

Abstract
Toll-like receptor 4 (TLR4) induces two distinct signaling pathways controlled by the TIRAP-MyD88 and TRAM-TRIF pairs of adaptor proteins, which elicit the production of proinflammatory cytokines and type I interferons, respectively. How TLR4 coordinates the activation of these two pathways is unknown. Here we show that TLR4 activated these two signaling pathways sequentially in a process organized around endocytosis of the TLR4 complex. We propose that TLR4 first induces TIRAP-MyD88 signaling at the plasma membrane and is then endocytosed and activates TRAM-TRIF signaling from early endosomes. Our data emphasize a unifying theme in innate immune recognition whereby all type I interferon–inducing receptors signal from an intracellular location.
The mammalian immune system is notable for its ability to sense and respond to a wide range of infectious microorganisms. Detection of microbes is mediated by several families of pattern-recognition receptors (PRRs), and this recognition leads to the activation of innate and adaptive immune responses1. Toll-like receptors (TLRs) are the best characterized PRRs and are linked to the control of bacterial and viral infection2. After microbial detection, one or more adaptor protein(s) containing a Toll–interleukin 1 (IL-1) receptor (TIR) domain bind(s) to the cytosolic TIR domains of TLRs. Four TIR domain–containing adaptors are involved in propagating TLR signaling: MyD88, TIRAP (also called Mal), TRAM and TRIF. These adaptors link activated TLRs with ‘downstream’ kinases of the IL-1 receptor–associated kinase and mitogen-activated protein kinase families, as well as with members of the TRAF family of E3 ubiquitin ligases. Activation of these enzymes leads to the activation of transcriptional regulators such as NF-κB, AP-1 and several interferon-regulatory factors (IRFs), which induce hundreds of genes involved in immune defense3. Notably, not all TLRs use the same set of adaptors, and adaptor ‘choice’ determines the transcriptional response induced after microbial detection. For example, the use of TIRAP and MyD88 by some TLRs induces the production of proinflammatory cytokines, whereas the use of TRAM and TRIF elicits a type I interferon response4–7.
Studies have identified a notable aspect of TLR biology: a link between receptor localization, the type of transcriptional response induced and the class of microbe detected1,8,9. For example, TLR2, TLR4 (A002296) and TLR5 all recognize different components of a bacterial cell wall and, appropriately, are found on the cell surface, where they induce the production of proinflammatory cytokines after the detection of microbes10,11. In contrast, TLR3, TLR7 and TLR9 detect viral nucleic acids and are found in endolysosomal compartments, where they are poised to detect nucleic acids released after viral degradation12–14. The latter group of TLRs induces a potent type I interferon response that is critical for antiviral defense15. TLR7 and TLR9 signal from an endolysosomal compartment, where they use MyD88 to activate IRF7 (refs. 16,17), whereas TLR3 signals from a distinct endosomal compartment, where it activates IRF3 through a TRIF-dependent pathway18,19.
In several ways, TLR4 is unique among TLRs. First, TLR4 is the only known TLR able to activate both MyD88-dependent induction of genes encoding inflammatory molecules and TRIF-dependent production of type I interferon20. Second, with the exception of TLR4, all other known receptors that induce the production of type I interferon are sensors of nucleic acids and induce activation of IRF3 or IRF7 from intracellular compartments. TLR3, TLR7 and TLR9 signal from endolysosomes, whereas the RNA helicases RIG-I and MDA-5 and the DNA sensor DAI signal from the cytosolic compartment21,22. Finally, TLR4 is the only known TLR that engages all four TIR domain– containing adaptors23. Those puzzling observations led us to question some of the prevailing assumptions regarding the mechanism of TLR4 signaling.
It is generally believed that TLR4 induces the TIRAP-MyD88– dependent and TRAM-TRIF–dependent signaling pathways simultaneously from the plasma membrane. Unlike the signaling adaptor MyD88, TIRAP functions as a sorting adaptor that recruits MyD88 to TLR4 through its ability to interact with phosphatidylinositol-4, 5-bisphosphate (PtdIns(4,5)P2)24. TIRAP also functions to recruit MyD88 to TLR2 (ref. 25). Thus, by analogy, TRAM may also function as a sorting adaptor to recruit TRIF to TLR4 (ref. 24). TRAM is targeted to the plasma membrane by myristoylation, and the present model of TRAM function is that it recruits TRIF to the plasma membrane, where TLR4 is located26,27.
Here we investigate the mechanism of TRAM function and find that, similar to TIRAP, TRAM indeed functions as a sorting adaptor. We show that TRAM contains a bipartite sorting signal that controls its trafficking between the plasma membrane and endosomes. Notably, we demonstrate that TRAM did not induce TRIF-dependent signaling from the plasma membrane. Instead, delivery of TLR4 and TRAM to endosomes was necessary for activation of the IRF3 signaling pathway. Our results suggest a new model of TLR4 signaling whereby TLR4 activates the two signaling pathways in a sequential way from distinct subcellular compartments. The TIRAP-MyD88 pathway is induced from the plasma membrane, whereas the TRAM-TRIF pathway is induced from endosomes. Our findings suggest that a unifying feature of all PRRs that trigger the type I interferon response is their localization in intracellular compartments. We suggest a possible explanation for this notable phenomenon.
