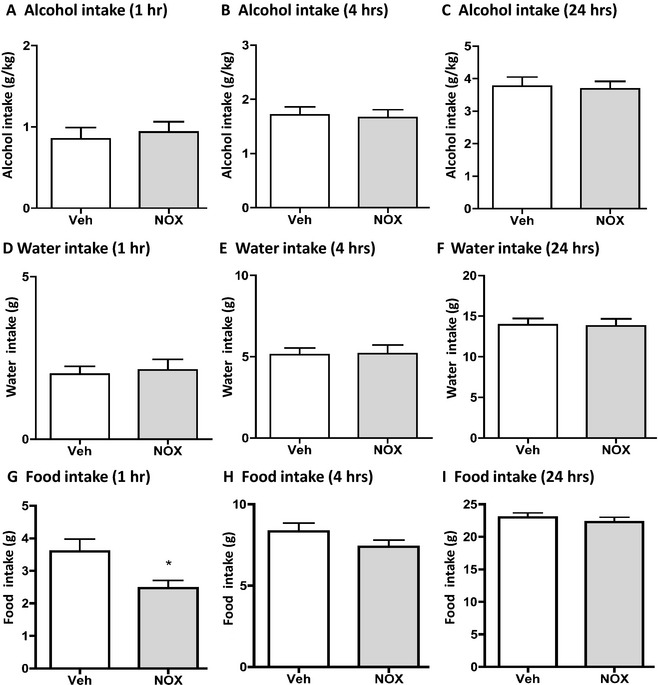
NOX‐B11‐2 does not affect alcohol or water intake, but reduced food intake in rats. NOX‐B11‐2 (NOX, 20 mg/kg intraperitoneal [IP]) did not affect alcohol intake (g/kg) at any time point measured (1 (A), 4 (B) or 24 (C) hours) compared to vehicle treatment in rats that had voluntarily consumed alcohol for 9 weeks before the treatment. NOX‐B11‐2 (NOX, 20 mg/kg IP) did not affect water intake (g/kg) at any time point measured (1 (D), 4 (E) or 24 (F) hours) compared to vehicle treatment in rats that had voluntarily consumed alcohol for 9 weeks before the treatment. NOX‐B11‐2 (NOX, 20 mg/kg IP) reduced food intake (g/kg) at the 1‐hour time point (G) but not at any other time point measured (4 (H) or 24 (I) hours) compared to vehicle treatment in rats that had voluntarily consumed alcohol for 9 weeks before the treatment. All values represent mean ± SEM (n = 15, *p < 0.05, compared to vehicle, paired t‐test).
To evaluate whether NOX‐B11‐2 treatment affected water consumption after the treatment was terminated, the water intake during the drinking session initiated 24 hours after NOX‐B11‐2 and vehicle administration was evaluated. No effect on the water intake was found during the first drinking session following the NOX‐B11‐2 treatment (24 hours: 25.2 ± 1.3 g for vehicle and 25.6 ± 0.8 g for NOX‐B11‐2, p = 0.743).
Effect of NOX‐B11‐2 on Blood Alcohol Concentration in Mice
NOX‐B11‐2 treatment (n = 9) had no effect on the blood alcohol concentration compared to vehicle treatment (n = 7) (42.9 ± 0.9 mM for NOX‐B11‐2, p = 0.629 and 43.6 ± 1.0 mM for vehicle).
Effects of NOX‐B11‐2 on Preference for Nonethanol Tastants (Saccharin as well as Quinine) in Mice
NOX‐B11‐2 treatment (n = 4) had no effect on the preference for saccharin compared to vehicle treatment (n = 4) (65.4 ± 12.0% for vehicle and 61.3 ± 14.9% for NOX‐B11‐2, p = 0.840). Nor had NOX‐B11‐2 treatment (n = 4) an effect on the preference for quinine compared to vehicle treatment (n = 4) (22.9 ± 8.4% for NOX‐B11‐2, p = 0.134 and 52.2 ± 14.7% for vehicle).
Discussion
The present study provides novel data indicating that circulating endogenous ghrelin is not important for alcohol reinforcement. Indeed, we showed that NOX‐B11‐2, which binds to acylated ghrelin in the periphery and thereby prevents its brain access, does not attenuate the alcohol‐induced locomotor activity, accumbal dopamine release, and expression of conditioned place preference in mice. Moreover, NOX‐B11‐2 did not affect alcohol intake using the intermittent access 20% alcohol 2‐bottle‐choice drinking paradigm in rats. In addition, we showed that NOX‐B11‐2 did not affect the blood alcohol concentration in mice. Finally, NOX‐B11‐2 did not affect the preference for saccharin nor for quinine. The present finding supports a role for circulating ghrelin as a physiological regulator of food intake as we here showed that NOX‐B11‐2 reduced food intake in rats.
