

Abstract
Multiple studies have demonstrated alterations in the intestinal microbial community (termed the microbiome) in Crohn’s disease (CD) and several lines of evidence suggest these changes may have a significant role in disease pathogenesis. In active and quiescent disease, both the faecal and mucosa-associated microbiome are discordant with matched controls with reduced biodiversity, changes in dominant organisms and increased temporal variation described. Mucosa-associated adherent, invasive Escherichia coli (E. coli) (AIEC), pro-inflammatory and resistant to killing by mucosal macrophages, appear to be particularly important. AIEC possess several virulence factors which may confer pathogenic potential in CD. Type-1 pili (FimH) allow adherence to intestinal cells via cell-surface carcinoembryonic antigen-related cell adhesion molecules and possession of long polar fimbrae promotes translocation across the intestinal mucosa via microfold (M)-cells of the follicle-associated epithelium. Resistance to stress genes (htrA, dsbA and hfq) and tolerance of an acidic pH may contribute to survival within the phagolysosomal environment. Here we review the current understanding of the role of mucosa-associated E. coli in Crohn’s pathogenesis, the role of the innate immune system, factors which may contribute to prolonged bacterial survival and therapeutic strategies to target intracellular E. coli.
Keywords: Crohn’s disease, Inflammatory bowel disease, Escherichia coli, Intra-macrophage survival and replication, Phagolysosome, Autophagy
Core tip: There is significant evidence implicating adherent, invasive mucosa-associated Escherichia coli (AIEC) in the pathogenesis of Crohn’s disease. AIEC translocate M-cells of Peyer’s patches and lymphoid follicles of the colon, and then to survive and replicate within underlying mucosal macrophages. How Crohn’s AIEC resist killing and adapt to the environment within the phagolysosme to survive and grow within macrophages is still poorly understood. Here we review the current understanding of the role of AIEC in Crohn’s pathogenesis, the role of the innate immune system, factors which may contribute to prolonged bacterial survival and therapeutic strategies to target intracellular AIEC.
INTRODUCTION
Crohn’s disease (CD) is a chronic relapsing inflammatory bowel disease (IBD) of multifactorial aetiology, affecting any part of the gastrointestinal tract from mouth to anus. Patients typically suffer from abdominal pain, diarrhoea and weight loss which may be associated with extra-intestinal manifestations including erythema nodosum, iritis and arthritis. The intestinal pathological findings are characterised by transmural inflammation, deep mucosal ulcers, abscesses, fissures and granuloma formation[1]. These chronic inflammatory lesions are proposed to develop due to a disrupted intestinal barrier, Paneth cell dysfunction and a disturbed innate immune response, resulting in the accumulation of antigen-presenting cells (such as dendritic cells and macrophages), lymphocytes and plasma cells within the intestinal mucosal layer[1,2]. Pathological characteristics resemble the mucosal lesions and intestinal inflammation elicited by known enteric gut pathogens such as Shigella and Salmonella spp[3].
CD is classically described to have a bimodal incidence with the highest rates seen in adolescents and young adults and a second peak in later years, although this has recently been questioned[4]. It is associated with a small increase in mortality (standardised mortality ratio 1.52) but very considerable morbidity, disrupting work, study and family life[5]. Historically approximately 80% of cases needed surgery at some time[6] but the use of immunosuppressants and biologics has increased and is associated with a reduced 5 years risk of major surgery[7]. The condition is more common in Europe and North America[8]. However, incidence is rapidly increasing worldwide particularly in developed nations adopting a western style diet, as seen in Japan[9]. Likewise, those emigrating from poor and developing nations to the West, within a few years of moving are at increased risk of developing CD presumably due to a key change in their lifestyle and environment[10].
The gut microbiota plays an essential role in the shaping of the intestinal immune response in healthy individuals[11]. There is now very strong evidence that both a reduction in the numbers of beneficial bacteria and increases in numbers of harmful bacteria living naturally in the gut are present in CD[12] although it is less clear which of these changes might be causative and which might be a consequence of inflammation. Several independent groups have consistently shown changes in both the faecal and mucosa-associated microbiome in Crohn’s patients and unaffected relatives[13-15], an imbalance referred to as “dysbiosis” (Figure 1). Changes are typified by reduced biodiversity and alterations in the dominant organisms, specifically reduction in beneficial firmicutes and increase in numbers of proteobacteria [including Escherichia coli (E. coli)][14,16,17].
Figure 1.
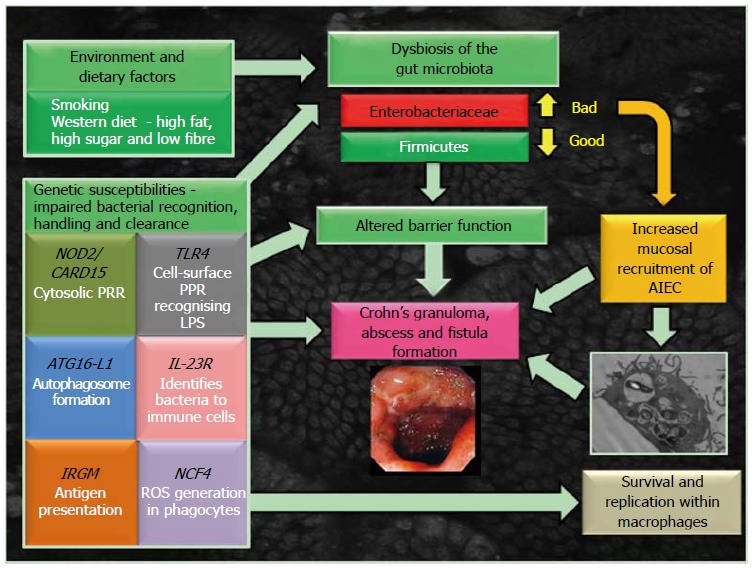
Model for the development of Crohn’s disease. AIEC: Adherent, invasive Escherichia coli; ATG16L1: Autophagy-related 16-like 1; CARD15/NOD2: Caspase-recruitment domain 15/nucleotide-binding oligomerization domain-containing-2 receptor; IL-23R: Interleukin-23 receptor; IRGM: Immunity-related GTPase M; LPS: Lipopolysaccharide; NCF4: Neutrophil cytosolic factor-4 gene; PRR: Pathogen recognition receptor; ROS: Reactive oxygen species; TLR4: Toll-like receptor 4.
There is also clear evidence to suggest that a number of lifestyle factors contribute to the dysbiosis of gut microbiota observed in CD (see Figure 1). This includes key environmental triggers such as smoking[18], with cessation abrogating the observed dysbiosis[19]. Also a key risk factor in CD is a intake of a “westernised” diet, high in fat and sugar, low in fruit and vegetable fibre[20]. In a mouse model with a humanised microbiota, a switch to a high fat, high sugar diet altered the microbiome within 1 d[21]. A similar diet has also been observed to increase numbers of Proteobacteria, such as Bilophila wadsworthia[22] and mucosally adherent, invasive E. coli (AIEC)[23].
GENETIC SUSCEPTIBILITIES IN BACTERIAL RECOGNITION, AUTOPHAGY AND PHAGOCYTE-SPECIFIC GENES IN CD
The recent identification of genes associated with CD has been informative in improving our understanding of its pathogenesis, highlighting impairment of genetic components essential for innate immunity, intestinal barrier integrity and in microbial recognition and clearance[24] (see Figure 1). Following on from earlier work[25,26], Genome-wide association studies have now identified 163 IBD risk loci, 30 of which are CD specific and 110 shared between ulcerative colitis and Crohn’s[27]. Identified polymorphisms in the innate immune system of Crohn’s patients include genes that are linked to processes such as pathogen recognition [nucleotide-binding oligomerization domain-containing-2 (NOD2)/Crohn’s-associated gene identified was Caspase-recruitment domain 15 (CARD15) and interleukin 23 receptor (IL23R)] and autophagy [immunity-related GTPase M (IRGM) and autophagy-related 16-like 1 (ATG16L1)], all relevant to killing of bacteria within macrophages[24-26].