Results
Dynamin controls TLR4 endocytosis
Although extensive functional studies have been done of TLR4, the subcellular sites of TLR4-induced signal transduction remain unclear. To assess this issue, we examined the localization of a tagged construct of TLR4 in which the ectodomain of the receptor was replaced with the ectodomain of CD4. TLR4 was present at the plasma membrane and on intracellular vesicles that costained with the early endosomal marker Rab5 (Fig. 1a). We obtained similar results with a full-length hemagglutinin-tagged construct of TLR4 (Supplementary Fig. 1a). We did not detect TLR4 staining on other subcellular organelles such as the Golgi complex or the endoplasmic reticulum (data not shown).
Figure 1.

Inhibition of TLR4 endocytosis selectively disrupts TRAM-TRIF signaling. (a) Fluorescence microscopy of macrophages transfected with plasmid encoding a TLR4 construct with the ectodomain of the receptor replaced with the ectodomain of CD4 (CD4-TLR4) or Rab5 tagged with red fluorescent protein (RFP-Rab5). Outlined areas are enlarged in bottom left corners. Scale bars, 5 μm. Data are representative of at least three independent experiments with over 500 cells per condition, in which over 95% of the cells had similar staining patterns. (b) Flow cytometry of TLR4 surface staining on wild-type or TLR4-deficient (TLR4-KO) macrophages left untreated (left) or treated with dynasore (right) and left unstimulated (Untreated) or stimulated with LPS (100 ng/ml), assessed with the TLR4-specific antibody Sa15-21. PE, phycoerythrin. Data are representative of two independent experiments. (c) Immunoblot analysis of macrophages treated for 0, 20 or 40 min with LPS (100 ng/ml) and either dimethyl sulfoxide (DMSO) or 80 μM dynasore and probed for phosphorylated (p-) or total IRF3 or p38. Data are representative of two independent experiments. (d,e) RT-PCR analysis (d) and ELISA (e) of LPS-induced gene expression in macrophages left untreated or treated with dynasore. Data are representative of three independent experiments.
Published studies have established that lipopolysaccharide (LPS) and CD14 are internalized from the plasma membrane into early endosomes by a process dependent on the GTPase dynamin28. Dynamin GTPases regulate endocytosis by controlling the ‘pinching off’ of plasma membrane invaginations, which creates early endosomes29. To determine whether TLR4 is also internalized by a dynamin-dependent process, we used an antibody (Sa15-21) that recognizes TLR4 in the presence or absence of LPS30 and a highly specific inhibitor of dynamin called ‘dynasore’31. The use of dynasore, rather than dominant negative dynamin mutants, allowed us to bypass the compensatory upregulation of other endocytic processes that can occur after long-term inhibition of dynamin, a situation that has confounded experimental interpretations in other systems32–34. These reagents allowed us to examine the activity of endogenous TLR4 in primary cells. The antibody Sa15-21 specifically recognized TLR4, as staining was lost in TLR4-deficient cells (Fig. 1b). Consistent with published reports28,30, within 30 min of LPS treatment, TLR4 cell surface staining was lost, which indicated that TLR4 is internalized after ligand binding. In contrast, treatment with dynasore prevented the LPS-induced internalization of TLR4 (Fig. 1b). These data indicate that like CD14 and LPS, TLR4 is internalized by a dynamin-dependent process.
TRAM-TRIF signaling requires TLR4 endocytosis
LPS-induced TLR4 internalization is thought to result in downregulation of TLR4 signaling and, consequently, macrophage desensitization28,30. Alternatively, the internalization could be a requisite step in TLR4 signaling. To determine the functional consequence of disrupting the LPS-induced internalization of TLR4, we examined signaling events dependent on either TRAM-TRIF or TIRAP-MyD88 signaling. LPS-induced TRAM-TRIF-dependent phosphorylation of IRF3, a transcription factor necessary for the expression of type I interferon4,5,18,19,26, was abolished in dynasore-treated cells (Fig. 1c). In contrast, LPS-induced phosphorylation of the mitogen-activated protein kinase p38 and degradation of the inhibitor protein IκBα, both TIRAP-MyD88–dependent events6,7, proceeded normally in dynasore-treated macrophages (Fig. 1c and Supplementary Fig. 1b).
To corroborate those findings, we examined the effect of inhibiting TLR4 endocytosis on LPS-induced transcriptional responses. Dynasore treatment abolished the LPS-induced expression of the TRAM-TRIF–dependent genes encoding interferon-β (IFN-β), the chemokine RANTES (CCL5) and IL-6 (refs. 4,5; Fig. 1d,e). In contrast, IκBα expression, which can be controlled by either the TIRAP-MyD88 or the TRAM-TRIF pathway3, was still detectable, albeit to a diminished degree (Fig. 1d).
The results presented above collectively suggested that rather than simply serving the function of desensitizing cells, LPS- and dynamin-dependent endocytosis of TLR4 is needed to induce TRAM-TRIF– dependent signaling and production of type I interferon. In contrast, dynasore did not interfere with p38 phosphorylation or IκBα expression, consistent with the idea that TLR4 induces the two signaling pathways from two different locations: TIRAP-MyD88 from the plasma membrane, and TRAM-TRIF from an intracellular compartment.
Next we sought to understand the mechanisms by which TLR4 triggers the production of type I interferon from endosomes. The ability of TLR4 to induce TIRAP-MyD88–dependent signaling is linked to the localization of TIRAP to PtdIns(4,5)P2-rich regions of the plasma membrane by virtue of its amino-terminal PtdIns(4,5)P2-binding domain24. Thus, we considered the possibility that the ability of TLR4 to induce TRAM-TRIF–dependent signaling would be linked to TRAM localization. Examination of the subcellular distribution of TRAM showed that like TLR4, this adaptor was present at both the plasma membrane and in Rab5+ early endosomes (Fig. 2a). In contrast, TIRAP, although present at the cell surface, was not detectable on early endosomes (Fig. 2b).
