

Abstract
AIM: To test the role of mast cells in gut inflammation and colitis using interleukin (IL)-10-deficient mice as an experimental model.
METHODS: Mast cell-deficient (KitW-sh/W-sh) mice were crossbred with IL-10-deficient mice to obtain double knockout (DKO) mice. The growth, mucosal damage and colitis status of DKO mice were compared with their IL-10-deficient littermates.
RESULTS: DKO mice exhibited exacerbated colitis compared with their IL-10-deficient littermates, as shown by increased pathological score, higher myeloperoxidase content, enhanced Th1 type pro-inflammatory cytokines and inflammatory signaling, elevated oxidative stress, as well as pronounced goblet cell loss. In addition, deficiency in mast cells resulted in enhanced mucosal damage, increased gut permeability, and impaired epithelial tight junctions. Mast cell deficiency was also linked to systemic inflammation, as demonstrated by higher serum levels of tumor necrosis factor α and interferon γ in DKO mice than that in IL-10-deficient mice.
CONCLUSION: Mast cell deficiency in IL-10-deficient mice resulted in systematic and gut inflammation, impaired gut barrier function, and severer Th1-mediated colitis when compared to mice with only IL-10-deficiency. Inflammation and impaired gut epithelial barrier function likely form a vicious cycle to worsen colitis in the DKO mice.
Keywords: Colitis, Interleukin-10, Inflammation, Inflammatory bowel disease, Mast cells, Mice
Core tip: Colitis is characterized by chronic inflammation and mast cells accumulate at the pathological sites, implicating their mediating roles, but the exact roles of mast cells in colitis remain poorly defined and controversial. In this study, the authors cross-bred mast cell-deficient mice with interleukin-10-deficient mice to investigate the role of mast cells in gut inflammation and the onset of colitis. Data show that mast cells have protective roles in the development of colitis by suppressing Th1 type immune response and inflammation, altering gut microbiota composition, improving gut epithelial barrier function, and reducing epithelial damage.
INTRODUCTION
Inflammatory bowel disease (IBD), primarily Crohn’s disease and ulcerative colitis, is one of the most common gastrointestinal diseases. Our understanding of IBD etiology, however, is far from complete. Interleukin (IL)-10-deficient mice develop IBD spontaneously after 3 mo of age, which is a common model for studying the etiology of Crohn’s disease. The onset of colitis in these mice is associated with enhanced CD4+ Th1/Th17 mediated inflammatory responses[1].
Mast cells are generated from bone marrow derived hematopoietic progenitor cells that migrate into vascularized tissues, where they undergo final maturation[2]. The resident mast cells comprise about 2%-3% of mucosal cells in the healthy gut[3], but they can be recruited in large numbers in response to an array of stimuli. They regulate epithelial barrier function and inflammation through: (1) affecting the expression and distribution of tight junction proteins[4,5]; (2) regulating enteric nervous system by secretion of neurotransmitters[6]; and (3) recruiting and activating other immune cells by releasing cytokines[7].
The density of mast cells increases in the gastrointestinal tract of IBD patients, indicating the likely involvement of mast cells in the etiology of IBD[8-10]. It has been reported that mast cells potentiated inflammation in Dextran Sodium Sulfate (DSS) induced colitis, since mast cell deficiency dampened DSS-induced body weight loss and attenuated colonic hypersensitivity[11]. In stress-induced gut inflammation, mast cells mediated epithelial barrier dysfunction in rats[12,13], and mast cell-deficient mice had decreased basal jejunum permeability ex vivo[14]. However, mast cells have also been documented to have a protective role in colonic colitis; deletion of mast cells in IL-10-deficient mice resulted in enhanced mucosal epithelial permeability[15], while mast cell deficiency has no inhibitory role in helicobacter induced gut inflammatory response in IL-10-deficient mice[15]. These data indicate a complex nature of the role of mast cells in gut inflammation and IBD pathogenesis, which possibly depends on the genotype and physiological status of mice as well as environmental factors. Here in this study, we cross-bred mast cell-deficient mice with IL-10-deficient mice to investigate the role of mast cells in gut inflammation and the onset of colitis, and further explored underlying mechanisms.
MATERIALS AND METHODS
Animal care and experimental design
All animal procedures were approved by the University of Wyoming Animal Care and Use Committee. IL-10-deficient mice (B6.129P2-IL-10tm1Cgn/J; stock #002251) and mast cell-deficient mice (STOCK KitW-sh/HNihrJaeBsmJ; stock #005051) were obtained from the Jackson Laboratory (Bar Harbor, ME). Both strains are on the C57/BL6 background. IL-10-deficient mice and mast cell-deficient mice were cross-bred for two generations to obtain mast cell heterozygous IL-10-deficient mice. At five weeks of age, mast cell heterozygous, IL-10-deficient female mice were fed either with a control diet (D12450B, 10% energy from fat, Research Diets Inc.) or a high energy diet (D12451, 45% energy from fat) for 3 mo and then bred with the same genotype male mice fed with the control diet. Offspring with both mast cell-deficient and IL-10-deficient (double deficient, for simplicity, we called double knockout, DKO) mice and only IL-10-deficient mice from the same litter were obtained and used for further studies. All mice were housed in sterile high-efficiency particulate air filter cages, with access to food and water ad libitum. However, for unknown reasons, very few or no viable neonatal DKO mice could be obtained from mothers fed the control diet. Therefore, only IL-10-deficient and DKO offspring from mothers fed the high energy diet were used for further studies. All mice were sacrificed at 10 wk of age.