The first CARD15 encoding the NOD2 receptor[28,29]. Mutations in this gene probably account for about 15% of Crohn’s causation in the West although there are geographical variations with a lesser effect in northern European countries and no apparent impact on CD causation in Japan[30]. The NOD2/CARD15 protein is part of the innate immune system and is expressed in the cytoplasm of macrophages and Paneth cells[31]. CD-associated mutations in NOD2/CARD15 affect the leucine-rich domain recognising the bacterial cell wall peptidoglycan component, muramyl dipeptide (MDP), of both Gram-positive and Gram-negative bacteria. After recognition, NOD2 activates nuclear factor kappa B and induces the production and release of proinflammatory cytokines. Crohn’s-associated NOD2/CARD15 mutations are considered to be loss of function mutations with evidence for reduced production of anti-bacterial defensins by Paneth cells and for a reduced IL-8 response to MDP by macrophages[32]. In association with NOD2/CARD15 mutations, polymorphism in genes SLC22A4 and SLC22A5, encoding the organic cation transporters OCTN1 and OCTN2 have also been identified with variants expressed in intestinal epithelial cells, T cells and macrophages[33]. In addition, a mutation in two haplotypes of DLG5, encoding scaffolding protein, has also been confirmed to be associated with NOD2/CARD15 mutations in Crohn’s patients[34].
Two other key genes associated with Crohn’s are ATG16L1 and IRGM[35-37]. Both encode proteins that play a key role in autophagy, a cellular process facilitate not only disposal of protein aggregates, DNA, lipids and damaged organelles but also an integral step in the mechanism by which macrophages degrade, kill and clear invading phagocytosed bacteria (a process also termed xenophagy), including Mycobacteria and Salmonellae[38-40].
Additional Crohn’s susceptibility loci relevant to aberrant microbial recognition and handling and/or phagocyte function include toll-like receptor 4 (TLR4), leucine-rich repeat serine, threonine protein kinase-2 (LRRK2), neutrophil cytosolic factor-4 (NCF4) and IL-23R.
TLR4 is an apical cell-surface pathogen recognition receptor on intestinal epithelial cells, macrophages and dendritic cells, key in detection of lipopolysaccharide (LPS) presented on the outer-membrane surface of Gram-negative bacteria, with polymorphism of TLR4 at D299G leading to hypo-responsiveness to LPS[41]. LRRK2 has been linked to CD through the association of a single-nucleotide polymorphism on chromosome 12q12[26] and in murine studies where LRRK2-deficiency resulted in increased inflammation and significantly poorer clinical outcomes following administration of dextran sodium sulphate to induce colitis[42]. The identification of NCF4 as a Crohn’s susceptibility gene is also important[36]. NCF4 encodes the p40-phox subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase crucial for reactive oxygen species (ROS) production by phagocytic cells in response to microbial infection, with molecular defects in NADPH oxidase already established to result in chronic granulomatous disease[43]. Key studies show that altered neutrophil recruitment, along with an abnormal production of cytokines and reduced bacterial clearance, follow either acute trauma to the rectum and ileum[44], or subcutaneous injection of heat-killed E. coli in Crohn’s patients[45]; (see Figure 2). Whilst these studies suggest macrophages may be involved in a key step of the observed immune dysfunction in CD, it is not yet clear whether this represents an inherent defect in macrophage function.
Figure 2.
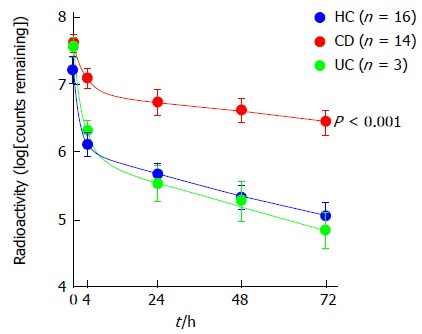
Patients with Crohn’s disease exhibit reduced bacterial clearance of subcutaneously injected 32P-labelled heat-killed Escherichia coli relative to healthy controls and patients with ulcerative colitis. Reproduced with permission. © 2009 Rockefeller University Press. Originally published in Journal of Experimental Medicine 206: 1883-1897[45]. CD: Crohn’s disease; HC: Healthy controls; UC: Ulcerative colitis.
Variants of the IL-23R gene have also been linked to Crohn’s[46]. IL-23R is expressed by activated dendritic cells and macrophages, and IL-23 can induce production of inflammatory cytokines that may contribute to intestinal inflammation[47].
SPECIFIC BACTERIA IN THE PATHOGENESIS OF CD
There have been a number of distinctive studies that strongly favour the hypothesis that a specific bacterium plays a pivotal role in the initiation of chronic inflammation and development of CD. Early serological and culture studies suggested that Mycobacterium avium subspecies paratuberculosis (MAP), an obligate intracellular bacterium causing a chronic intestinal inflammatory disease in cattle (Johne’s disease), was more prevalent in Crohn’s patients[48,49]. A study by Ryan and colleagues[50] also confirmed the presence of MAP DNA in granulomatous lesions of CD patients. MAP-reactive CD4 T cells have also been found in patients with Crohn’s[51]. Even though, MAP has been hypothesised to be as contributing agent for Crohn’s pathogenesis, there is still great controversy, and absence of conclusive evidence, to fully supporting this hypothesis[52]. Our own studies have suggested perhaps that microbial mannan (present in yeast cell walls and Mycobacterium species such as MAP) may be a key environmental factor to suppress macrophage killing of intracellular bacteria[53]. The shared susceptibility association of NOD2 and IL-23R polymorphisms seen in both CD and Mycobacterial disease suggests MAP may yet be important in CD pathogenesis[54].
Faecalibacterium prausnitzii may also be important with low levels strongly associated with early disease recurrence after intestinal surgery[55]. This effect may be due to bacterial production of anti-inflammatory molecules with culture supernatant shown to reduce the severity of colitis in an animal model.
The finding of increased mucosa-associated E. coli in the sub-mucus niche or within the mucosa itself has proved particularly consistent in CD[12]. Early serological studies described high antibody titres against E. coli in Crohn’s patients compared to unaffected controls[56] and this was later supported by immunohistochemical studies demonstrating E. coli antigens within macrophages in CD tissue[57]. Many groups, including our own, have shown an increase in mucosa-associated E. coli in CD, both in the ileum and in the colorectum[58-64]. We ourselves observed that aerobic culture of colonoscopic biopsies after removal of the mucus layer with dithiothreitol is often sterile in control colons whereas the colon in CD and colon cancer contains increased bacterial numbers in this sub-mucus niche, more than half of which are E. coli[60], even though these organisms account for less than 1% of the faecal microbiota[65]. Poor correlation between site of inflammation and presence of E. coli[63] and tendency to show that the same organisms can be identified from various sites within the same colon[60,66] are compatible with the organisms having a causative role in the inflammation rather than merely colonising inflamed mucosa. Evidence for a primary pathogenic role is also given by their presence within granulomas[67], the histological hallmark of CD, by their ability to induce granuloma formation in vitro[68] and ability for similar E. coli to cause granulomatous colitis in dogs[69], and potentially in cats and swine too[70].
These E. coli pathovars associated with CD have been designated AIEC based on their ability to adhere to, and invade into, intestinal epithelial cell-lines, induce release of pro-inflammatory cytokines, and possess an ability to survive and replicate with intestinal macrophages[71]. Phylogenetic analysis shows that most mucosa-associated E. coli isolated from the tissue of Crohn’s patients belong to groups B2 and D[65] as per extra-intestinal isolates, whereas most commensal E. coli strains would belong to group A[72].