Figure 2.

Distinct subcellular distribution of TRAM and TIRAP. (a,b) Fluorescence microscopy of macrophages transfected with plasmids encoding GFP-TRAM (a) or GFP-TIRAP (b) and RFP-Rab5. (c,d) Fluorescence microscopy of macrophages transfected with plasmid encoding GFP-TRAM (c) or CD4-TLR4 (d) and treated for 30 min with dextran labeled with Texas red. Outlined areas are enlarged in bottom left corners. Scale bars, 5 μm. Data are representative of at least three independent experiments with over 500 cells per condition, in which over 95% of the cells had similar staining patterns.
In macrophages, true endosomes are able to rapidly accumulate material internalized from the extracellular milieu35. To determine if the TRAM+ vesicles were able to incorporate internalized materials, we supplemented the macrophage culture medium with fluorescence-labeled dextran and within minutes noted prominent accumulation of these tracers in TRAM+ compartments (Fig. 2c). Similarly, TLR4+ vesicles costained with dextran (Fig. 2d). Thus, in contrast to TIRAP, both TRAM and TLR4 were present at the plasma membrane and on a true endosomal compartment. Notably, although dynasore interfered with the endocytosis of TLR4, the localization of TRAM was insensitive to dynasore (data not shown), which suggested that TRAM can target endosomes directly, bypassing the plasma membrane. That observation suggests that TLR4 and TRAM can traffic to endosomes by different mechanisms and therefore may not need to be transported together after internalization.
TRAM contains a bipartite localization motif
Next we did deletion analysis of TRAM to identify the cis-acting regulatory sequences responsible for membrane targeting. A TRAM mutant lacking the TIR domain had a subcellular distribution similar to that of the wild-type protein (Fig. 3a,b). We obtained similar results with TRAM mutants lacking all but the first 20 residues, which indicated that although TRAM localized together with TLR4, it did so independently of TIR-dependent interactions with this receptor (Fig. 3c). These results confirm the idea that TRAM and TLR4 can use distinct transport pathways to gain access to the organelles in which they both reside. Notably, TRAM mutants containing only the first seven amino acids had a unique subcellular distribution. Instead of residing mainly at the plasma membrane with a minor population on endosomes, such mutants were located exclusively on endosomes (Fig. 3d). Thus, the minimum localization domain of TRAM is defined as amino acids 1–20, but a subdomain from residue 1 to residue 7 is sufficient for endosomal targeting. Examination of the properties of the minimum localization motif showed a domain with a higher isoelectric point than that of full-length TRAM. Residues 1–20 had a isoelectric point of nearly 10, whereas each successive set of 20 residues throughout the protein had a isoelectric point of less than 5 (Fig. 3e). The domain of residues 1–20 contains the previously characterized myristoylation motif of residues 1–7 (ref. 27). The region of positions 8–20 is enriched for basic and aromatic residues, which explains the high isoelectric point of this domain (Fig. 3f).
Figure 3.

A bipartite localization motif regulates the localization of TRAM. (a–d) Fluorescence microscopy of macrophages transfected with plasmids encoding chimeric proteins consisting of full-length TRAM (a) or various amino acids (in parentheses) of TRAM (b–d) fused in-frame to GFP. Outlined areas are enlarged in bottom left corners. Scale bars, 5 μm. Data are representative of at least three independent experiments with over 500 cells per condition, in which over 95% of the cells had similar staining patterns (unless stated otherwise elsewhere). (e) Isoelectric points of sequential 20–amino acid segments of TRAM. Data are representative of experiments done two times. (f) Primary structure of mouse TRAM. Below, bipartite localization motif (amino acids 1–20), with the myristoylation motif (red letters) followed by the polybasic motif (substituted amino acids are underlined). (g) Amino acid alignment of human proteins identified as containing a bipartite localization motif, with putative myristoylation motifs (underlined residues) followed by the polybasic motif, and with basic and aromatic residues indicated by red lettering.
To determine if this bipartite localization motif (consisting of an amino-terminal myristate group followed by a polybasic domain) is found in other proteins of known function, we searched the human genome for proteins that share these physiochemical properties. This analysis yielded 36 proteins whose first 20 amino acid residues have properties similar to those of TRAM and whose remaining residues have a difference in isoelectric point of over 4 units relative to that of the first 20 residues. The proteins identified by these criteria do not contain a conserved sequence motif (except for the myristoylation consensus; Fig. 3g), yet all share the common physiochemical properties of the bipartite motif of TRAM in that they contain a putative site of myristoylation followed by a region enriched for basic and aromatic residues. Indeed, even human and mouse sequences of TRAM did not align perfectly, yet both contain this motif (Fig. 3f,g).
Notably, this group of 36 proteins included many regulators of signal transduction that shuttle between the plasma membrane and endosomes, such as TRAM, Src, Yes-1 and MARCKS, as well as β-arrestins and members of the ARF family of GTPases. All of these proteins are known to cycle between the plasma membrane and an internal endosomal pool, regulating some aspect of signal transduc-tion36–38. We therefore hypothesized that this bipartite motif is a common localization domain used by proteins that control the organization of signaling pathways that operate at both the plasma membrane and an intracellular compartment.