Tissue collection
After euthanasia, the colonic tissue was dissected from surrounding tissue. A 5 mm section from the colonic tissue at constant location was fixed in 4% (w/v) paraformaldehyde, processed and embedded into paraffin. The remaining gut segments were cut opened, rinsed in PBS, frozen in liquid nitrogen, and stored at -80 °C till analysis.
In vivo intestinal permeability
Seven-week-old mice were fasted for 5 h with water provided, and then gavaged with FITC-dextran (Sigma, St Louis, MO) at 120 μg/kg body weight. Blood was collected 4 hours after gavage and centrifuged for 5 min at 4000 × g. The resulting serum was 1:5 diluted in PBS (pH 7.4), and the fluorescence intensity was measured at excitation 485 nm and emission 520 nm by a SpectraMax M5 Spectrophotometer (Molecular Device, Sunnyvale, CA)[16].
Glucose tolerance test
Mice at eight-week-old were subjected to intraperitoneal (i.p.) glucose tolerance test after overnight fasting with free access to water. D-glucose (2 mg/g body weight) was i.p. injected into mice. The blood glucose level was monitored at 0, 15, 30, 60 and 120 min after injection by tail tip bleeding using a Contour glucometer (Bayer Healthcare, Mishawaka, IN).
Measurement of GSH content
Glutathione vs glutathione disulfide ratio (GSH/GSSG) recycle assay was performed as previously described[17]. Briefly, 10 mg colon tissues were homogenized in 200 μL of l.3% picric acid solution (Sigma) and followed by sonication and centrifugation. The supernatant was assayed for total GSH and GSSG by incubation with 2-vinylpyridine (Sigma), which conjugates any GSH present in the sample so that only GSSG is recycled to GSH without interference by GSH. The GSSG (as GSH × 2) was then subtracted from the total GSH to calculate the level of GSH.
Serum tumor necrosis factor α and interferon-γ level
Serum levels of tumor necrosis factor (TNF)-α and interferon (IFN)-γ were analyzed by ELISA (eBiosciences, San Diego, CA) according to the manufacturer’s manual. The overall intra-assay and inter-assay coefficient of variation was < 5.0% and < 10%, respectively.
Histology
For pathobiological examination, embedded colonic tissue were cut into 5 μm thickness and subjected to hematoxylin-eosin (HE) staining. HE stained slides were scored using a scale as previously described[18]. Briefly, slides were scored for the presence of epithelial hyperplasia, the intensity and severity of inflammation. The maximum score of each colon section is 15. A higher number indicates more extensive/severe disease symptoms. To quantify the goblet cell density, the colonic tissue section was stained with alcian blue per published method[19]. The quantification of goblet cells (goblet cell area vs the tissue section area) of alcian blue stained section was performed using the Image J software (split color channels).
Quantitative reverse transcriptase PCR
Total RNA was extracted from colonic tissue using Trizol reagent (Sigma) and treated with DNase I (Qiagen, Valencia, CA) followed by purification with RNeasy® Mini Kit (Qiagen). The cDNA was synthesized with the iScript™ cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). Reverse transcriptase PCR (qRT-PCR) was conducted on a Bio-Rad CFX96 thermocycler using glyceraldehyde 3-phosphate dehydrogenase as the housekeeping gene. The primers are listed in Table 1. SYBR Green Master Mix (Bio-Rad) was used in all PCR reactions. The amplification efficiency was 0.90 to 0.99. The qRT-PCR conditions were 95 °C, 3 min; 35 cycles of 95 °C for 10 s, 56 °C for 10 s and 72 °C for 20 s. At the end of each run, dissociation melt curves were obtained to confirm the purity of PCR products, and the PCR products were electrophoresed to confirm the targeted sizes. Relative expression of mRNA was determined after normalization to GAPDH reference using ΔΔ-Ct method.
Table 1.
Primer sets used for quantitative reverse transcriptase-polymerase chain reaction analysis of mouse colonic tissue
| Gene name | Accession No. | Product size | Direction | Sequence (5’ to 3’) | Ref. |
| Claudin2 | NM_016675.4 | 120 bp | Forward | GGCGTCCAACTGGTGGGCTAC | [42] |
| Reverse | AACCGCCGTCACAATGCTGGC | ||||
| Claudin3 | NM_009902.4 | 132 bp | Forward | CAGGGGCAGTCTCTGTGCGAG | [42] |
| Reverse | GCCGCTGGACCTGGGAATCAAC | ||||
| GAPDH | NM_008084.2 | 132 bp | Forward | AACTTTGGCATTGTGGAAGG | [42] |
| Reverse | GGATGCAGGGATGATGTTCT | ||||
| IL-1β | NM_008361 | 73 bp | Forward | TCGCTCAGGGTCACAAGAAA | [43] |
| Reverse | CATCAGAGGCAAGGAGGAAAAC | ||||
| INFγ | NM_008337.3 | 93 bp | Forward | AGGTCCAGCGCCAAGCATTCAA | [42] |
| Reverse | AGCAGCGACTCCTTTTCCGCTT | ||||
| iNOS | U43428.1 | 76 bp | Forward | CAAAGTCTCAGACATGGCTTGC | This study |
| Reverse | TTCCTCTGTCAGGTCACTTTGG | ||||
| NOX1 | NM_172203.1 | 113 bp | Forward | CAGGCATCCTCATTTTGCGG | This study |
| Reverse | CCTTCTGCTGGGAGCGATAA | ||||
| T-bet | NM_019507.2 | 138 bp | Forward | CCACTGGATGCGCCAGGAAGTT | [42] |
| Reverse | TTCACCTCCACGATGTGCAGCC | ||||
| TNF-α | NM_013693.2 | 67 bp | Forward | TGGGACAGTGACCTGGACTGT | [43] |
| Reverse | TTCGGAAAGCCCATTTGAGT |
Gut microflora analysis
Bacterial genomic DNA was extracted from fecal samples using QIAamp® DNA Stool Mini Kit (Qiagen) per the manufacturer’s instruction. The abundance of specific intestinal bacterial groups was quantified by qPCR using Bio-Rad CFX96 thermocycler as stated above. Genus or species specific 16S rRNA gene primers were listed in Table 2. The 16S rRNA of Eubacteria was used as the internal control.