The ability of alcohol to induce a locomotor stimulation, dopamine release and conditioned place preference is reduced in ghrelin knockout mice (Jerlhag et al., 2011), implying that either centrally or peripherally produced ghrelin is of importance for alcohol reinforcement. The findings that IP injections of ghrelin does not alter alcohol intake in alcohol‐naïve rats (Lyons et al., 2008), that ghrelin is produced centrally (Cowley et al., 2003; Lu et al., 2002; Mondal et al., 2005), and that NOX‐B11‐2 does not alter alcohol reinforcement collectively suggest that centrally, rather than peripherally, produced ghrelin is of importance for alcohol intake and alcohol reward in rodents. Supportively, local administration of ghrelin into the reward nodes ventral tegmental area or laterodorsal tegmental area increases alcohol consumption in mice (Jerlhag et al., 2009). Another tentative explanation may be that the preprandial increase of ghrelin (for review, see Egecioglu et al., 2011), which is blocked by administration of NOX‐B11‐2, is too low to regulate brain‐mediated behaviors such as alcohol intake in rats. However, this appears less likely as NOX‐B11‐2 reduces food intake, which is mediated via hypothalamic GHS‐R1A, in our alcohol‐drinking rats.
Albeit, the present study shows that circulating ghrelin does not mediate alcohol reward and alcohol intake in rodents, previous clinical studies have found associations between plasma levels of ghrelin and alcohol use disorder in humans. Indeed, high plasma levels of ghrelin have been observed in abstinent alcoholics (Kim et al., 2005; Kraus et al., 2005) and have been associated with high craving scores in alcoholics (Addolorato et al., 2006; Hillemacher et al., 2007; Koopmann et al., 2012; Leggio et al., 2012). In addition, a recent longitudinal study showed that ghrelin levels increase when alcoholics abstain, while ghrelin levels decrease when alcoholics relapse in drinking (Leggio et al., 2012). A speculative explanation may be that the elevated peripheral ghrelin levels observed in craving alcoholics indirectly reflect the central ghrelin levels of those patients. It has also been found that the plasma levels of ghrelin are lower in active drinking alcoholics than in healthy controls (Addolorato et al., 2006; Badaoui et al., 2008; Calissendorff et al., 2006), which in all probability may be due to that acute alcohol intake reduces the plasma levels of ghrelin (Zimmermann et al., 2007). The possibility should therefore be considered that NOX‐B11‐2 did not affect alcohol intake in our rats since they were actively drinking and therefore may have had low plasma levels of ghrelin. However, this needs to be further elucidated. The findings that the cholinergic–dopaminergic reward link is activated by pharmacological‐induced hyperghrelinemia (Jerlhag, 2008; Jerlhag et al., 2012) and that elevated ghrelin levels associated with craving (Addolorato et al., 2006; Koopmann et al., 2012; Leggio et al., 2012), may imply that high plasma levels of ghrelin may be needed for reward interactions. Supportively, animal studies show that hyperghrelinemia is associated with cocaine seeking and that peripheral ghrelin administration augments the cocaine‐induced conditioned place preference and locomotor stimulation (Clifford et al., 2012; Davis et al., 2007; Tessari et al., 2007; Wellman et al., 2005, 2012). Conclusively, future studies on the role of peripheral versus central ghrelin in relation to alcohol reward, intake, and craving are warranted.
Herein we present novel data showing that NOX‐B11‐2, which prevents brain access for circulating ghrelin, does not affect the ability of alcohol to induce a locomotor stimulation, accumbal dopamine release, and expression of conditioned place preference in mice. It should be noted that this conditioned place preference design possibly does not allow to distinguishing whether alcohol works through increasing preference or through decreasing avoidance of the nonpreferred floor. The possibility that anxiolytic, rather than rewarding, effects of alcohol are studies should also be considered. However, we hypothesize that the rewarding properties of alcohol are measured as we also show that NOX‐B11‐2 does not interfere with parameters reflecting reward and activation of the mesolimbic dopamine system, namely the alcohol‐induced locomotor stimulation and accumbal dopamine release. In the present series of experiments, a dose of NOX‐B11‐2 that does not affect the locomotor activity, accumbal dopamine release, and conditioned place preference was used. Moreover, the selected dose did not affect the blood alcohol concentration in mice.