To assess the importance of the bipartite localization domain in TRAM, we selectively substituted either the glycine at position 2 (to eliminate myristoylation) or the polybasic motif by replacing three conserved basic or aromatic residues with glutamic acid (underlined residues, Fig. 3f; Fig. 4). As reported before27, a myristoylation-deficient TRAM mutant was completely mislocalized and was uniformly distributed throughout the cell (Fig. 4b). In contrast, a TRAM construct with substitutions in the polybasic domain had the same localization as the TRAM mutant containing only the first seven amino acids (Figs. 3d and 4a); both of these mutants lacked the polybasic domain (by either substitution or deletion) and both were exclusively located in early endosomes that costained with Rab5. These results indicate that both halves of the bipartite motif are necessary for plasma membrane targeting but only myristoylation is required for endosomal localization.
Figure 4.

TLR4 can induce the production of type I interferon from early endosomes. (a,b) Fluorescence microscopy of macrophages transfected with plasmids encoding a GFP-tagged TRAM construct with substitutions in the polybasic domain (TRAM3X) and RFP-Rab5 (a) or with a myristoylation-deficient TRAM mutant (TRAMG2A; b). Outlined areas are enlarged in the bottom left and top right corners (a). Scale bars, 5 μm. Data are representative of at least three independent experiments with over 500 cells per condition, in which over 95% of the cells had similar staining patterns. (c) ‘PIP strips’ of various lipids (identified at right) overlaid with GST-tagged wild-type TRAM (left) or TRAM with substitutions in the polybasic domain (middle); lipid binding was identified by standard protein-protein immunoblot techniques. LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine 1-phosphate; PA, phosphatidic acid; PS, phosphatidylserine. Data are representative of two independent experiments. (d) ELISA of IL-6 and RANTES in TRAM-deficient macrophages transduced with retroviral vectors encoding GFP-tagged TRAM mutants (horizontal axes) and challenged with 1, 10 or 100 ng/ml (key) of LPS. The transduction efficiency of each resulting macrophage population was determined to be 30–40% by flow cytometry and fluorescence microscopy. Data are representative of three independent experiments.
Because the first half of the bipartite motif is the site of myristate attachment, we investigated the biochemical interactions that occur with the polybasic region of this motif. Polybasic motifs are commonly found in proteins that are able to bind acidic phospholipids39. To assess the ability of TRAM to bind to membrane lipids, we used ‘PIP strips’, which are nitrocellulose filters that contain a variety of lipid species commonly found in mammalian cells. A fusion protein of glutathione S-transferase (GST) and TRAM bound detectably to many acidic phospholipids, including most phosphoinositides, as well as to phosphatidic acid (Fig. 4c). Liposome-binding assays further confirmed those results (data not shown). Notably, in neither assay did TRAM show ‘preference’ for any particular lipid species (Fig. 4c and data not shown). In contrast to wild-type TRAM, the TRAM construct with substitutions in the polybasic domain did not bind detectably to any lipid species, which indicated that the polybasic domain of TRAM is necessary for binding to acidic phospholipids (Fig. 4c). Such binding properties are distinct from results obtained before with the polybasic domain of TIRAP, which binds ‘preferentially’ to PtdIns(4,5)P2 (ref. 24). Because the polybasic regions of both TIRAP and TRAM are necessary for plasma membrane targeting, we hypothesize that the presence of the myristate group in TRAM alleviates the need for a specific phospholipid-binding domain, like that found in TIRAP. Thus, it is likely that both adaptors are recruited to the plasma membrane by interactions with acidic phospholipids but that the cis-acting sequences that mediate membrane targeting do so by distinct mechanisms.
Endosomal TRAM-TRIF signaling
The endosomal localization of the TRAM construct with substitutions in the polybasic domain provided us with a tool for studying the importance of endosomal residence of this adaptor in terms of TLR4 signal transduction. For this, we reconstituted TRAM-deficient mouse bone marrow–derived macrophages with retroviral vectors encoding wild-type TRAM, the myristoylation-deficient TRAM mutant or the TRAM construct with substitutions in the polybasic domain and assessed the ability of each construct to complement the signaling defect intrinsic to TRAM-deficient cells. Macrophages ‘mock’ infected or infected with virus encoding the myristoylation-deficient TRAM mutant were unable to produce the cytokines IL-6 and RANTES after LPS treatment, which indicated that membrane localization of TRAM is necessary for the initiation of signal transduction (Fig. 4d). In contrast, cells reconstituted with the TRAM construct with substitutions in the polybasic domain produced substantial amounts of those cytokines (Fig. 4d), which suggested that endosomal residence of TRAM is sufficient to induce TLR4-mediated TRAM-TRIF signaling. Similarly, a TRAM mutant in which the entire bipartite localization domain was replaced with the analogous region from Src was able to support TLR4 signaling when expressed in TRAM-deficient macrophages (Supplementary Fig. 1c,d). These data provide functional evidence of the importance of the proteins we identified containing bipartite localization motifs. To assess whether delivery of TRAM to endosomes is necessary for TLR4 signaling, we replaced the localization domain of TRAM with the plasma membrane– targeting domain from the tyrosine kinase Fyn. The localization motif of Fyn is a Src homology 4 domain, which differs from the localization domains of TRAM and Src because the Src homology 4 domain is a dual-acylation motif rather than a bipartite motif 38. Dual-acylation motifs bind tightly to the plasma membrane, which precludes their efficient delivery to endosomes. Notably, the chimera of the TRAM with its localization domain replaced with the plasma membrane–targeting domain of Fyn only weakly promoted TLR4 signaling in reconstituted macrophages (Supplementary Fig. 1c,d), which suggested that efficient endosomal delivery of TRAM is required for optimal TLR4 signaling. These data, along with the observation that dynasore disrupted TLR4 internalization and TRAM-TRIF–dependent signaling, show that residence of TRAM on endosomes is both necessary and sufficient to induce TLR4 signaling and support a model whereby LPS induces the internalization of TLR4 into endosomes where the TRAM-TRIF– dependent signaling pathway is activated.