Table 2.
Primers for quantitative polymerase chain reaction analysis of selected fecal microbiota
| Target organism | Primer set | Sequence (5’ to 3’) | Product size | Ref. |
| Bacteroides | BactF285 | GGTTCTGAGAGGAGGTCCC | 53 | [44] |
| UniR338 | GCTGCCTCCCGTAGGAGT | |||
| Ec-ssul | Ec-ssu1F | GGATAACACTTGGAAACAGG | 115 | [45] |
| Ec-ssu1R | TCCTTGTTCTTCTCTAACAA | |||
| Eubacteria | UniF340 | ACTCCTACGGGAGGCAGCAGT | 210 | [46] |
| UniR514 | ATTACCGCGGCTGCTGGC | |||
| Lactobacillus | LabF362 | AGCAGTAGGGAATCTTCCA | 315 | [44] |
| LabR677 | CACCGCTACACATGGAG | |||
| Ruminococcus albus (Ralb) | Ralb561F | CAGGTGTGAAATTTAGGGGC | 246 | [47] |
| Ralb807R | GTCAGTCCCCCCACACCTAG |
Immunoblotting
Immunoblot analysis was conducted according to the procedures previously described[17]. Briefly, protein extracts from colonic tissues were separated by 5%-15% gradient SDS-PAGE gels and transferred to nitrocellulose membranes for immunoblotting analyses. Antibodies against myeloperoxidase, phospho-p65 and p65 were purchased from Cell Signaling Technology (Beverly, MA). Claudin2 and Claudin3 were purchased from Invitrogen (Camarillo, CA). GAPDH was purchased from GeneTex (Irvine, CA). Band density was quantified by Image J software and normalized according to the GAPDH content.
Serum total free fatty acid colorimetric assay
Plasma total free fatty acid (FFA) content was analyzed colorimetrically following the previous published methods[20,21]. Total FFA concentration was calculated based on the standard curve. Each sample was analyzed in duplicate and mean values were reported.
Statistical analysis
Statistical analyses were conducted as previously described[17,21]. Data were analyzed as a complete randomized design using GLM (General Linear Model of Statistical Analysis System, SAS, 2000). Mean ± SE are reported. Statistical significance is considered as P < 0.05.
RESULTS
DKO mice showed aggravated colitis compared to their IL-10-deficient littermates
The severity of colitis was evaluated by examining pathological changes, goblet cell density, pro-inflammatory cytokine expression and neutrophil content. As shown in Figure 1A, colon section of DKO mice had a much higher pathological score than that of IL-10-deficient littermates. Meanwhile, the colonic tissue of DKO mice exhibited an increased expression of Th1 type inflammatory cytokines such as IL-1β and IFN-γ (Figure 1B, C), enhanced NF-κB inflammatory signaling (Figure 1D), and elevated neutrophil infiltration, as indicated by increased myeloperoxidase (MPO) content (Figure 1E). Being the major source of secreted mucin in the gastrointestinal tract, goblet cells play a vital role in regulating intestinal homeostasis. The depletion of goblet cells in the large intestine is another characterized feature of IBD. Alcian blue staining revealed that mast cell deletion resulted in decreased goblet cell staining in the colon of IL-10-deficient mice (Figure 1F).
Figure 1.
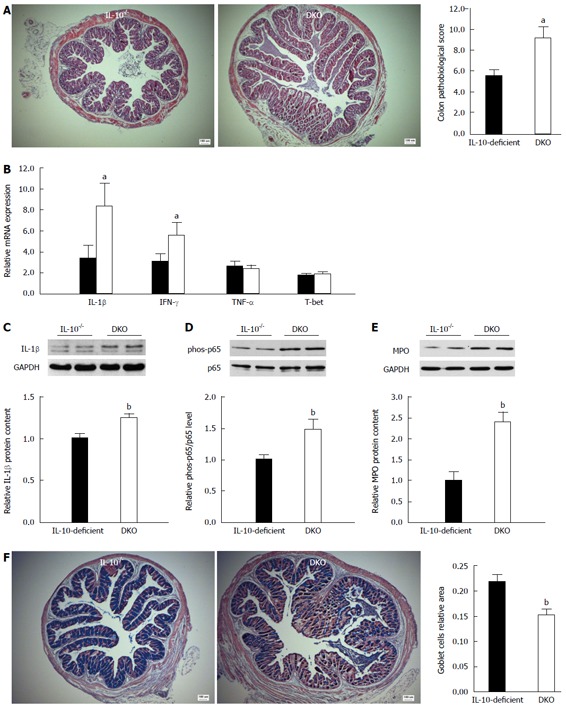
Mast cell deficiency aggravated colitis in the colon of interleukin-10-deficient mice. A: Pathological score; B: mRNA expression of inflammatory cytokines; C: Relative IL-1β protein content; D: NF-κB p65 inflammatory signaling; E: Relative myeloperoxidase content in colonic tissue; F: Goblet cell density. Mean ± SE, aP < 0.05, DKO vs IL-10-deficient, bP < 0.01, DKO vs IL-10-deficient, n = 8 for mRNA analysis, n = 10 for others. IL: Interleukin; DKO: Double knockout; MPO: Myeloperoxidase; NF-κB: Nuclear factor κB.