CROHN’S AIEC-HOST INTESTINAL MUCOSA INTERACTIONS
Aphthous ulcers of the “dome” or follicle-associated epithelium (FAE), overlying Peyer’s patches in the distal ileum and lymphoid follicles of the colon, are likely the initial mucosal lesions occurring in Crohn’s patients[73-75], and have been observed in patients using magnifying chromoendoscopy[76]. The FAE effectively forms the interface between the intestinal lymphoid system and the luminal environment. Specialized microfold (M) cells accounting for about 5% of cells in the FAE are optimized for antigen adherence and transport, and for immunological sampling of microorganisms[77]. Several invasive bacteria take advantage of the transcytotic characteristics of M cells to use them to cross the gut, including Yersinia, Salmonella and Shigella spp[78-80]. It was suspected that the portal of mucosal entry of AIEC was also likely through M cells[81] and own recent studies successfully modelling M cells in vitro, demonstrated that Crohn’s AIEC could indeed translocate through M cells (up to 20-fold compared with parent Caco2 cells) and through isolated human ileal FAE[82]. Adhesion and subsequent translocation of AIEC across murine and human Peyer’s patches, and across M cells in vitro, was observed to be dependent on possession of the lpf operon, encoding long polar fimbriae (Lpf) in AIEC[83]. Isolates expressing lpf have been found to be more prevalent in Crohn’s mucosae than that of non-IBD controls[84]. Ex vivo studies also indicate a defective mucosal barrier to bacteria in the Peyer’s patches from Crohn’s patients[85,86]. It is plausible therefore that increased bacterial load at M cells is important in the development of Crohn’s. A striking correlation also exists between the age-related incidence of CD and the number of Peyer’s patches in the small bowel, the latter peaking in late adolescence and then falling away[87].
Ileal AIEC isolates also typically express type-1 pili (FimH) on their surface supporting adherence to ileal enterocytes via interaction with carcinoembryonic antigen-related cell adhesion molecule-6 (CEACAM6) receptors known to be over expressed on the inflamed ileal (but not colonic) epithelium in Crohn’s[88]. Highly glycosylated CEACAMs have also been proposed as M cell microbial receptors[89]. It is plausible that one or more members of the CEACAM receptor family may play an important role in regulating endocytosis of CD mucosa-associated E. coli into host M cells. A recent study also reported that the glycoprotein 2 (GP2), specifically expressed on the apical plasma membrane of M cells among enterocytes, is recognized by FimH[90]. By an intriguing coincidence it has also recently been found that the same GP2 protein is the epitope for the “anti-pancreatic” antibody found in CD sera[91]. In addition, Crohn’s AIEC outer-membrane vesicles (OMV), also show ability to interact with enterocyte endoplasmic reticulum stress response glycoprotein 96 receptor, increased in expression on the inflamed intestinal epithelium[92]. These OMVs, in association with flagellin, also possess significant ability to evoke pro-inflammatory cytokine release[93]. Colonic mucosally associated AIEC isolates expressing afimbrial adhesin afa operon, more commonly associated with diarrhoeagenic diffusely adherent E. coli, have also been observed to be more prevalent in CD patients than in non-IBD controls[84]. The presence of the afa operon correlates with diffuse adherence to, and invasion of intestinal epithelial cells[84].
A summary of Crohn’s AIEC genotype relevant to host intestinal mucosa interactions is summarised in Figure 3.
Figure 3.
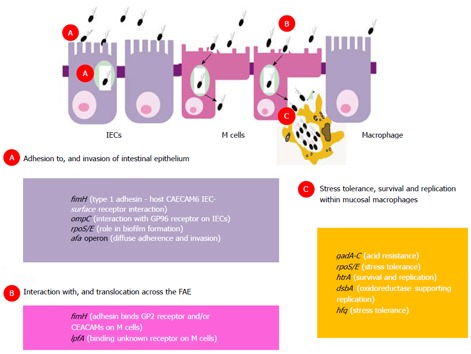
Crohn’s mucosally associated adherent, invasive Escherichia coli host mucosa interactions: genotype-phenotype relationships. A: Adhesion to, and invasion of intestinal epithelium; B: Mucosal entry across the follicle-associated epithelium; C: Tolerance to stress, habituation and replication within mucosal macrophages. afa: Operon encoding afimbrial adhesin; CEACAM: Carcinoembryonic antigen-related cell adhesion molecule; dsbA: Gene encoding bacterial disulfide oxidoreductase; fimH: Gene encoding bacterial type-1 fimbrial adhesin; gadA-C: Glutamate-dependent acid resistance genes; GP2: Glycoprotein 2 receptor; GP96: Endoplasmic reticulum stress response glycoprotein 96; hfq: Gene encoding RNA-binding host factor essential for replication of the bacteriophage Qβ; htrA: Gene encoding high temperature stress protein A; IECs: Intestinal epithelial cells; lpfA: Gene encoding long polar fimbriae adhesin; M cells: Microfold cells; ompC: Gene encoding outer-membrane vesicle protein C; rpoS/E: Genes encoding stress tolerance sigma factors.
VIRULENCE FACTORS SUPPORTING CROHN’S AIEC SURVIVAL AND REPLICATION WITHIN HOST MACROPHAGES
AIEC isolated from Crohn’s ileal and colonic biopsy tissue demonstrate ability to survive and replicate within phagolysosomes of host macrophages[94,95]; see Figure 4. However, they are not unique in this ability as other pathogens are also known to survive and replicate within macrophages, including Mycobacteria, Salmonella, Shigella, Coxiella, Brucella, Legionella and Listeria species. Key defence mechanisms adopted by these pathogens support their resistance to killing within the low pH, low nutrient environment, high oxidative and nitrosative stress environment of the phagolysosome. For example, Shigella and Listeria are able to escape from the mature phagolysosome, Salmonellae can inhibit fusion of phagosome with the lysosome, whilst Mycobacterium tuberculosis is able to modify the intra-phagolysosome environment[96]. Key genes supporting AIEC survival and replication within macrophages have been identified (see Figure 3) using isogenic mutants of the “paradigm” ileal AIEC LF82, including htrA (encoding high temperature stress protein), dsbA (encoding an oxidoreductase) and hfq (encoding a RNA chaperone important in mediating bacterial adaptation to chemical stress)[97-99]. However, HtrA and DsbA are fairly ubiquitous in E. coli, and it is likely that other unidentified factors are needed to support AIEC survival within the stressful conditions of the phagolysosome.
Figure 4.
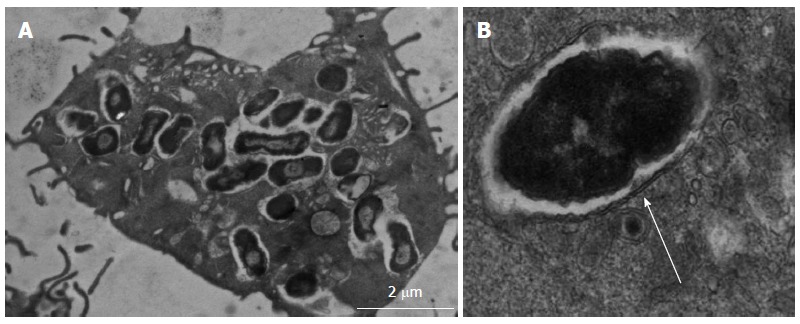
Transmission electron micrograph of adherent, invasive Escherichia coli within macrophages1. A: Crohn’s disease colonic mucosa-associated isolate HM605 surviving and replicating within vesicles of J774-A1 murine macrophages; B: Double membrane around intra-macrophage vesicle indicates bacteria are contained within phagolysosomes (arrow). 1Images courtesy of Dr. Carol L Roberts (University of Liverpool, United Kingdom).