In addition to central ghrelin production, a role for GHS‐R1A in alcohol‐mediated behaviors should be considered. Indeed, previous studies show that pharmacological (central or peripheral GHS‐R1A antagonist administration) and genetical suppression of GHS‐R1A signaling blocks the alcohol‐induced locomotor stimulation, accumbal dopamine release, and conditioned place preference in rodents (Jerlhag et al., 2009). In addition, GHS‐R1A antagonism reduces the intake and motivation to consume alcohol in rodents (Jerlhag et al., 2009; Kaur and Ryabinin, 2010; Landgren et al., 2012). Additionally, the expression of GHS‐R1A in the ventral tegmental area is higher in high‐ compared to low‐alcohol consuming rats (Landgren et al., 2011). Further support for a role of GHS‐R1A in reward regulation is the human genetic data showing that a single nucleotide polymorphism in the GHS‐R1A gene is associated with high alcohol consumption in humans (Landgren et al., 2008). Given that GHS‐R1A has been shown to alter the sensitivity of the mesolimbic dopamine system via its ability to heterodimerize with dopamine D1‐ and D2‐like receptors (Jiang et al., 2006; Kern et al., 2012) as well as via its constitutive activity (Holst et al., 2003), we hypothesize that ventral tegmental GHS‐R1A are important for reward processes and for development of alcohol use disorder. Given that ghrelin is an orexigenic peptide and that alcohol contains calories, it should be considered that the ability of GHS‐R1A antagonists to reduce alcohol intake is due to interference with alcohol's calorific value and rather than its rewarding properties. However, this appears less likely since animal studies show that the rewarding properties of rewards without caloric content, such as cocaine, amphetamine, and nicotine, are attenuated by GHS‐R1A antagonist treatment (Jerlhag and Engel, 2011; Jerlhag et al., 2010). In addition to drug reward, central ghrelin signaling also appears to be important for natural rewards such as palatable food, sucrose, and saccharine (for review, see Egecioglu et al., 2011). In the present series of experiments, we showed that NOX‐B11‐2 does not affect the preference for saccharin, supporting a role for central, rather than peripheral, ghrelin signaling for the intake of palatable rewards. Collectively, these data imply that GHS‐R1A have a general role in reward and addiction processes.
The present findings support a physiological role for circulating ghrelin in food intake, appetite as well as meal initiation (Egecioglu et al., 2011; Wren et al., 2000, 2001a,b), as we here showed that NOX‐B11‐2 reduced food intake in alcohol‐drinking rats. Supportively, previous studies show that NOX‐B11‐2 decreased food intake in rodents via ghrelin‐dependent mechanisms (Kobelt et al., 2006; Shearman et al., 2006). In both rodents and humans, the plasma levels of ghrelin increase preprandially and thereby induce food intake (for review, see Egecioglu et al., 2011). By administrating NOX‐B11‐2 before the dark phase in alcohol‐drinking rats, the preprandial increase of ghrelin is blunted, which, in all probability, may cause the observed reduction in food intake. Ghrelin‐induced food intake is, at least in part, mediated via hypothalamic circuits (Wren et al., 2000, 2001b), raising the possibility that circulating ghrelin may access the hypothalamus but not deeper brain areas such as the mesolimbic dopamine system. However, this appears less likely since peripheral ghrelin administration increases the locomotor activity, accumbal dopamine release, and conditioned place preference (Jerlhag, 2008). We therefore hypothesize that circulating endogenous ghrelin is a physiological regulator of food intake and meal initiation, rather than a mediator of alcohol reinforcement. The results regarding food intake may be of clinical interest as prevention of ghrelin's brain access tentatively could be used as treatment of compulsive overeating.
Collectively, it may be implied that central ghrelin signaling, specifically via GHS‐R1A on reward nodes, may be a potential target for pharmacological treatment of alcohol dependence.