DKO mice experienced increased oxidative stress in the colon compared to their IL-10-deficient littermates
Oxidative stress has arisen to be another crucial etiological event in colitis progression[22]. Consistent with aggravated colitis, GSH/GSSG recycle assay demonstrated that mast cell deficiency resulted in a marked decrease of GSH content (Figure 2A). Meanwhile, mRNA expression of NADPH oxidase 1 (NOX1) was increased in the colon of DKO mice compared to that of their IL-10-deficient littermates, but iNOS expression was unchanged (Figure 2B). These data indicated mast cell deletion resulted in a more severe oxidative stress in the colon of IL-10-deficient mice, which was consistent with the enhanced inflammatory responses observed in the colon of DKO mice.
Figure 2.
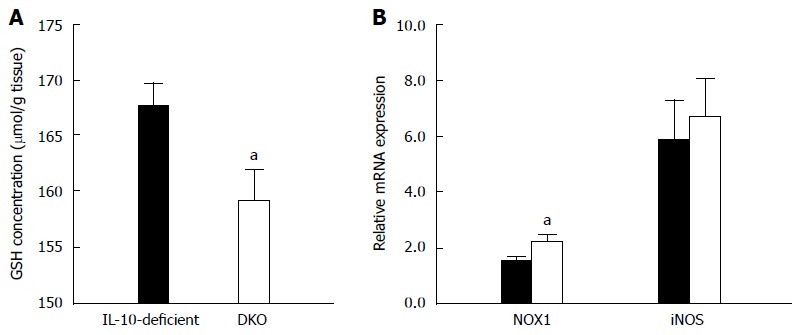
Oxidative stress in the colon tissue of interleukin-10-deficient and double knockout mice. A: Colonic glutathione concentration was measured by GSH/GSSG recycling assay; B: mRNA expression of NADPH oxidase 1 (NOX1) and inducible NO synthase (iNOS). Mean ± SE, aP < 0.05, DKO vs IL-10-deficient, n = 8. IL: Interleukin; DKO: Double knockout.
DKO mice exhibited more mucosal damage than their IL-10-deficient littermates
Increased intestinal permeability is an important etiological event in the development of colitis in IL-10-deficient mice[23]. Consistent with aggravated colitis, the in vivo intestinal permeability of DKO mice was higher (P < 0.01) than that of their IL-10-deficient littermates (Figure 3A), indicating escalated mucosal barrier damage. In agreement with impaired intestinal permeability, mast cell deficiency decreased claudin-3 mRNA expression (Figure 3B) while increased “channel forming” claudin-2 protein content (Figure 3C). In addition, the myosin light chain 2 (MLC-2) phosphorylation and CK2α protein content were enhanced in the colon of DKO mice (Figure 3C).
Figure 3.
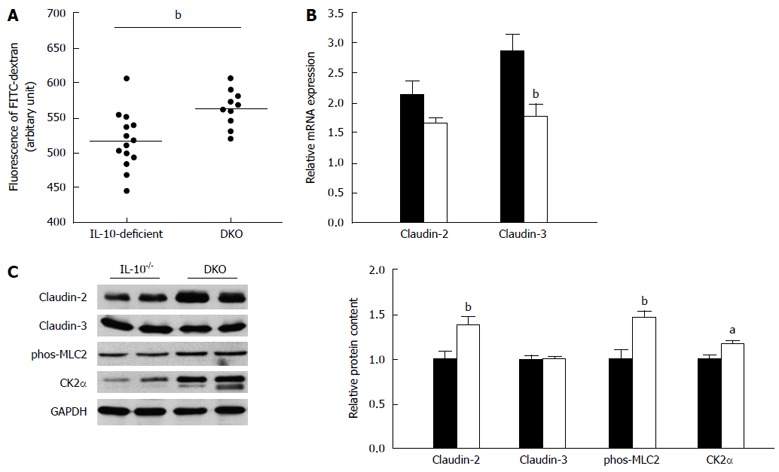
Mast cell deficiency exaggerated mucosal damage in interleukin-10-deficient. A: In vivo intestinal paracellular permeability; B: mRNA expression of Claudin-2 and Claudin-3; C: Relative protein content of Claudin-2, Claudin-3, phosphorylation of MLC-2 and CK2α in colonic tissues. Mean ± SE, aP < 0.05, bP < 0.01, DKO vs IL-10-deficient, n = 8 for mRNA analysis, n = 10 for western blotting analysis. IL: Interleukin; DKO: Double knockout.
Alteration of gut microflora composition
We further evaluated whether gut microflora could be a factor contributing to the enhanced inflammation in the DKO gut. Using genus or species specific 16s rRNA primers, quantitative PCR indicated that DKO mice had decreased Ruminococcus albus (P < 0.05) but no change in Bacteroides, Lactic acid bacteria, Clostridium perfringens, Enterococcus and Faecalibacterium prausnitzii compared to their IL-10-deficient littermates (Figure 4).
Figure 4.