Acid stress is the antimicrobial environment likely encountered by active enteric bacteria within the phagolysosome. Salmonella spp., Shigella spp. and E. coli have all been reported to possess a repertoire of low pH inducible systems that support resistance, tolerance and habituation during environmental acid stress. Likewise, AIEC certainly appear to be tolerant of the low pH intra-phagolysosome environment[97]. E. coli is notable due to its possession of four known acid resistance systems. The first system requires sigma factor RpoS and the cyclic AMP receptor protein CRP, with RpoS functioning as a major environmental stress response regulator in both E. coli and Salmonellae[100]. Deletion of RpoS from a Crohn’s AIEC (strain O83:H1) has been observed to increase sensitivity of this clinical isolate to oxidative stress[101]. The second system requires extracellular glutamate. The components of glutamate-dependent acid response are two isoforms of glutamate decarboxylase encoded by gadA and gadB, and a glutamate-γ-aminobutyric acid antiporter encoded by gadC[102,103]. Murine AIEC have been observed to respond to chronic intestinal inflammation by up-regulating expression of gadA and gadB[104]. The third acid resistance system requires is arginine-dependent utilising of arginine decarboxylase (AdiA and AdiC) antiporter[100] and the fourth is lysine dependent, involving lysine decarboxylase[103]. In addition, E. coli also harbour specific mechanisms that enable them to resist high levels of ROS that form the oxidative and super-oxidative response to phagocytosed pathogens. These defensive resources have recently been found to be grouped particularly into two regulated sets of genes soxRS and oxyR regulons[105,106].
DEFECTIVE AUTOPHAGY AND LACK OF CLEARANCE OF AIEC
ATG16L1 and IRGM function in autophagosome formation and evidence from our own studies supports a role for autophagy as an antimicrobial mechanism downstream of toll-like receptor and NOD-like receptor signalling. Activation of NOD2 by MDP induces autophagy in antigen-presenting cells (such as dendritic cells and macrophages) in a receptor-interacting serine-threonine kinase-2 dependent manner[107]. Knock-down of ATG16L1 and IRGM using siRNA approaches results in defective recognition and clearance of Crohn’s mucosa-associated E. coli within host epithelial cells and macrophages[108]. However, deficiency in either gene did not interfere with the replication and survival ability of other non-pathogenic, environmental, commensal, or gastroenteritis-inducing E. coli, suggesting a specific role for autophagy in restraining AIEC. Similarly, expression of the Crohn’s variant ATG16L1*300A in intestinal Caco2 epithelial cells impairs their ability to capture internalized Salmonella spp. within autophagosomes[109] and is also associated with abnormalities in Paneth cell granule exocytosis[110], impaired production of antimicrobial α-defensins[111], and increased production of pro-inflammatory cytokines IL-1β and IL-18 by macrophages in response to LPS[112].
STRATEGIES TO TARGET INTRA-MACROPHAGE AIEC IN CD
If AIEC have a primary pathogenic role then it follows that targeted treatment should lead to clinical benefit. This hypothesis is supported by studies in Boxer dogs which develop a granulomatous colitis following infection with an AIEC strain[69], with subsequent clinical resolution following treatment with the 4-quinolone antibiotics, enrofloxacin[113]. However bacterial antibiotic resistance is common both in animal and human studies and is associated with poor clinical outcome[114]. Trials of antibiotics in the treatment of active CD have been disappointing to date with good evidence only for their use in the prevention of post-operative disease recurrence[115,116]. A large metanalysis recently failed to show any clear benefit for their use in maintenance of remission or in the treatment of active luminal or peri-anal disease[117]. In some trials, early open label studies were positive only for later randomised trials to fail to show clear benefit[118,119], which may, in part, be due to the development of antibiotic resistance. In vitro, quinolone-based antibiotics regimens to target intra-macrophage Crohn’s AIEC isolates are effective[95] but again single antibiotic use likely increases the risk of drug resistance, a problem highlighted by a recent study in which multidrug resistance was seen in 61.5% of Crohn’s AIEC isolates[120]. Triple antibiotic regimens are superior to ciprofloxacin mono-therapy and reduce intra-macrophage AIEC survival to 3% relative to untreated controls[95]. Unfortunately significant drug-drug interactions occur with some antibiotics and azathioprine which have limited the use of triple combinations to date. Consequently, alternative strategies are being explored including using adjuvant agents to manipulate the phagolysosomal environment to support microbial phagocytosis.
A more promising strategy may be to alter phagolysosomal pH to aid bacterial killing within macrophages. It has already been shown that AIEC are dependent on an acidic environment for survival[97] and that alkalinisation leads to reduced survival. Hydroxychloroquine, a weak base able to increase phagolysosomal pH, is known to improve killing of bacteria where intra-macrophage survival plays a key step in disease pathogenesis[119]. For example, Coxiella burnetii the agent of Q fever, maintains an intracellular lifestyle through adaptation to survival at an acidic pH[121,122]. Coxiella survival was significantly reduced in vitro by hydroxychloroquine treatment and this benefit translated into clinical response in a randomised trial[123,124]. Hydroxychloroquine in combination with antibiotics, is also now standard therapy for treatment of Whipple’s disease, where replication of Tropheryma whipplei within tissue macrophages is a central part of the pathogenesis[125]. Similarly, our own recent studies have shown that dose-dependent enhancement of macrophage killing of Crohn’s AIEC can be seen with hydroxychloroquine treatment and synergy with standard antibiotics is also observed[126].
Vitamin D supplementation also enhances killing of intracellular AIEC in both murine and human macrophages[127]. This may be due to enhancement of the respiratory burst but effects are likely to be multimodal with influences on several intracellular pathways. Cellular production of the antimicrobial peptides, such as cathelicidin antimicrobial peptide (CAMP) and β2 defensin, follows stimulation of toll-like receptors in the presence of vitamin D and conversely, vitamin D deficiency leads to impaired macrophage function due to defective defensin production[128]. This has significance in CD, where muramyl dipeptide stimulation in the presence of vitamin D leads to increased CAMP expression. Futhermore, vitamin D stimulates NOD2 expression and leads to downstream β2 defensin production[129]. Vitamin D deficiency is common in CD with up to 70% of patients affected, even in quiescent disease[130,131]. This now appears to have clinical consequence with several studies demonstrating a correlation between serum levels and disease behaviour. In a large prospective cohort study with nearly 1.5 m patient years of follow up, a validated method for predicting vitamin D levels was used to compare the incidence of CD in the lowest quartile relative to the highest quartile, finding the highest risk associated with the lowest Vitamin D levels[132]. This correlation is not limited to the relative disease risk and recent studies now show a clear correlation between disease behaviour and serum concentrations. CD activity, defined both by CDAI and CRP level, has been shown to be inversely correlated with Vitamin D levels, with greatest activity seen in those with the lowest levels[133]. Furthermore, in a retrospective study of 3217 patients, a lower likelihood of requiring surgery for Crohn’s was seen with higher vitamin D levels, when using a cut off of 30 ng/mL[134]. Given these findings we might therefore expect a clinical effect from Vitamin D supplementation. This question was addressed in a randomised double-blind placebo-controlled trial in which a trend was seen towards lower relapse rates in patients treated with 1200 U/d of Vitamin D, although this did not quite reach significance[135]. However a significant reduction in risk of requiring surgery was seen for deficient patients who normalised their vitamin D levels with supplementation[134]. Overall these data suggest a clinical role for vitamin D supplementation in CD although further clinical trials are required. Whilst no data yet exists for the effect of vitamin D on AIEC-macrophage interactions in vivo, it appears that supplementation may hold promise as a clinical strategy for targeting Crohn’s mucosa-associated E. coli.
Smoking has long been associated with disease activity and leads to greater treatment requirements, more stricturing disease, more peri-anal disease and shorter disease free survival[135,136]. These affects are likely to be multimodal in origin with effects seen on macrophage function, gut microbiota and vitamin D levels[137-139]. Interventional studies clearly show benefit from smoking cessation[140] and that this is an achievable therapeutic aim[141]. There are some data to support a hypothesis that this may in part be due to recovery of immune cell function but to date this has not been systematically studied in CD[142].