Acknowledgments
Britt‐Mari Larsson and Kenn Johannessen are gratefully acknowledged for expert and valuable technical assistance. Axel Vater is an employee of NOXXON Pharma AG, which is the company that provided NOX‐B11‐2, but has no competing interests in regard to the present manuscript. However, this does not alter the author's adherence to any policies on sharing data and materials. The research was supported by grants from the Swedish Research Council (grant nos. K2006‐21X‐04247‐33‐3, 2009‐2782, and K2010‐80X‐21496‐01‐6), The Swedish brain foundation, LUA/ALF (grant no. 148251) from the Sahlgrenska University Hospital, Alcohol research council of the Swedish alcohol retailing monopoly and the foundations of Adlerbertska, Fredrik and Ingrid Thuring, Tore Nilsson, Längmanska, Torsten and Ragnar Söderberg, Wilhelm and Martina Lundgren, NovoNordisk, Knut and Alice Wallenberg, Magnus Bergvall, Anérs, Jeansons, Åke Wiberg, the Swedish Society of Medicine, Swedish Society for Medical Research.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschop MH, Gao XB, Horvath TL (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116:3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addolorato G, Capristo E, Leggio L, Ferrulli A, Abenavoli L, Malandrino N, Farnetti S, Domenicali M, D'Angelo C, Vonghia L, Mirijello A, Cardone S, Gasbarrini G (2006) Relationship between ghrelin levels, alcohol craving, and nutritional status in current alcoholic patients. Alcohol Clin Exp Res 30:1933–1937 [DOI] [PubMed] [Google Scholar]
- Badaoui A, De Saeger C, Duchemin J, Gihousse D, de Timary P, Starkel P (2008) Alcohol dependence is associated with reduced plasma and fundic ghrelin levels. Eur J Clin Invest 38:397–403 [DOI] [PubMed] [Google Scholar]
- Calissendorff J, Danielsson O, Brismar K, Rojdmark S (2006) Alcohol ingestion does not affect serum levels of peptide YY but decreases both total and octanoylated ghrelin levels in healthy subjects. Metabolism 55:1625–1629 [DOI] [PubMed] [Google Scholar]
- Carnicella S, Amamoto R, Ron D (2009) Excessive alcohol consumption is blocked by glial cell line‐derived neurotrophic factor. Alcohol 43:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS, Rodriguez J, Schul D, Hughes S, Kniffin T, Hart N, Eitan S, Brunel L, Fehrentz JA, Martinez J, Wellman PJ (2012) Attenuation of cocaine‐induced locomotor sensitization in rats sustaining genetic or pharmacologic antagonism of ghrelin receptors. Addict Biol 17:956–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia‐Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu HY, Pinto S, Colmers WF, Cone RD, Horvath TL (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37:649–661 [DOI] [PubMed] [Google Scholar]
- Davis KW, Wellman PJ, Clifford PS (2007) Augmented cocaine conditioned place preference in rats pretreated with systemic ghrelin. Regul Pept 140:148–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson SL, Egecioglu E, Landgren S, Skibicka KP, Engel JA, Jerlhag E (2011) The role of the central ghrelin system in reward from food and chemical drugs. Mol Cell Endocrinol 340:80–87 [DOI] [PubMed] [Google Scholar]
- Egecioglu E, Skibicka KP, Hansson C, Alvarez‐Crespo M, Friberg PA, Jerlhag E, Engel JA, Dickson SL (2011) Hedonic and incentive signals for body weight control. Rev Endocr Metab Disord 12:141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1996) The Mouse Brain in Stereotaxic Coordinates. Academic Press, New York, NY [Google Scholar]
- Hillemacher T, Kraus T, Rauh J, Weiss J, Schanze A, Frieling H, Wilhelm J, Heberlein A, Groschl M, Sperling W, Kornhuber J, Bleich S (2007) Role of appetite‐regulating peptides in alcohol craving: an analysis in respect to subtypes and different consumption patterns in alcoholism. Alcohol Clin Exp Res 31:950–954 [DOI] [PubMed] [Google Scholar]
- Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW (2003) High constitutive signaling of the ghrelin receptor — identification of a potent inverse agonist. Mol Endocrinol 17:2201–2210 [DOI] [PubMed] [Google Scholar]