TRAF3 localization dictates interferon-producing ability
Before this report, TLR4 was the only apparent exception to the rule that induction of type I interferon by PRRs occurs from the intracellular compartments and not from the plasma membrane. Our results, however, indicated that TLR4 triggers TRAM-TRIF–dependent signaling for IFN-β induction from an endosomal compartment. Thus, no known PRR can trigger the production of type I interferon from the cell surface. The mechanistic explanation for this phenomenon is unknown at present. One possibility is that a key regulator of the production of type I interferon is anchored in an intracellular location and is therefore inefficiently delivered to the plasma membrane. The requirement for intracellular localization applies to TLR7 and TLR9, which signal by MyD88-dependent IRF7 activation16,17, and to TLR3 and TLR4, which signal by TRIF-dependent IRF3 activation18,19. The only known component shared by these two pathways is TRAF3 (A002309), which is critically involved in the induction of type I interferon by both mechanisms40,41. Therefore, we considered the possibility that accessibility to TRAF3 may account for the requirement for intracellular localization for the induction of type I interferon. Unlike TIRAP and TRAM, we did not detect TRAF3 at the plasma membrane; instead, pleomorphic structures scattered through-out the cytosol were enriched for TRAF3 (Fig. 5a). Treatment with LPS did not induce any detectable change in the subcellular distribution of TRAF3 (data not shown).
Figure 5.
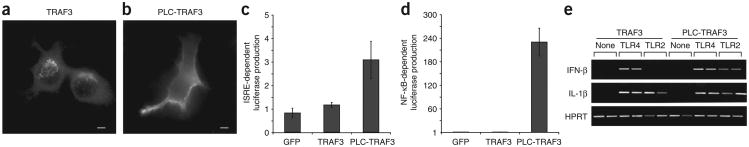
The subcellular localization of TRAF3 dictates the ability of TLRs to produce type I interferon. (a,b) Fluorescence microscopy of macrophages transfected with plasmids encoding hemagglutinin-tagged constructs of wild-type TRAF3 (a) or a chimera of the pleckstrin homology domain of PLC-δ1 and TRAF3 (PLC-TRAF3; b). Scale bars, 5 μm. Data are representative of at least three independent experiments with over 500 cells per condition, in which over 95% of the cells had similar staining patterns. (c,d) Luciferase production by 293T cells 24 h after transfection with expression vectors encoding GFP only, wild-type TRAF3 or the chimera of PLC-δ1 and TRAF3, plus a luciferase reporter dependent on ISRE (c) or NF-κB (d). Values are relative to those of the GFP vector, set as 1. Data are representative of three independent experiments. (e) RT-PCR analysis of the expression of genes encoding IFN-β, IL-1β or HPRT by RAW267 macrophage-like cells stably expressing either wild-type TRAF3 (left) or the chimera of PLC-δ1 and TRAF3 (right), left untreated (None) or treated for 4 h with the TLR4 agonist LPS (100 ng/ml; TLR4) or the TLR2 agonist PAM3CSK (1 μg/ml; TLR2). Data are representative of three independent experiments done in duplicate.
Either MyD88 or TRIF can engage TRAF3 (ref. 41), which suggests that any TLR should be able to induce expression of type I interferon through MyD88. However, only intracellular TLRs induce TRAF3-dependent type I interferon. If one of the reasons why plasma membrane–resident TLRs are unable to induce type I interferon is the spatial separation of these receptors from TRAF3, then targeting TRAF3 to the plasma membrane should permit TLRs located in the plasma membrane, such as TLR2, to induce the production of type I interferon. To test that prediction, we generated a plasma membrane– targeted mutant of TRAF3 by attaching the pleckstrin homology domain of phospholipase C-δ1 (PLC-δ1) to the amino terminus of TRAF3. This chimeric protein efficiently localized to the plasma membrane of transfected primary macrophages (Fig. 5b). Although many TRAF family members can induce the activation of NF-κB-dependent reporter genes when overexpressed, TRAF3 is unable to do so42. Notably, whereas wild-type TRAF3 was unable to induce expression of luciferase reporter constructs driven by NF-κB or an interferon-stimulated response element (ISRE), the chimera of PLC-δ1 and TRAF3 induced the activation of each reporter after over-expression (Fig. 5c,d). Thus, simple alteration of the subcellular distribution of TRAF3 can alter signaling potential. To determine if targeting TRAF3 to the plasma membrane could promote the production of type I interferon by TLR2, which does not normally induce such production11, we generated stable RAW264.7 macrophage cell lines expressing either wild-type TRAF3 or the chimera of PLC-δ1 and TRAF3. Both LPS and the TLR2 ligand PAM3CSK induced robust expression of a MyD88-dependent gene encoding IL-1β in cells expressing either TRAF3 construct. Notably, TLR2 induced IFN-β in cells transfected with the PLC-d1–TRAF3 chimera but not those transfected with wild-type TRAF3 (Fig. 5e). Thus, relocalization of TRAF3 to the plasma membrane allowed TLR2-mediated induction of IFN-β expression, which indicates that localization TRAF3 is one of the critical factors responsible for dictating the requirement for intracellular localization of type I interferon–inducing PRRs.