CONCLUSION
Based on the findings of a diversity of individual studies, there has been accumulating evidence proving the implication of bacteria such as AIEC in the pathogenesis of CD, a chronic-relapsing IBD. AIEC have been shown to translocate M cells of Peyer’s patches and lymphoid follicles of the colon, and then to survive and replicate within underlying mucosal macrophages and dendritic cells. However, the mechanism of how Crohn’s AIEC resist killing process and adapt to the environment within the phagolysosme to survive and grow within macrophages without inducing cell death is still poorly understood. There is no doubt that further investigation is warranted to characterise and identify the key virulence factors relevant to AIEC phenotype, supporting current and novel, targeted treatments for future clinical benefit.
Footnotes
Supported by Award from the Ministry of Higher Education and Scientific Research/Cultural Attache - London Libyan Embassy, No. UM873-611-23962; National Institute for Health Research (NIHR) Biomedical Research Fellowship, No. BRF-2011-025; Shire Innovation Fund for Specialist Registrars; and funding from Crohn’s amd Colitis United Kingdom, No. M-08-1/M-13-2; Liverpool NIHR-Biomedical Research Centre for Microbial Diseases (01CD1); and Support of the European Science Foundation, in the framework of the Research Networking Programme, The European Network for Gastrointestinal Health Research
P- Reviewers: D'Elios MM, Ghigo E S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 2.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 3.Campieri M, Gionchetti P. Bacteria as the cause of ulcerative colitis. Gut. 2001;48:132–135. doi: 10.1136/gut.48.1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Val JH. Old-age inflammatory bowel disease onset: a different problem? World J Gastroenterol. 2011;17:2734–2739. doi: 10.3748/wjg.v17.i22.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canavan C, Abrams KR, Mayberry JF. Meta-analysis: mortality in Crohn’s disease. Aliment Pharmacol Ther. 2007;25:861–870. doi: 10.1111/j.1365-2036.2007.03276.x. [DOI] [PubMed] [Google Scholar]
- 6.Vermeire S, van Assche G, Rutgeerts P. Review article: Altering the natural history of Crohn’s disease--evidence for and against current therapies. Aliment Pharmacol Ther. 2007;25:3–12. doi: 10.1111/j.1365-2036.2006.03134.x. [DOI] [PubMed] [Google Scholar]
- 7.Rungoe C, Langholz E, Andersson M, Basit S, Nielsen NM, Wohlfahrt J, Jess T. Changes in medical treatment and surgery rates in inflammatory bowel disease: a nationwide cohort study 1979-2011. Gut. 2013:Epub ahead of print. doi: 10.1136/gutjnl-2013-305607. [DOI] [PubMed] [Google Scholar]
- 8.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42; quiz e30. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am J Clin Nutr. 1996;63:741–745. doi: 10.1093/ajcn/63.5.741. [DOI] [PubMed] [Google Scholar]
- 10.Barreiro-de Acosta M, Alvarez Castro A, Souto R, Iglesias M, Lorenzo A, Dominguez-Muñoz JE. Emigration to western industrialized countries: A risk factor for developing inflammatory bowel disease. J Crohns Colitis. 2011;5:566–569. doi: 10.1016/j.crohns.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK. Host-bacterial symbiosis in health and disease. Adv Immunol. 2010;107:243–274. doi: 10.1016/B978-0-12-381300-8.00008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanagan P, Campbell BJ, Rhodes JM. Bacteria in the pathogenesis of inflammatory bowel disease. Biochem Soc Trans. 2011;39:1067–1072. doi: 10.1042/BST0391067. [DOI] [PubMed] [Google Scholar]
- 13.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 14.Willing B, Halfvarson J, Dicksved J, Rosenquist M, Järnerot G, Engstrand L, Tysk C, Jansson JK. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–660. doi: 10.1002/ibd.20783. [DOI] [PubMed] [Google Scholar]
- 15.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 16.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mondot S, Kang S, Furet JP, Aguirre de Carcer D, McSweeney C, Morrison M, Marteau P, Doré J, Leclerc M. Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm Bowel Dis. 2011;17:185–192. doi: 10.1002/ibd.21436. [DOI] [PubMed] [Google Scholar]
- 18.Mahid SS, Minor KS, Stevens PL, Galandiuk S. The role of smoking in Crohn’s disease as defined by clinical variables. Dig Dis Sci. 2007;52:2897–2903. doi: 10.1007/s10620-006-9624-0. [DOI] [PubMed] [Google Scholar]
- 19.Biedermann L, Zeitz J, Mwinyi J, Sutter-Minder E, Rehman A, Ott SJ, Steurer-Stey C, Frei A, Frei P, Scharl M, et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One. 2013;8:e59260. doi: 10.1371/journal.pone.0059260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapman-Kiddell CA, Davies PS, Gillen L, Radford-Smith GL. Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:137–151. doi: 10.1002/ibd.20968. [DOI] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, Darfeuille-Michaud A, Barnich N. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 2014;63:116–124. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 24.Lee JC, Parkes M. Genome-wide association studies and Crohn’s disease. Brief Funct Genomics. 2011;10:71–76. doi: 10.1093/bfgp/elr009. [DOI] [PubMed] [Google Scholar]
- 25.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ; NIDDK IBD Genetics Consortium, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E; Belgian-French IBD Consortium; Wellcome Trust Case Control Consortium, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Büning C, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H; International IBD Genetics Consortium (IIBDGC), Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 29.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 30.Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. 2005;6:376–388. doi: 10.1038/nrg1607. [DOI] [PubMed] [Google Scholar]
- 31.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 33.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 34.Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, Rosenstiel P, Albrecht M, Croucher PJ, Seegert D, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476–480. doi: 10.1038/ng1345. [DOI] [PubMed] [Google Scholar]
- 35.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 36.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 39.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 40.Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy. 2006;2:156–158. doi: 10.4161/auto.2825. [DOI] [PubMed] [Google Scholar]
- 41.Ouburg S, Mallant-Hent R, Crusius JB, van Bodegraven AA, Mulder CJ, Linskens R, Peña AS, Morré SA. The toll-like receptor 4 (TLR4) Asp299Gly polymorphism is associated with colonic localisation of Crohn’s disease without a major role for the Saccharomyces cerevisiae mannan-LBP-CD14-TLR4 pathway. Gut. 2005;54:439–440. doi: 10.1136/gut.2004.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volpp BD, Nauseef WM, Clark RA. Two cytosolic neutrophil oxidase components absent in autosomal chronic granulomatous disease. Science. 1988;242:1295–1297. doi: 10.1126/science.2848318. [DOI] [PubMed] [Google Scholar]