- Jerlhag E (2008) Systemic administration of ghrelin induces conditioned place preference and stimulates accumbal dopamine. Addict Biol 13:358–363 [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA (2006a) Ghrelin stimulates locomotor activity and accumbal dopamine‐overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol 11:45–54 [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA (2007) Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol 12:6–16 [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Dickson SL, Engel JA (2010) Ghrelin receptor antagonism attenuates cocaine‐ and amphetamine‐induced locomotor stimulation, accumbal dopamine release, and conditioned place preference. Psychopharmacology 211:415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Egecioglu E, Landgren S, Salome N, Heilig M, Moechars D, Datta R, Perrissoud D, Dickson SL, Engel JA (2009) Requirement of central ghrelin signaling for alcohol reward. Proc Natl Acad Sci USA 106:11318–11323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Engel JA (2011) Ghrelin receptor antagonism attenuates nicotine‐induced locomotor stimulation, accumbal dopamine release and conditioned place preference in mice. Drug Alcohol Depend 117:126–131 [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Grotli M, Luthman K, Svensson L, Engel JA (2006b) Role of the subunit composition of central nicotinic acetylcholine receptors for the stimulatory and dopamine‐enhancing effects of ethanol. Alcohol Alcohol 41:486–493 [DOI] [PubMed] [Google Scholar]
- Jerlhag E, Janson A‐C, Waters S, Engel JA (2012) Concomitant release of ventral tegmental acetylcholine and acumbal dopamine release by ghrelin in rats. Plos One 7:e49557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlhag E, Landgren S, Egecioglu E, Dickson SL, Engel JA (2011) The alcohol‐induced locomotor stimulation and accumbal dopamine release is suppressed in ghrelin knockout mice. Alcohol 45:341–347 [DOI] [PubMed] [Google Scholar]
- Jiang H, Betancourt L, Smith RG (2006) Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol 20:1772–1785 [DOI] [PubMed] [Google Scholar]
- Kaur S, Ryabinin AE (2010) Ghrelin receptor antagonism decreases alcohol consumption and activation of perioculomotor urocortin‐containing neurons. Alcohol Clin Exp Res 34:1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Albarran‐Zeckler R, Walsh HE, Smith RG (2012) Apo‐ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 73:317–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, Yoon SJ, Choi B, Kim TS, Woo YS, Kim W, Myrick H, Peterson BS, Bin Choi Y, Kim YK, Jeong J (2005) Increased fasting plasma ghrelin levels during alcohol abstinence. Alcohol Alcohol 40:76–79 [DOI] [PubMed] [Google Scholar]
- Kobelt P, Helmling S, Stengel A, Wlotzka B, Andresen V, Klapp BF, Wiedenmann B, Klussmann S, Monnikes H (2006) Anti‐ghrelin Spiegelmer NOX‐B11 inhibits neurostimulatory and orexigenic effects of peripheral ghrelin in rats. Gut 55:788–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth‐hormone‐releasing acylated peptide from stomach. Nature 402:656–660 [DOI] [PubMed] [Google Scholar]
- Koopmann A, von der Goltz C, Grosshans M, Dinter C, Vitale M, Wiedemann K, Kiefer F (2012) The association of the appetitive peptide acetylated ghrelin with alcohol craving in early abstinent alcohol dependent individuals. Psychoneuroendocrinology 37:980–986 [DOI] [PubMed] [Google Scholar]
- Kraus T, Schanze A, Groschl M, Bayerlein K, Hillemacher T, Reulbach U, Kornhuber J, Bleich S (2005) Ghrelin levels are increased in alcoholism. Alcohol Clin Exp Res 29:2154–2157 [DOI] [PubMed] [Google Scholar]
- Landgren S, Engel JA, Hyytia P, Zetterberg H, Blennow K, Jerlhag E (2011) Expression of the gene encoding the ghrelin receptor in rats selected for differential alcohol preference. Behav Brain Res 221:182–188 [DOI] [PubMed] [Google Scholar]
- Landgren S, Jerlhag E, Zetterberg H, Gonzalez‐Quintela A, Campos J, Olofsson U, Nilsson S, Blennow K, Engel JA (2008) Association of pro‐ghrelin and GHS‐R1A gene polymorphisms and haplotypes with heavy alcohol use and body mass. Alcohol Clin Exp Res 32:2054–2061 [DOI] [PubMed] [Google Scholar]
- Landgren S, Simms JA, Hyytia P, Engel JA, Bartlett SE, Jerlhag E (2012) Ghrelin receptor (GHS‐R1A) antagonism suppresses both operant alcohol self‐administration and high alcohol consumption in rats. Addict Biol 17:86–94 [DOI] [PubMed] [Google Scholar]
- Larsson A, Engel JA (2004) Neurochemical and behavioral studies on ethanol and nicotine interactions. Neurosci Biobehav Rev 27:713–720 [DOI] [PubMed] [Google Scholar]
- Larsson A, Jerlhag E, Svensson L, Soderpalm B, Engel JA (2004) Is an alpha‐conotoxin MII‐sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol 34:239–250 [DOI] [PubMed] [Google Scholar]