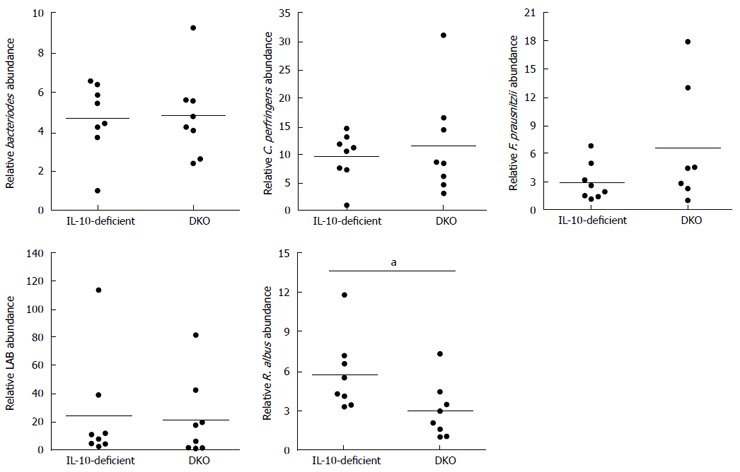
Abundance of selected fecal microflora in interleukin-10-deficient and double knockout mice. Mean ± SE, aP < 0.05, DKO vs IL-10-deficient, n = 8. IL: Interleukin; DKO: Double knockout.
Mast cell deficiency in IL-10-deficient mice led to systemic inflammation
Besides exasperated colitis, DKO mice exhibited lower wean (4-wk-old) body weight (Figure 5A) compared to that of their IL-10-deficient littermates. Upon necropsy (10-wk-old), DKO mice showed higher spleen and liver weight (Table 3), associated with enhanced serum TNF-α and IFN-γ levels (Figure 5B). In addition, mast cell deficiency dramatically impeded systemic glucose tolerance in IL-10-deficient mice at 15 min, 30 min and 60 min post injection of glucose (Figure 5C).
Figure 5.
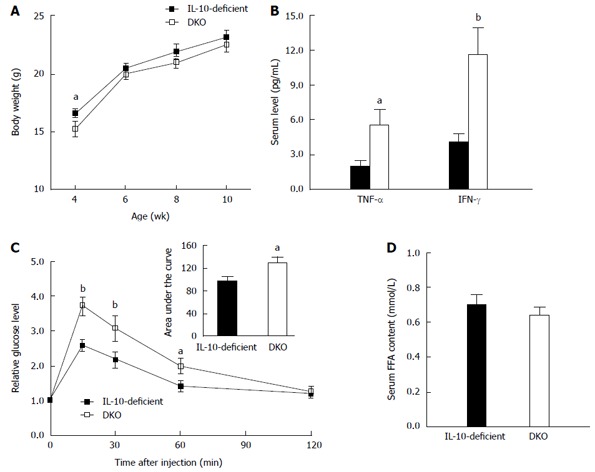
Systemic inflammation in interleukin-10-deficient and double knockout mice. A: Body weight; B: Serum tumor necrosis factor (TNF)-α and interferon (IFN)-γ level; C: Glucose tolerance test; D: Circulatory free fatty acids. Mean ± SE, aP < 0.05, bP < 0.01, n = 8. IL: Interleukin; DKO: Double knockout; FFA: Free fatty acid.
Table 3.
Organ weights of 10-week-old interleukin-10-deficient and double knockout mice at necropsy
| Organ weight (g) | IL-10-deficient | DKO | P value |
| Liver | 1.12 ± 0.041 | 1.26 ± 0.03 | 0.0098 |
| Heart | 0.13 ± 0.01 | 0.13 ± 0.01 | NS |
| Spleen | 0.09 ± 0.01 | 0.16 ± 0.02 | 0.0006 |
| Vastus muscle | 0.24 ± 0.01 | 0.23 ± 0.02 | NS |
| Gastrocnemius muscle | 0.27 ± 0.01 | 0.25 ± 0.01 | NS |
| Tibials muscle | 0.081 ± 0.003 | 0.076 ± 0.003 | NS |
| Subcutaneous fat | 0.31 ± 0.03 | 0.21 ± 0.02 | 0.0052 |
| Gonadal fat | 0.33 ± 0.04 | 0.27 ± 0.03 | NS |
1Mean ± SE, n = 10. IL: Interleukin; DKO: Double knockout.
Interestingly, the subcutaneous fat weight of DKO mice at necropsy was 32.3% ± 8.1% less than that of their IL-10-deficient littermates, while there was no difference in gonadal fat weight (Table 3). Subcutaneous fat is proposed to be the “sink” for free fatty acids (FFA)[24]. Therefore, we further analyzed the serum FFA level, which, however, did not differ between DKO mice and their IL-10-deficient littermates (Figure 5D).