- 44.Marks DJ, Harbord MW, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, Lees W, Novelli M, Bloom S, Segal AW. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet. 2006;367:668–678. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 45.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJ, Sewell GW, Palmer CD, Wilde J, Foxwell BM, Gloger IS, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–1897. doi: 10.1084/jem.20091233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut. 2007;56:1333–1336. doi: 10.1136/gut.2006.115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, Pfyffer GE, Jemmi T, Baumgartner A, Egger M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: a systematic review and meta-analysis. Lancet Infect Dis. 2007;7:607–613. doi: 10.1016/S1473-3099(07)70211-6. [DOI] [PubMed] [Google Scholar]
- 49.Naser SA, Ghobrial G, Romero C, Valentine JF. Culture of Mycobacterium avium subspecies paratuberculosis from the blood of patients with Crohn’s disease. Lancet. 2004;364:1039–1044. doi: 10.1016/S0140-6736(04)17058-X. [DOI] [PubMed] [Google Scholar]
- 50.Ryan P, Bennett MW, Aarons S, Lee G, Collins JK, O’Sullivan GC, O’Connell J, Shanahan F. PCR detection of Mycobacterium paratuberculosis in Crohn’s disease granulomas isolated by laser capture microdissection. Gut. 2002;51:665–670. doi: 10.1136/gut.51.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olsen I, Tollefsen S, Aagaard C, Reitan LJ, Bannantine JP, Andersen P, Sollid LM, Lundin KE. Isolation of Mycobacterium avium subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of Crohn’s disease patients. PLoS One. 2009;4:e5641. doi: 10.1371/journal.pone.0005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendoza JL, San-Pedro A, Culebras E, Cíes R, Taxonera C, Lana R, Urcelay E, de la Torre F, Picazo JJ, Díaz-Rubio M. High prevalence of viable Mycobacterium avium subspecies paratuberculosis in Crohn’s disease. World J Gastroenterol. 2010;16:4558–4563. doi: 10.3748/wjg.v16.i36.4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mpofu CM, Campbell BJ, Subramanian S, Marshall-Clarke S, Hart CA, Cross A, Roberts CL, McGoldrick A, Edwards SW, Rhodes JM. Microbial mannan inhibits bacterial killing by macrophages: a possible pathogenic mechanism for Crohn’s disease. Gastroenterology. 2007;133:1487–1498. doi: 10.1053/j.gastro.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 54.Zhang F, Liu H, Chen S, Low H, Sun L, Cui Y, Chu T, Li Y, Fu X, Yu Y, et al. Identification of two new loci at IL23R and RAB32 that influence susceptibility to leprosy. Nat Genet. 2011;43:1247–1251. doi: 10.1038/ng.973. [DOI] [PubMed] [Google Scholar]
- 55.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tabaqchali S, O’Donoghue DP, Bettelheim KA. Escherichia coli antibodies in patients with inflammatory bowel disease. Gut. 1978;19:108–113. doi: 10.1136/gut.19.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, van Kruiningen HJ, West AB, Cartun RW, Cortot A, Colombel JF. Immunocytochemical evidence of Listeria, Escherichia coli, and Streptococcus antigens in Crohn’s disease. Gastroenterology. 1995;108:1396–1404. doi: 10.1016/0016-5085(95)90687-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 59.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 60.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 61.Mylonaki M, Rayment NB, Rampton DS, Hudspith BN, Brostoff J. Molecular characterization of rectal mucosa-associated bacterial flora in inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:481–487. doi: 10.1097/01.mib.0000159663.62651.4f. [DOI] [PubMed] [Google Scholar]
- 62.Sasaki M, Sitaraman SV, Babbin BA, Gerner-Smidt P, Ribot EM, Garrett N, Alpern JA, Akyildiz A, Theiss AL, Nusrat A, et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab Invest. 2007;87:1042–1054. doi: 10.1038/labinvest.3700661. [DOI] [PubMed] [Google Scholar]
- 63.Kotlowski R, Bernstein CN, Sepehri S, Krause DO. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut. 2007;56:669–675. doi: 10.1136/gut.2006.099796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez-Medina M, Aldeguer X, Lopez-Siles M, González-Huix F, López-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15:872–882. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 65.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115:281–286. doi: 10.1016/s0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 67.Ryan P, Kelly RG, Lee G, Collins JK, O’Sullivan GC, O’Connell J, Shanahan F. Bacterial DNA within granulomas of patients with Crohn’s disease--detection by laser capture microdissection and PCR. Am J Gastroenterol. 2004;99:1539–1543. doi: 10.1111/j.1572-0241.2004.40103.x. [DOI] [PubMed] [Google Scholar]
- 68.Meconi S, Vercellone A, Levillain F, Payré B, Al Saati T, Capilla F, Desreumaux P, Darfeuille-Michaud A, Altare F. Adherent-invasive Escherichia coli isolated from Crohn’s disease patients induce granulomas in vitro. Cell Microbiol. 2007;9:1252–1261. doi: 10.1111/j.1462-5822.2006.00868.x. [DOI] [PubMed] [Google Scholar]
- 69.Simpson KW, Dogan B, Rishniw M, Goldstein RE, Klaessig S, McDonough PL, German AJ, Yates RM, Russell DG, Johnson SE, et al. Adherent and invasive Escherichia coli is associated with granulomatous colitis in boxer dogs. Infect Immun. 2006;74:4778–4792. doi: 10.1128/IAI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martinez-Medina M, Garcia-Gil J, Barnich N, Wieler LH, Ewers C. Adherent-invasive Escherichia coli phenotype displayed by intestinal pathogenic E. coli strains from cats, dogs, and swine. Appl Environ Microbiol. 2011;77:5813–5817. doi: 10.1128/AEM.02614-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Darfeuille-Michaud A. Adherent-invasive Escherichia coli: a putative new E. coli pathotype associated with Crohn’s disease. Int J Med Microbiol. 2002;292:185–193. doi: 10.1078/1438-4221-00201. [DOI] [PubMed] [Google Scholar]
- 72.Boyd EF, Hartl DL. Chromosomal regions specific to pathogenic isolates of Escherichia coli have a phylogenetically clustered distribution. J Bacteriol. 1998;180:1159–1165. doi: 10.1128/jb.180.5.1159-1165.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morson BC. The early histological lesion of Crohn’s disease. Proc R Soc Med. 1972;65:71–72. [PMC free article] [PubMed] [Google Scholar]
- 74.Rickert RR, Carter HW. The “early” ulcerative lesion of Crohn’s disease: correlative light- and scanning electron-microscopic studies. J Clin Gastroenterol. 1980;2:11–19. [PubMed] [Google Scholar]
- 75.Fujimura Y, Kamoi R, Iida M. Pathogenesis of aphthoid ulcers in Crohn’s disease: correlative findings by magnifying colonoscopy, electron microscopy, and immunohistochemistry. Gut. 1996;38:724–732. doi: 10.1136/gut.38.5.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shikuwa S, Isomoto H, Mizuta Y, Suematsu T, Ito M, Kohno S. Magnifying videoendoscopic findings of Peyer’s patches in the terminal ileum of Crohn’s disease. Gut. 2007;56:894–895. doi: 10.1136/gut.2007.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kraehenbuhl JP, Neutra MR. Epithelial M cells: differentiation and function. Annu Rev Cell Dev Biol. 2000;16:301–332. doi: 10.1146/annurev.cellbio.16.1.301. [DOI] [PubMed] [Google Scholar]
- 78.Marra A, Isberg RR. Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer’s patch intestinal epithelium. Infect Immun. 1997;65:3412–3421. doi: 10.1128/iai.65.8.3412-3421.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med. 1994;180:15–23. doi: 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sansonetti PJ, Arondel J, Cantey JR, Prévost MC, Huerre M. Infection of rabbit Peyer’s patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect Immun. 1996;64:2752–2764. doi: 10.1128/iai.64.7.2752-2764.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gullberg E, Söderholm JD. Peyer’s patches and M cells as potential sites of the inflammatory onset in Crohn’s disease. Ann N Y Acad Sci. 2006;1072:218–232. doi: 10.1196/annals.1326.028. [DOI] [PubMed] [Google Scholar]