Discussion
The identification of many families of microbe-detection receptors has led to an explosion of interest in understanding the organization of these innate immune signaling pathways. Although genetic analysis of innate immunity has progressed rapidly, much is unclear about how these pathways have been integrated into the more general cellular infrastructure in which they operate. TLR4 represents a good model system for understanding the cellular organization of the initiation of signal transduction, as TLR4 signaling is well characterized genetically, yet many cell-biological questions remain unanswered.
TLR4 induces two independent signaling pathways that are regulated by the TIRAP-MyD88 and TRAM-TRIF adaptor pairs1,23. The TIRAP-MyD88–dependent signaling pathway induces the rapid activation of serine-threonine kinases such as p38 and IKKβ, whereas the TRAM-TRIF pathway induces the activation of interferon-regulatory factors such as IRF3. It is generally believed that the two signaling pathways are induced simultaneously and from the plasma membrane. Here we have made a side-by-side comparison of the activity of these pathways and found that the TRAM-TRIF–dependent signaling pathway was induced mainly from an endosomal location, after LPS-induced endocytosis of TLR4. Two independent lines of evidence supported that conclusion. First, disrupting TLR4 endocytosis by interfering with dynamin GTPases disrupted the TRAM-TRIF–depen-dent phosphorylation of IRF3 and, consequently, IFN-β expression. Second, TRAM mutants that resided specifically on early endosomes (and were thus absent from the cell surface) retained the ability to transmit TLR4 signals resulting in type I interferon induction. The simplest interpretation of these two complementary experiments is that although TLR4 and TRAM can be found with TIRAP at the cell surface, TRAM-TRIF–dependent signaling occurs after internalization. The observation that TIRAP-MyD88–dependent signaling seemed intact in dynasore-treated cells is consistent with work showing that TIRAP recruits MyD88 to PtdIns(4,5)P2-rich regions of the plasma membrane, where signaling is initiated24.
These results suggest a new model of the initiation of TLR4 signaling. We propose that LPS induces assembly of the ligand-binding complex consisting of CD14, MD-2 and TLR4 at the plasma membrane. It is at this initial site of ligand binding that the TIRAP-MyD88 complex interacts ‘preferentially’ with the TIR domain of TLR4. From this location, which is probably a PtdIns(4,5)P2-rich subdomain of the plasma membrane, signaling is initiated and the receptor is endocytosed by a dynamin-dependent process. During endocytosis, PtdIns(4,5)P2 concentrations on the invaginating membrane drop precipitously43, thereby releasing the TIRAP-MyD88 complex from the invaginating membrane, which will ultimately become an early endosome. Loss of the TIRAP-MyD88 complex allows the TRAM-TRIF complex to engage the TIR domain of TLR4 on early endosomes and induce the second phase of signaling from an intracellular location, ultimately leading to the induction of the gene encoding IFN-β. This model helps to provide a plausible explanation for published data suggesting that both adaptor pairs can bind to the same site on the TIR domain of TLR4 (ref. 44), as these adaptor pairs do not bind simultaneously but instead bind sequentially. The sequential triggering of these two pathways is coordinated around endocytosis of the receptor, which raises many questions about the regulation of this system that should be the focus of future investigations.
The spatial separation of signaling pathway engagement is not unique to TLR4 but is noted for other signaling receptors such as the epidermal growth factor receptor, the tumor necrosis factor receptor and several G protein–coupled receptors37,45,46. Notably, cytosolic regulators of epidermal growth factor receptor and G protein–coupled receptor signaling, such as c-Src and β-arrestins, contain a bipartite localization motif similar to that present in TRAM. The function of the bipartite localization motif is to control the distribution of TRAM between the plasma membrane and endosomes. We have shown here that disruption of either the first (myristate group) or second (phosphoinositide-binding) half of this motif substantially altered the subcellular distribution of TRAM. The finding that the bipartite motif controls the localization of TRAM raises the possibility that natural alterations in this domain (such as those induced by other signals) would alter the localization and signaling potential of TRAM. We propose that the use of bipartite localization modules may be a general strategy for controlling receptors capable of inducing signaling pathways from distinct subcellular locations.
Our conclusion that TLR4 induces TRAM-TRIF–dependent signaling from an intracellular location eliminates the only PRR thought to activate the production of type I interferon from the plasma membrane. Although it is unclear why PRRs should induce type I interferon exclusively from intracellular compartments, our proof-of-principle experiments with TRAF3 have provided a potential mechanistic explanation for how type I interferon–inducing ability is restrained. TRAF3 is somehow unable to be recruited to the cell surface to participate in TLR signaling, which precludes it from interacting with TLR-MyD88 complexes at the cell surface. In support of that hypothesis, bypassing that restriction by targeting TRAF3 to the plasma membrane was sufficient to allow TLR2 to induce IFN-β expression. A likely explanation for why TLR2 does not normally induce IFN-β is that its expression is restricted to the plasma membrane, where TRAF3 is absent. Furthermore, even if TLR2 were to be internalized during ligand binding, we expect that the TIRAP-MyD88 adaptor pair would be lost from the receptor complex as it reached an endosomal location. TLR4 seems to have evolved a way of bypassing this problem by engaging the TRAM-TRIF complex after receptor internalization; this complex, like MyD88, can interact with TRAF3. This model also readily explains how TLR3, which is located in endosomes, can trigger the TRIF pathway without the requirement for TRAM. Indeed, TRAM seems to be necessary for coupling of the TLR4 located on the cell surface to the endosomal activation of TRIF-TRAF3. We note, however, that it is unlikely that TRAF3 is completely incapable of being recruited to the plasma membrane, as it seems to function at the plasma membrane ‘downstream’ of CD40 activation47,48. Notably, however, CD40 does not induce the production of type I interferon47,48, which suggests that TRAF3 engaged by CD40 has a function distinct from that of TRAF3 engaged by TLRs. Nevertheless, because the main site of TRAF3 residence is an intracellular locale, it is unclear at present what regulates the recruitment of TRAF3 to the cell surface or any other subcellular location. In conclusion, our study has established a connection between the subcellular localization and signaling specificity of TLR4 and has identified a bipartite sorting motif in TRAM. Thus, like TIRAP, TRAM functions as a sorting adaptor that controls the initiation of TRIF-dependent signaling pathway from an endosomal compartment.