DISCUSSION
Mast cells play a crucial role in innate immune responses and IBD pathogenesis. Deletion of mast cells markedly attenuated multiple organ injury and damped systematic inflammation in response to trauma[25]. In chemical induced colitis, mast cells act as an initiator of innate immune response and likely aggravate disease indices[10]. However, our data showed that lack of mast cells exacerbated colitis in IL-10-deficient mice, associated with impaired mucosal barrier function, which was consistent with a previous study, where deletion of mast cell resulted in earlier onset of spontaneous colitis and associated with increased intestinal permeability in IL-10-deficient mice[15]. We speculate that mast cells might have bilateral roles under different circumstances. Mast cells may act as inflammatory mediators in intact immune system, but serve as sentinels under immune compromised conditions, which is currently underappreciated. As an anti-inflammatory cytokine, IL-10 plays a substantial role in intestinal immune regulation and homeostasis. The level of IL-10 was negatively correlated with the mucosal infiltration of inflammatory cells and the severity of IBD in the colon[26]. Loss of IL-10 signaling by itself is sufficient to drive changes in pro-inflammatory gene expression, but other existing endogenous compensatory mechanisms may be able to prevent robust inflammation. Indeed, inflammatory TLR4 signaling functions to maintain Treg cell populations and intestinal epithelial homeostasis in IL-10-deficient mice[27]. In this regard, mice lacking mast cells in addition to IL-10 deficiency would lose their immune regulation ability thus resulted in exasperated immune deregulation and aggravated colitis. In agreement, we found that mast cell deficiency induced exaggerated inflammatory responses in the gut as indicated by increased expression of inflammatory cytokines, enhanced NF-κB inflammatory signaling and elevated neutrophil infiltration. Such inflammation might directly contribute to the far severe colitis pathological changes observed in the colon of DKO mice. Aligned with enhanced gut inflammation, we also detected more severe oxidative stress in colonic tissues, which is another possible etiological factor in the initiation or progression of IBD[28].
Impairment of the epithelial barrier function allows the transmission of antigens, viruses and bacteria, which aggravates inflammation and forms a vicious circle to induce colitis. In IL-10-deficient mice, high intestinal permeability preceded the development of colitis, whereas improved epithelial barrier function alleviated colitis[23], clearly indicating that intestinal permeability is an important etiological factor in the development of colitis in IL-10-deficient mice. In alignment with enhanced gut inflammation, mast cell deficiency markedly enhanced the gut permeability in IL-10-deficient mice, which might propel the progress of colitis. Consistent with the enhanced gut permeability, we observed that IL-10-deficiency increased the “pore forming” Claudin-2 protein content, while decreased mRNA expression of barrier sealing protein, Claudin-3 in colonic tissue of DKO. Myosin light-chain kinase (MLCK) phosphorylates the regulatory light chain of myosin 2 (MLC2) and regulates actin-myosin contraction and further impairs tight junction formation to enhance paracellular permeability[29]. The dramatic increased phosphorylation of MLC2 in our study might serve as one of mechanisms for the impaired epithelial barrier function in DKO mice. Casein kinase 2 (CK2) is a key regulator of intestinal epithelial homeostasis in chronic intestinal inflammation, and enhanced intestinal epithelial cell CK2 protein content was observed in chronic experimental induced colitis[30]. Consistent with the previous report[30] and enhanced colitis, DKO mice showed increased CK2α protein content in the colon tissue. The impairment of gut epithelial barrier function and exaggerated gut inflammatory responses might reinforce each other to deteriorate colitis symptoms.
Besides local gut inflammation, we also observed systemic inflammation in DKO mice, as evidenced by splenomegaly and hepatomegaly, as well as elevated serum TNF-α and IFN-γ levels. We speculated that splenomegaly and hepatomegaly were mediated by increased serum pro-inflammatory cytokines, which accompanied colitis as previously reported[31]. Increased serum pro-inflammatory cytokines could enhance glucose metabolism disorder[32]. Consistently, DKO mice showed glucose intolerance compared with their IL-10-deficient littermates, which could be attributed to the increased serum TNF-α and IFN-γ levels in DKO[32-34], but the exact mechanism remains to be determined.
It has been reported that mast cells are necessary for adipogenesis, while deficiency in mast cells reduces fat mass[35,36]. In the lean mice, mast cells are more prevalent in subcutaneous fat than visceral fat[37]. Indeed, we observed decreased subcutaneous fat in DKO mice while no change in gonadal fat. Because subcutaneous fat functions as FFA buffer[24], insufficient subcutaneous fat might limit the ability of absorbing circulating FFA, leading to elevated serum FFA level and systemic inflammation[24,38]. However, we did not observe significant difference in the serum FFA level between mice with/without mast cells, ruling out FFA as a source of observed exacerbated inflammatory response in DKO mice.
Gut microbiota is increasingly recognized as an important player in gut inflammation and IBD[39,40]. The current results showed increased Ruminococcus albus in DKO compared to their IL-10-deficient littermates. In support to our result, a previous report indicated that IBD patients had decreased Ruminococcus albus content in the gut[41]. The significance of Ruminococcus albus in colitis development needs to be further defined.
In conclusion, mast cell deficiency resulted in exaggerated colitis in IL-10-deficient mice, which was associated with enhanced gut and systematic inflammation, oxidative stress, altered gut microbiota and impaired gut barrier function. Enhanced inflammation and gut permeability likely form a vicious cycle to propel the aggravation of colitis in DKO mice. Our data suggest a protective role of mast cells in the development of colitis in IL-10-deficient mice through a balance of multiple factors.
COMMENTS
Background
Colitis is characterized by chronic inflammation and mast cells accumulate at the pathological sites, implicating their mediating roles, but the exact roles of mast cells in colitis remain poorly defined and controversial. The interleukin-10-deficient mice (IL-10-/-) are one of the most frequently used models for studying inflammatory bowel diseases, which will be used to assess the role of mast cells in gut inflammation and colitis.
Research frontiers
In this study, the authors cross-bred mast cell-deficient mice with IL-10-deficient mice to investigate the role of mast cells in gut inflammation and the onset of colitis. Data show that mast cells have protective roles in the development of colitis by suppressing Th1 type immune response and inflammation, altering gut microbiota composition, improving gut epithelial barrier function, and reducing epithelial damage in IL-10-deficient mice.