- 82.Roberts CL, Keita AV, Duncan SH, O’Kennedy N, Söderholm JD, Rhodes JM, Campbell BJ. Translocation of Crohn’s disease Escherichia coli across M-cells: contrasting effects of soluble plant fibres and emulsifiers. Gut. 2010;59:1331–1339. doi: 10.1136/gut.2009.195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chassaing B, Rolhion N, de Vallée A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Söderholm JD, Hugot JP, Colombel JF, et al. Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest. 2011;121:966–975. doi: 10.1172/JCI44632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, Knight P, Codling C, Marchesi JR, Winstanley C, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2014;63:761–770. doi: 10.1136/gutjnl-2013-304739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keita AV, Salim SY, Jiang T, Yang PC, Franzén L, Söderkvist P, Magnusson KE, Söderholm JD. Increased uptake of non-pathogenic E. coli via the follicle-associated epithelium in longstanding ileal Crohn’s disease. J Pathol. 2008;215:135–144. doi: 10.1002/path.2337. [DOI] [PubMed] [Google Scholar]
- 86.Salim SY, Silva MA, Keita AV, Larsson M, Andersson P, Magnusson KE, Perdue MH, Söderholm JD. CD83+CCR7- dendritic cells accumulate in the subepithelial dome and internalize translocated Escherichia coli HB101 in the Peyer’s patches of ileal Crohn’s disease. Am J Pathol. 2009;174:82–90. doi: 10.2353/ajpath.2009.080273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Kruiningen HJ, West AB, Freda BJ, Holmes KA. Distribution of Peyer’s patches in the distal ileum. Inflamm Bowel Dis. 2002;8:180–185. doi: 10.1097/00054725-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 88.Barnich N, Darfeuille-Michaud A. Abnormal CEACAM6 expression in Crohn disease patients favors gut colonization and inflammation by adherent-invasive E. coli. Virulence. 2010;1:281–282. doi: 10.4161/viru.1.4.11510. [DOI] [PubMed] [Google Scholar]
- 89.Baranov V, Hammarström S. Carcinoembryonic antigen (CEA) and CEA-related cell adhesion molecule 1 (CEACAM1), apically expressed on human colonic M cells, are potential receptors for microbial adhesion. Histochem Cell Biol. 2004;121:83–89. doi: 10.1007/s00418-003-0613-5. [DOI] [PubMed] [Google Scholar]
- 90.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. 2009;462:226–230. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 91.Roggenbuck D, Hausdorf G, Martinez-Gamboa L, Reinhold D, Büttner T, Jungblut PR, Porstmann T, Laass MW, Henker J, Büning C, et al. Identification of GP2, the major zymogen granule membrane glycoprotein, as the autoantigen of pancreatic antibodies in Crohn’s disease. Gut. 2009;58:1620–1628. doi: 10.1136/gut.2008.162495. [DOI] [PubMed] [Google Scholar]
- 92.Rolhion N, Barnich N, Bringer MA, Glasser AL, Ranc J, Hébuterne X, Hofman P, Darfeuille-Michaud A. Abnormally expressed ER stress response chaperone Gp96 in CD favours adherent-invasive Escherichia coli invasion. Gut. 2010;59:1355–1362. doi: 10.1136/gut.2010.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Subramanian S, Rhodes JM, Hart CA, Tam B, Roberts CL, Smith SL, Corkill JE, Winstanley C, Virji M, Campbell BJ. Characterization of epithelial IL-8 response to inflammatory bowel disease mucosal E. coli and its inhibition by mesalamine. Inflamm Bowel Dis. 2008;14:162–175. doi: 10.1002/ibd.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bringer MA, Glasser AL, Tung CH, Méresse S, Darfeuille-Michaud A. The Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 replicates in mature phagolysosomes within J774 macrophages. Cell Microbiol. 2006;8:471–484. doi: 10.1111/j.1462-5822.2005.00639.x. [DOI] [PubMed] [Google Scholar]
- 95.Subramanian S, Roberts CL, Hart CA, Martin HM, Edwards SW, Rhodes JM, Campbell BJ. Replication of Colonic Crohn’s Disease Mucosal Escherichia coli Isolates within Macrophages and Their Susceptibility to Antibiotics. Antimicrob Agents Chemother. 2008;52:427–434. doi: 10.1128/AAC.00375-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Amano A, Nakagawa I, Yoshimori T. Autophagy in innate immunity against intracellular bacteria. J Biochem. 2006;140:161–166. doi: 10.1093/jb/mvj162. [DOI] [PubMed] [Google Scholar]
- 97.Bringer MA, Barnich N, Glasser AL, Bardot O, Darfeuille-Michaud A. HtrA stress protein is involved in intramacrophagic replication of adherent and invasive Escherichia coli strain LF82 isolated from a patient with Crohn’s disease. Infect Immun. 2005;73:712–721. doi: 10.1128/IAI.73.2.712-721.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bringer MA, Rolhion N, Glasser AL, Darfeuille-Michaud A. The oxidoreductase DsbA plays a key role in the ability of the Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 to resist macrophage killing. J Bacteriol. 2007;189:4860–4871. doi: 10.1128/JB.00233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Simonsen KT, Nielsen G, Bjerrum JV, Kruse T, Kallipolitis BH, Møller-Jensen J. A role for the RNA chaperone Hfq in controlling adherent-invasive Escherichia coli colonization and virulence. PLoS One. 2011;6:e16387. doi: 10.1371/journal.pone.0016387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Castanie-Cornet MP, Penfound TA, Smith D, Elliott JF, Foster JW. Control of acid resistance in Escherichia coli. J Bacteriol. 1999;181:3525–3535. doi: 10.1128/jb.181.11.3525-3535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Allen CA, Niesel DW, Torres AG. The effects of low-shear stress on Adherent-invasive Escherichia coli. Environ Microbiol. 2008;10:1512–1525. doi: 10.1111/j.1462-2920.2008.01567.x. [DOI] [PubMed] [Google Scholar]
- 102.Foster JW. Escherichia coli acid resistance: tales of an amateur acidophile. Nat Rev Microbiol. 2004;2:898–907. doi: 10.1038/nrmicro1021. [DOI] [PubMed] [Google Scholar]
- 103.Iyer R, Williams C, Miller C. Arginine-agmatine antiporter in extreme acid resistance in Escherichia coli. J Bacteriol. 2003;185:6556–6561. doi: 10.1128/JB.185.22.6556-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tchaptchet S, Fan TJ, Goeser L, Schoenborn A, Gulati AS, Sartor RB, Hansen JJ. Inflammation-induced acid tolerance genes gadAB in luminal commensal Escherichia coli attenuate experimental colitis. Infect Immun. 2013;81:3662–3671. doi: 10.1128/IAI.00355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pomposiello PJ, Bennik MH, Demple B. Genome-wide transcriptional profiling of the Escherichia coli responses to superoxide stress and sodium salicylate. J Bacteriol. 2001;183:3890–3902. doi: 10.1128/JB.183.13.3890-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–776. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 108.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 113.Mansfield CS, James FE, Craven M, Davies DR, O’Hara AJ, Nicholls PK, Dogan B, MacDonough SP, Simpson KW. Remission of histiocytic ulcerative colitis in Boxer dogs correlates with eradication of invasive intramucosal Escherichia coli. J Vet Intern Med. 2009;23:964–969. doi: 10.1111/j.1939-1676.2009.0363.x. [DOI] [PubMed] [Google Scholar]
- 114.Craven M, Dogan B, Schukken A, Volkman M, Chandler A, McDonough PL, Simpson KW. Antimicrobial resistance impacts clinical outcome of granulomatous colitis in boxer dogs. J Vet Intern Med. 2010;24:819–824. doi: 10.1111/j.1939-1676.2010.0527.x. [DOI] [PubMed] [Google Scholar]