Methods
Cell culture, immunofluorescence and transfection
Fugene-6 was used according to the manufacturer's instructions (Roche) to transfect human embryonic kidney 293T cells; cells were incubated for 24 h at 37 °C. Luciferase production by transfected 293T cells was measured with the Luciferase Assay System according to the manufacturer's instructions (Promega). Macrophages were transfected by nucleofection (AMAXA) with the mouse macrophage transfection reagent or by retroviral transduction as described49. Macrophages were stained 4 h after nucleofection. Where indicated, dextan proteins labeled with Texas red (10 μg/ml; Molecular Probes) were added to the cells 30 min before fixation. For immunostaining, cells were fixed for 20 min at 25 °C in 2% (vol/vol) paraformaldehyde and were made permeable for 10 min with 50 μM digitonin. Samples were treated for 30 min with blocking buffer (1% (wt/vol) BSA and 50 mM ammonium chloride in PBS) and were treated with the appropriate antibodies diluted in blocking buffer. Hemagglutinin-specific anti-bodies (3F10) were from Roche; CD4-specific antibodies (L3T4) were from R&D Systems; and Sa15-21, an antibody that recognizes TLR4, was provided by K. Miyake. For microscopy, antibody binding was detected with Alexa Fluor 488– conjugated antibody to mouse immunoglobulin G (anti–mouse IgG; A-21202) or anti–rat IgG (A-11006; both from Molecular Probes) and was visualized with an Axioplan 2 epifluorescence microscope (Carl Zeiss). Images of sections 0.1 mm in thickness were captured with a Axiocam HRm digital camera and were processed with Adobe Photoshop. Bone marrow–derived macrophages were prepared as described24. C57BL/6 mice were from Jackson Labs, and TRAM-knockout mice were provided by S. Akira. Mice were bred and maintained at the Yale Animal Resources Center at Yale University and all experiments were done with the approval of and in accordance with regulatory guidelines and standards set by the Institutional Animal Care and Use Committee of Yale University.
Dynasore treatment
Primary macrophages at day 7 or 8 were plated at 70% confluency in 96-well tissue culture plates for enzyme-linked immunosorbent assay (ELISA), in 6-well tissue culture plates for biochemical studies, or in 24-well plates for RT-PCR analysis. At 24 h after being replated, cells were washed once with PBS to remove serum and were ‘refreshed’ for 2 h in serum-free RPMI medium. The medium was replaced again with serum-free RPMI medium containing recombinant LPS-binding protein and 80 μM dynasore (provided by T. Kirchhausen) or dimethyl sulfoxide (control). Cells were incubated for 30 min and were treated with various concentrations of LPS for various times.
Flow cytometry
Wild-type or TLR4-deficient bone marrow–derived macro-phages, pretreated with dynasore or not, were left unstimulated or were stimulated for 30 min at 37 °C with LPS, then were washed with flow cytometry buffer (0.1% (vol/vol) FCS and 2 mM EDTA in PBS). Sa15-21 conjugated to biotin (EZ-Link NHS-SS-Biotin; Pierce) was used for staining. Cells were incubated for 20 min on ice with the primary antibody, followed by streptavidin-phycoerythrin, and staining was assessed with a FACSCalibur (Becton Dickinson).
Lipid-binding assays
‘PIP strips’ (Echelon Biosciences) were immersed for 1 h in blocking buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% (vol/vol) Tween-20 and 0.1% (wt/vol) ovalbumin). Strips were probed for 2 h at 25 °C with GST fusion protein (50 ng/ml) in the presence of the GST-specific antibody GST-2 (Sigma). Blots were then washed in blocking buffer three times for 10 min each and were probed for 30 min in blocking buffer with horseradish peroxidase– conjugated anti–mouse IgG (NXA931; Amersham). Bound protein was detected with ECL (Amersham).
Plasmids and protein purification
Plasmids encoding the TLR4 construct with the ectodomain of the receptor replaced with the ectodomain of CD4 and the luciferase reporters dependent on NF-κB or ISRE have been described50. TRAF3 or TRAM cDNA was isolated from a mouse cDNA library and was used as a template to clone TRAF3 into pCDNA3-flu or TRAM into pEGFP-N1 (Clontech). All mutants of TRAM were generated by PCR with green fluorescent protein–tagged TRAM (GFP-TRAM) as a template and cloning of the product into pEGFP-N1. Fusions of GST with mutant TRAM were generated by cloning of cDNA from pEGFP-N1 into pGEX-4Ti (Amersham). Retroviral vectors encoding various TRAM cDNA constructs were generated by subcloning of the GFP-tagged cDNA from pEGFP-N1 into pMSCV2.2. Rab5 plasmids were provided by C. Roy. The PLC-δ1 cDNA has been described24. GST fusion proteins were purified from the BL21 derivative of Escherichia coli with glutathione Sepharose 4B according to the manufacturer's instructions (Amersham). The purity of each GST preparation was confirmed by SDS-PAGE and silver staining.