Innovations and breakthroughs
Up to now, the roles of mast cells in the development of colitis remain poorly defined and controversial. This study shows that mast cells protect gut epithelium from the development of colitis. Mast cell deficiency in IL-10-deficient mice resulted in systematic and gut inflammation, impaired gut barrier function, and severer Th1-mediated colitis when compared to mice with only IL-10 deficiency. Inflammation and impaired gut epithelial barrier function likely form a vicious cycle to worsen colitis in the double knockout mice. Therefore, both excess and deficiency of mast cells appear to be detrimental for the incidence of colitis.
Applications
The data suggest a protective role of mast cells in the development of colitis in IL-10-deficient mice through a balance of multiple factors. Thus mast cells likely provide a clinical target to mitigate the symptoms of inflammatory bowel disease.
Peer review
The authors are describing interesting results about the effects of mast cell deficiency on colitis in a double knockout mouse model obtained by cross-breeding mast cell-deficient mice with IL-10-deficient mice. The paper is well-written, the methods used are sound, and results established background for future research in this area.
Footnotes
Supported by USDA-AFRI, No. 2009-65203-05716; NIH, No. 1R15HD073864; and NIH-INBRE, No. P20RR016474
P- Reviewers: Hewicker-Trautwein M, Ribatti D, Singer SM S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
- 1.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okayama Y, Kawakami T. Development, migration, and survival of mast cells. Immunol Res. 2006;34:97–115. doi: 10.1385/IR:34:2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischoff SC, Wedemeyer J, Herrmann A, Meier PN, Trautwein C, Cetin Y, Maschek H, Stolte M, Gebel M, Manns MP. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology. 1996;28:1–13. doi: 10.1046/j.1365-2559.1996.262309.x. [DOI] [PubMed] [Google Scholar]
- 4.Jacob C, Yang PC, Darmoul D, Amadesi S, Saito T, Cottrell GS, Coelho AM, Singh P, Grady EF, Perdue M, et al. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and beta-arrestins. J Biol Chem. 2005;280:31936–31948. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 5.McDermott JR, Bartram RE, Knight PA, Miller HR, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci USA. 2003;100:7761–7766. doi: 10.1073/pnas.1231488100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konturek PC, Brzozowski T, Konturek SJ. Stress and the gut: pathophysiology, clinical consequences, diagnostic approach and treatment options. J Physiol Pharmacol. 2011;62:591–599. [PubMed] [Google Scholar]
- 7.Bischoff SC. Physiological and pathophysiological functions of intestinal mast cells. Semin Immunopathol. 2009;31:185–205. doi: 10.1007/s00281-009-0165-4. [DOI] [PubMed] [Google Scholar]
- 8.De Winter BY, van den Wijngaard RM, de Jonge WJ. Intestinal mast cells in gut inflammation and motility disturbances. Biochim Biophys Acta. 2012;1822:66–73. doi: 10.1016/j.bbadis.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Stoyanova II, Gulubova MV. Mast cells and inflammatory mediators in chronic ulcerative colitis. Acta Histochem. 2002;104:185–192. doi: 10.1078/0065-1281-00641. [DOI] [PubMed] [Google Scholar]
- 10.Hamilton MJ, Sinnamon MJ, Lyng GD, Glickman JN, Wang X, Xing W, Krilis SA, Blumberg RS, Adachi R, Lee DM, et al. Essential role for mast cell tryptase in acute experimental colitis. Proc Natl Acad Sci USA. 2011;108:290–295. doi: 10.1073/pnas.1005758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rijnierse A, Koster AS, Nijkamp FP, Kraneveld AD. Critical role for mast cells in the pathogenesis of 2,4-dinitrobenzene-induced murine colonic hypersensitivity reaction. J Immunol. 2006;176:4375–4384. doi: 10.4049/jimmunol.176.7.4375. [DOI] [PubMed] [Google Scholar]
- 12.Vicario M, Guilarte M, Alonso C, Yang P, Martínez C, Ramos L, Lobo B, González A, Guilà M, Pigrau M, et al. Chronological assessment of mast cell-mediated gut dysfunction and mucosal inflammation in a rat model of chronic psychosocial stress. Brain Behav Immun. 2010;24:1166–1175. doi: 10.1016/j.bbi.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Söderholm JD, Yang PC, Ceponis P, Vohra A, Riddell R, Sherman PM, Perdue MH. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology. 2002;123:1099–1108. doi: 10.1053/gast.2002.36019. [DOI] [PubMed] [Google Scholar]
- 14.Groschwitz KR, Ahrens R, Osterfeld H, Gurish MF, Han X, Abrink M, Finkelman FD, Pejler G, Hogan SP. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/Mcpt4-dependent mechanism. Proc Natl Acad Sci USA. 2009;106:22381–22386. doi: 10.1073/pnas.0906372106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chichlowski M, Westwood GS, Abraham SN, Hale LP. Role of mast cells in inflammatory bowel disease and inflammation-associated colorectal neoplasia in IL-10-deficient mice. PLoS One. 2010;5:e12220. doi: 10.1371/journal.pone.0012220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu MJ, Han B, Tong J, Ma C, Kimzey JM, Underwood KR, Xiao Y, Hess BW, Ford SP, Nathanielsz PW, et al. AMP-activated protein kinase signalling pathways are down regulated and skeletal muscle development impaired in fetuses of obese, over-nourished sheep. J Physiol. 2008;586:2651–2664. doi: 10.1113/jphysiol.2007.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burich A, Hershberg R, Waggie K, Zeng W, Brabb T, Westrich G, Viney JL, Maggio-Price L. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G764–G778. doi: 10.1152/ajpgi.2001.281.3.G764. [DOI] [PubMed] [Google Scholar]