- 115.D'Haens GR, Vermeire S, Van Assche G, Noman M, Aerden I, Van Olmen G, Rutgeerts P. Therapy of metronidazole with azathioprine to prevent postoperative recurrence of Crohn’s disease: a controlled randomized trial. Gastroenterology. 2008;135:1123–1129. doi: 10.1053/j.gastro.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 116.Rutgeerts P, Van Assche G, Vermeire S, D’Haens G, Baert F, Noman M, Aerden I, De Hertogh G, Geboes K, Hiele M, et al. Ornidazole for prophylaxis of postoperative Crohn’s disease recurrence: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2005;128:856–861. doi: 10.1053/j.gastro.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 117.Khan KJ, Ullman TA, Ford AC, Abreu MT, Abadir A, Marshall JK, Talley NJ, Moayyedi P. Antibiotic therapy in inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011;106:661–673. doi: 10.1038/ajg.2011.72. [DOI] [PubMed] [Google Scholar]
- 118.Leiper K, Martin K, Ellis A, Watson AJ, Morris AI, Rhodes JM. Clinical trial: randomized study of clarithromycin versus placebo in active Crohn’s disease. Aliment Pharmacol Ther. 2008;27:1233–1239. doi: 10.1111/j.1365-2036.2008.03661.x. [DOI] [PubMed] [Google Scholar]
- 119.Leiper K, Morris AI, Rhodes JM. Open label trial of oral clarithromycin in active Crohn’s disease. Aliment Pharmacol Ther. 2000;14:801–806. doi: 10.1046/j.1365-2036.2000.00753.x. [DOI] [PubMed] [Google Scholar]
- 120.Dogan B, Scherl E, Bosworth B, Yantiss R, Altier C, McDonough PL, Jiang ZD, Dupont HL, Garneau P, Harel J, et al. Multidrug resistance is common in Escherichia coli associated with ileal Crohn’s disease. Inflamm Bowel Dis. 2013;19:141–150. doi: 10.1002/ibd.22971. [DOI] [PubMed] [Google Scholar]
- 121.Rolain JM, Colson P, Raoult D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int J Antimicrob Agents. 2007;30:297–308. doi: 10.1016/j.ijantimicag.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maurin M, Benoliel AM, Bongrand P, Raoult D. Phagolysosomes of Coxiella burnetii-infected cell lines maintain an acidic pH during persistent infection. Infect Immun. 1992;60:5013–5016. doi: 10.1128/iai.60.12.5013-5016.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maurin M, Benoliel AM, Bongrand P, Raoult D. Phagolysosomal alkalinization and the bactericidal effect of antibiotics: the Coxiella burnetii paradigm. J Infect Dis. 1992;166:1097–1102. doi: 10.1093/infdis/166.5.1097. [DOI] [PubMed] [Google Scholar]
- 124.Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med. 1999;159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 125.Boulos A, Rolain JM, Raoult D. Antibiotic susceptibility of Tropheryma whipplei in MRC5 cells. Antimicrob Agents Chemother. 2004;48:747–752. doi: 10.1128/AAC.48.3.747-752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Flanagan PK, Campbell BJ, Rhodes JM. Hydroxychloroquine inhibits intra-macrophage replication of Crohn's disease E. coli and enhances the antimicrobial effect of antibiotics doxycycline and ciprofloxacin. Gut. 2011;60(Suppl 3):A210. [Google Scholar]
- 127.Flanagan PK, Campbell BJ, Rhodes JM. Vitamin D enhances macrophage function and improves killing of Crohn’s associated E. coli. J Crohns Colitis. 2012;7(Supp 1):S20. [Google Scholar]
- 128.Hewison M. Vitamin D and the immune system: new perspectives on an old theme. Endocrinol Metab Clin North Am. 2010;39:365–379, table of contents. doi: 10.1016/j.ecl.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang TT, Dabbas B, Laperriere D, Bitton AJ, Soualhine H, Tavera-Mendoza LE, Dionne S, Servant MJ, Bitton A, Seidman EG, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-defensin beta2 innate immune pathway defective in Crohn disease. J Biol Chem. 2010;285:2227–2231. doi: 10.1074/jbc.C109.071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Farraye FA, Nimitphong H, Stucchi A, Dendrinos K, Boulanger AB, Vijjeswarapu A, Tanennbaum A, Biancuzzo R, Chen TC, Holick MF. Use of a novel vitamin D bioavailability test demonstrates that vitamin D absorption is decreased in patients with quiescent Crohn’s disease. Inflamm Bowel Dis. 2011;17:2116–2121. doi: 10.1002/ibd.21595. [DOI] [PubMed] [Google Scholar]
- 131.Suibhne TN, Cox G, Healy M, O’Morain C, O’Sullivan M. Vitamin D deficiency in Crohn’s disease: prevalence, risk factors and supplement use in an outpatient setting. J Crohns Colitis. 2012;6:182–188. doi: 10.1016/j.crohns.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 132.Ananthakrishnan AN, Khalili H, Higuchi LM, Bao Y, Korzenik JR, Giovannucci EL, Richter JM, Fuchs CS, Chan AT. Higher predicted vitamin D status is associated with reduced risk of Crohn’s disease. Gastroenterology. 2012;142:482–489. doi: 10.1053/j.gastro.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jørgensen SP, Hvas CL, Agnholt J, Christensen LA, Heickendorff L, Dahlerup JF. Active Crohn’s disease is associated with low vitamin D levels. J Crohns Colitis. 2013;7:e407–e413. doi: 10.1016/j.crohns.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 134.Ananthakrishnan AN, Cagan A, Gainer VS, Cai T, Cheng SC, Savova G, Chen P, Szolovits P, Xia Z, De Jager PL, et al. Normalization of plasma 25-hydroxy vitamin D is associated with reduced risk of surgery in Crohn’s disease. Inflamm Bowel Dis. 2013;19:1921–1927. doi: 10.1097/MIB.0b013e3182902ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jørgensen SP, Agnholt J, Glerup H, Lyhne S, Villadsen GE, Hvas CL, Bartels LE, Kelsen J, Christensen LA, Dahlerup JF. Clinical trial: vitamin D3 treatment in Crohn’s disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. doi: 10.1111/j.1365-2036.2010.04355.x. [DOI] [PubMed] [Google Scholar]
- 136.Nunes T, Etchevers MJ, Domènech E, García-Sánchez V, Ber Y, Peñalva M, Merino O, Nos P, Garcia-Planella E, Casbas AG, et al. Smoking does influence disease behaviour and impacts the need for therapy in Crohn’s disease in the biologic era. Aliment Pharmacol Ther. 2013;38:752–760. doi: 10.1111/apt.12440. [DOI] [PubMed] [Google Scholar]
- 137.Tobin MV, Logan RF, Langman MJ, McConnell RB, Gilmore IT. Cigarette smoking and inflammatory bowel disease. Gastroenterology. 1987;93:316–321. doi: 10.1016/0016-5085(87)91021-3. [DOI] [PubMed] [Google Scholar]
- 138.Benjamin JL, Hedin CR, Koutsoumpas A, Ng SC, McCarthy NE, Prescott NJ, Pessoa-Lopes P, Mathew CG, Sanderson J, Hart AL, et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm Bowel Dis. 2012;18:1092–1100. doi: 10.1002/ibd.21864. [DOI] [PubMed] [Google Scholar]
- 139.King TE, Savici D, Campbell PA. Phagocytosis and killing of Listeria monocytogenes by alveolar macrophages: smokers versus nonsmokers. J Infect Dis. 1988;158:1309–1316. doi: 10.1093/infdis/158.6.1309. [DOI] [PubMed] [Google Scholar]
- 140.Cosnes J, Beaugerie L, Carbonnel F, Gendre JP. Smoking cessation and the course of Crohn’s disease: an intervention study. Gastroenterology. 2001;120:1093–1099. doi: 10.1053/gast.2001.23231. [DOI] [PubMed] [Google Scholar]
- 141.Nunes T, Etchevers MJ, Merino O, Gallego S, García-Sánchez V, Marín-Jiménez I, Menchén L, Barreiro-de Acosta M, Bastida G, García S, et al. High smoking cessation rate in Crohn’s disease patients after physician advice--the TABACROHN Study. J Crohns Colitis. 2013;7:202–207. doi: 10.1016/j.crohns.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 142.Kotani N, Kushikata T, Hashimoto H, Sessler DI, Muraoka M, Matsuki A. Recovery of intraoperative microbicidal and inflammatory functions of alveolar immune cells after a tobacco smoke-free period. Anesthesiology. 2001;94:999–1006. doi: 10.1097/00000542-200106000-00013. [DOI] [PubMed] [Google Scholar]