RT-PCR and ELISA
For RT-PCR, cells were collected into RNA-Bee (Teltest). RNA was reverse-transcribed with Superscript II (Invitrogen) and cDNA was used for PCR. The abundance of each cytokine mRNA was compared with the expression of Hprt1 (‘housekeeping’ gene encoding hypoxanthine guanine phosphoribosyl transferase), which was used confirm that similar amounts of RNA were used for each sample. Macrophage supernatants from a 96-well tissue culture plate were used for ELISA to detect expression of IL-6 or RANTES with reagents from R&D Systems; this was done according to the manufacturer's instructions.
Supplementary Material
Acknowledgments
We thank K. Miyake (Institute for Medical Sciences, University of Toyko) for Sa15-21; S. Akira (Osaka University) for TRAM-KO mice; C. Roy (Yale University) for Rab5 plasmids; T. Kirchhausen (Immune Disease Institute and Harvard Medical School) for dynasore; L. Marek, D. Hargreaves and C. Sokol for discussions; and T. Medjitov for help with bioinformatics analysis. Supported by the National Institutes of Health (1K99AI072955-01 to J.C.K., and R37 AI046688, P01 AI44220 and AI 061360 to R.M.) and the Howard Hughes Medical Institute (R.M.).
Footnotes
Accession codes. UCSD-Nature Signaling Gateway (http://www.signaling-gateway.org): A002296 and A002309.
Note: Supplementary information is available on the Nature Immunology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 3.Hirotani T, et al. Regulation of lipopolysaccharide-inducible genes by MyD88 and Toll/IL-1 domain containing adaptor inducing IFN-β. Biochem Biophys Res Commun. 2005;328:383–392. doi: 10.1016/j.bbrc.2004.12.184. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto M, et al. Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto M, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto M, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 7.Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature. 2002;420:329–333. doi: 10.1038/nature01180. [DOI] [PubMed] [Google Scholar]
- 8.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 9.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi F, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 11.Toshchakov V, et al. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 12.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 13.Lund JM, et al. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 16.Honda K, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, et al. Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 18.Doyle S, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 19.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-β induction. Nat Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 20.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka A, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 22.Kato H, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 23.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 24.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 25.Ulrichts P, Peelman F, Beyaert R, Tavernier J. MAPPIT analysis of TLR adaptor complexes. FEBS Lett. 2007;581:629–636. doi: 10.1016/j.febslet.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 26.Oshiumi H, et al. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-β. J Biol Chem. 2003;278:49751–49762. doi: 10.1074/jbc.M305820200. [DOI] [PubMed] [Google Scholar]
- 27.Rowe DC, et al. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc Natl Acad Sci USA. 2006;103:6299–6304. doi: 10.1073/pnas.0510041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Husebye H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 30.Akashi S, et al. Lipopolysaccharide interaction with cell surface Toll-like receptor 4-MD-2: higher affinity than that with MD-2 or CD14. J Exp Med. 2003;198:1035–1042. doi: 10.1084/jem.20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macia E, et al. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Boll W, Ehrlich M, Collier RJ, Kirchhausen T. Effects of dynamin inactivation on pathways of anthrax toxin uptake. Eur J Cell Biol. 2004;83:281–288. doi: 10.1078/0171-9335-00373. [DOI] [PubMed] [Google Scholar]
- 33.Damke H, Baba T, van der Bliek AM, Schmid SL. Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J Cell Biol. 1995;131:69–80. doi: 10.1083/jcb.131.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seeger M, Payne GS. A role for clathrin in the sorting of vacuolar proteins in the Golgi complex of yeast. EMBO J. 1992;11:2811–2818. doi: 10.1002/j.1460-2075.1992.tb05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Racoosin EL, Swanson JA. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J Cell Biol. 1993;121:1011–1020. doi: 10.1083/jcb.121.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald PH, et al. β-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 38.Sandilands E, Brunton VG, Frame MC. The membrane targeting and spatial activation of Src, Yes and Fyn is influenced by palmitoylation and distinct RhoB/RhoD endosome requirements. J Cell Sci. 2007;120:2555–2564. doi: 10.1242/jcs.003657. [DOI] [PubMed] [Google Scholar]
- 39.Martin TF. Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol. 1998;14:231–264. doi: 10.1146/annurev.cellbio.14.1.231. [DOI] [PubMed] [Google Scholar]
- 40.Oganesyan G, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 41.Hacker H, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 42.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-κB by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 43.Botelho RJ, et al. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nunez Miguel R, et al. A dimer of the Toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE. 2007;2:e788. doi: 10.1371/journal.pone.0000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 46.Schneider-Brachert W, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, Cheng G, Baltimore D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity. 1996;5:407–415. doi: 10.1016/s1074-7613(00)80497-5. [DOI] [PubMed] [Google Scholar]
- 48.Hostager BS, Catlett IM, Bishop GA. Recruitment of CD40 and tumor necrosis factor receptor-associated factors 2 and 3 to membrane microdomains during CD40 signaling. J Biol Chem. 2000;275:15392–15398. doi: 10.1074/jbc.M909520199. [DOI] [PubMed] [Google Scholar]
- 49.Kagan JC, Roy CR. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat Cell Biol. 2002;4:945–954. doi: 10.1038/ncb883. [DOI] [PubMed] [Google Scholar]
- 50.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.