- 19.Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J, Radtke F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011;140:1230–1240.e1-7. doi: 10.1053/j.gastro.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tinnikov AA, Boonstra R. Colorimetric micro-determination of free fatty acids in plasma using microplate readers. Clin Chim Acta. 1999;281:159–162. doi: 10.1016/s0009-8981(98)00216-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhu MJ, Du M, Nathanielsz PW, Ford SP. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta. 2010;31:387–391. doi: 10.1016/j.placenta.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Kamizato M, Nishida K, Masuda K, Takeo K, Yamamoto Y, Kawai T, Teshima-Kondo S, Tanahashi T, Rokutan K. Interleukin 10 inhibits interferon gamma- and tumor necrosis factor alpha-stimulated activation of NADPH oxidase 1 in human colonic epithelial cells and the mouse colon. J Gastroenterol. 2009;44:1172–1184. doi: 10.1007/s00535-009-0119-6. [DOI] [PubMed] [Google Scholar]
- 23.Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huffman DM, Barzilai N. Contribution of adipose tissue to health span and longevity. Interdiscip Top Gerontol. 2010;37:1–19. doi: 10.1159/000319991. [DOI] [PubMed] [Google Scholar]
- 25.Cai C, Cao Z, Loughran PA, Kim S, Darwiche S, Korff S, Billiar TR. Mast cells play a critical role in the systemic inflammatory response and end-organ injury resulting from trauma. J Am Coll Surg. 2011;213:604–615. doi: 10.1016/j.jamcollsurg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Feng BS, Yang SB, Chen X, Su J, Yang PC. Interleukin (IL)-23 suppresses IL-10 in inflammatory bowel disease. J Biol Chem. 2012;287:3591–3597. doi: 10.1074/jbc.M111.304949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matharu KS, Mizoguchi E, Cotoner CA, Nguyen DD, Mingle B, Iweala OI, McBee ME, Stefka AT, Prioult G, Haigis KM, et al. Toll-like receptor 4-mediated regulation of spontaneous Helicobacter-dependent colitis in IL-10-deficient mice. Gastroenterology. 2009;137:1380–1390.e1-3. doi: 10.1053/j.gastro.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 2012;237:474–480. doi: 10.1258/ebm.2011.011358. [DOI] [PubMed] [Google Scholar]
- 29.Turner JR, Rill BK, Carlson SL, Carnes D, Kerner R, Mrsny RJ, Madara JL. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378–C1385. doi: 10.1152/ajpcell.1997.273.4.C1378. [DOI] [PubMed] [Google Scholar]
- 30.Koch S, Capaldo CT, Hilgarth RS, Fournier B, Parkos CA, Nusrat A. Protein kinase CK2 is a critical regulator of epithelial homeostasis in chronic intestinal inflammation. Mucosal Immunol. 2013;6:136–145. doi: 10.1038/mi.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- 32.Barbosa-da-Silva S, Fraulob-Aquino JC, Lopes JR, Mandarim-de-Lacerda CA, Aguila MB. Weight cycling enhances adipose tissue inflammatory responses in male mice. PLoS One. 2012;7:e39837. doi: 10.1371/journal.pone.0039837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez DA, Puglisi MJ, Hasty AH. Impact of increased adipose tissue mass on inflammation, insulin resistance, and dyslipidemia. Curr Diab Rep. 2009;9:26–32. doi: 10.1007/s11892-009-0006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr Metab Cardiovasc Dis. 2009;19:146–152. doi: 10.1016/j.numecd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–945. doi: 10.1038/nm.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka A, Nomura Y, Matsuda A, Ohmori K, Matsuda H. Mast cells function as an alternative modulator of adipogenesis through 15-deoxy-delta-12, 14-prostaglandin J2. Am J Physiol Cell Physiol. 2011;301:C1360–C1367. doi: 10.1152/ajpcell.00514.2010. [DOI] [PubMed] [Google Scholar]
- 37.Altintas MM, Azad A, Nayer B, Contreras G, Zaias J, Faul C, Reiser J, Nayer A. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J Lipid Res. 2011;52:480–488. doi: 10.1194/jlr.M011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arsenault BJ, Beaumont EP, Després JP, Larose E. Mapping body fat distribution: a key step towards the identification of the vulnerable patient? Ann Med. 2012;44:758–772. doi: 10.3109/07853890.2011.605387. [DOI] [PubMed] [Google Scholar]
- 39.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 40.Albenberg LG, Lewis JD, Wu GD. Food and the gut microbiota in inflammatory bowel diseases: a critical connection. Curr Opin Gastroenterol. 2012;28:314–320. doi: 10.1097/MOG.0b013e328354586f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Xue Y, Zhang H, Huang Y, Yang G, Du M, Zhu MJ. Dietary grape seed extract ameliorates symptoms of inflammatory bowel disease in IL10-deficient mice. Mol Nutr Food Res. 2013;57:2253–2257. doi: 10.1002/mnfr.201300146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 44.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76:907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuda K, Tsuji H, Asahara T, Kado Y, Nomoto K. Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl Environ Microbiol. 2007;73:32–39. doi: 10.1128/AEM.01224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol. 1990;56:1919–1925. doi: 10.1128/aem.56.6.1919-1925.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartosch S, Fite A, Macfarlane GT, McMurdo ME. Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl Environ Microbiol. 2004;70:3575–3581. doi: 10.1128/AEM.70.6.3575-3581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]