- Leggio L, Ferrulli A, Cardone S, Nesci A, Miceli A, Malandrino N, Capristo E, Canestrelli B, Monteleone P, Kenna GA, Swift RM, Addolorato G (2012) Ghrelin system in alcohol‐dependent subjects: role of plasma ghrelin levels in alcohol drinking and craving. Addict Biol 17:452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Guan JL, Wang QP, Uehara K, Yamada S, Goto N, Date Y, Nakazato M, Kojima M, Kangawa K, Shioda S (2002) Immunocytochemical observation of ghrelin‐containing neurons in the rat arcuate nucleus. Neurosci Lett 321:157–160 [DOI] [PubMed] [Google Scholar]
- Lyons AM, Lowery EG, Sparta DR, Thiele TE (2008) Effects of food availability and administration of orexigenic and anorectic agents on elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res 32:1962–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee SA, Harrison EL, O'Malley SS, Krishnan‐Sarin S, Shi J, Tetrault JM, Picciotto MR, Petrakis IL, Estevez N, Balchunas E (2009) Varenicline reduces alcohol self‐administration in heavy‐drinking smokers. Biol Psychiatry 66:185–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JM, Teague CH, Kayser AS, Bartlett SE, Fields HL (2012) Varenicline decreases alcohol consumption in heavy‐drinking smokers. Psychopharmacology 223:299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal MS, Date Y, Yamaguchi H, Toshinai K, Tsuruta T, Kangawa K, Nakazato M (2005) Identification of ghrelin and its receptor in neurons of the rat arcuate nucleus. Regul Pept 126:55–59 [DOI] [PubMed] [Google Scholar]
- Shearman LP, Wang SP, Helmling S, Stribling DS, Mazur P, Ge L, Wang L, Klussmann S, Macintyre DE, Howard AD, Strack AM (2006) Ghrelin neutralization by a ribonucleic acid‐SPM ameliorates obesity in diet‐induced obese mice. Endocrinology 147:1517–1526 [DOI] [PubMed] [Google Scholar]
- Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, Wise R, Bartlett SE (2008) Intermittent access to 20% ethanol induces high ethanol consumption in Long‐Evans and Wistar rats. Alcohol Clin Exp Res 32:1816–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderpalm B, Lof E, Ericson M (2009) Mechanistic studies of ethanol's interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry 42(Suppl 1):S87–S94 [DOI] [PubMed] [Google Scholar]
- Steensland P, Fredriksson I, Holst S, Feltmann K, Franck J, Schilstrom B, Carlsson A (2012) The monoamine stabilizer (‐)‐OSU6162 attenuates voluntary ethanol intake and ethanol‐induced dopamine output in nucleus accumbens. Biol Psychiatry 72:823–331 [DOI] [PubMed] [Google Scholar]
- Steensland P, Simms JA, Holgate J, Richards JK, Bartlett SE (2007) Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci USA 104:12518–12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessari M, Catalano A, Pellitteri M, Di Francesco C, Marini F, Gerrard PA, Heidbreder CA, Melotto S (2007) Correlation between serum ghrelin levels and cocaine‐seeking behaviour triggered by cocaine‐associated conditioned stimuli in rats. Addict Biol 12:22–29 [DOI] [PubMed] [Google Scholar]
- Tschöp M, Smiley DL, Heiman ML (2000) Ghrelin induces adiposity in rodents. Nature 407:908–913 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Li TK (2004) Drug addiction: the neurobiology of behaviour gone awry. Nat Rev Neurosci 5:963–970 [DOI] [PubMed] [Google Scholar]
- Wellman PJ, Clifford PS, Rodriguez JA, Hughes S, Di Francesco C, Melotto S, Tessari M, Corsi M, Bifone A, Gozzi A (2012) Brain reinforcement system function is ghrelin dependent: studies in the rat using pharmacological fMRI and intracranial self‐stimulation. Addict Biol 17:908–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman PJ, Davis KW, Nation JR (2005) Augmentation of cocaine hyperactivity in rats by systemic ghrelin. Regul Pept 125:151–154 [DOI] [PubMed] [Google Scholar]
- Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR (2001a) Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 86:5992–5995 [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA, Bloom SR (2001b) Ghrelin causes hyperphagia and obesity in rats. Diabetes 50:2540–2547 [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DGA, Ghatei MA, Bloom SR (2000) The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141:4325–4328 [DOI] [PubMed] [Google Scholar]
- Zimmermann US, Buchmann A, Steffin B, Dieterle C, Uhr M (2007) Alcohol administration acutely inhibits ghrelin secretion in an experiment involving psychosocial stress. Addict Biol 12:17–21 [DOI] [PubMed] [Google Scholar]