

Abstract
Merkel cell carcinoma (MCC) is an aggressive skin malignancy with a high mortality rate and an increasing incidence. The recent discovery of Merkel cell polyomavirus has revolutionized our understanding of MCC pathogenesis. Viral oncoproteins appear to play a critical role in tumor progression and are expressed in the majority of MCC tumors. Virus-specific humoral and cellular immune responses are detectable in MCC patients and are linked to the natural history of the disease. Despite persistent expression of immunogenic viral proteins, however, MCC tumors are able to evade the immune system. Understanding of the mechanisms of immune evasion employed by MCC tumors is rapidly increasing and offers opportunities for development of rational immune therapies to improve patient outcomes. Here we review recent discoveries in MCC with a special focus on the pathogenic role of Merkel cell polyomavirus and the immunobiology of this virus-associated disease.
Keywords: Merkel cell carcinoma, immunotherapy, Merkel cell polyomavirus, MCV, MCpyV, cancer virus, viral cancer, immune evasion, immune escape, MHC, tumor immunology, tumor infiltrating lymphocytes, TILs, viral oncoproteins, T-antigen, immune suppression
Introduction
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine skin cancer with a disease-associated mortality three times that of malignant melanoma (46% vs. 15% respectively) [1]. MCC is an uncommon cancer with an estimated 1600 cases/year in the US [2, 3]. The reported incidence has more than tripled over the past 20 years [3, 4] and the health impact of MCC is growing rapidly with the proportional increase in the aging population [2, 3]. This increasing incidence is in part due to improved detection following availability of a specific immunohistochemical marker, cytokeratin-20 [5], but is also likely due to the higher prevalence of known risk factors for MCC: T cell immune suppression and Caucasian over 50 years of age with extensive prior sun exposure [6]. MCC now kills more patients than cutaneous T cell lymphoma and a similar number as chronic myelogenous leukemia, both well-known and frequently studied cancers [2, 7, 8].
MCC is an aggressive cancer with prognosis dependent on the stage at presentation. Stages I and II represent low-risk and high-risk primary disease, respectively, while stages III and IV represent the presence of nodal and distant metastases, respectively. The reported 5-year relative survival for patients with local, nodal and metastatic disease is 64%, 39% and 18% respectively [1]. Although surgery and/or radiation therapy (RT) may be curative for patients with loco-regional MCC without distant metastases, relapses are common and often incurable. There is no established adjuvant therapy after definitive management. For patients with distant metastatic disease, systemic chemotherapy is considered. The objective response rate (ORR) with platinum-based chemotherapy regimens is around 60 percent [9]; however, responses are usually short-lived and the impact on survival is unclear. Also, the chemotherapy regimens are associated with significant toxicity and may not be suitable for many MCC patients who usually tend to be older with multiple co-morbidities. There are no established second-line treatments for patients who have progressed on initial systemic chemotherapy regimens. There is therefore a strong and unmet need for novel, biology-driven therapies in this disease.
Fortunately, rapid strides are being made in our understanding of the biology of MCC that have opened up new avenues for investigation of rational therapies in this aggressive disease. We review the recent discoveries in MCC with a special focus on the emerging importance of immune mechanisms in the pathogenesis of this disease.
Link with immune suppression leads to discovery of Merkel cell polyomavirus
Epidemiologic data suggest a strong link between MCC and the immune system. Individuals with T cell dysfunction (solid organ transplant recipients [10, 11], HIV-infected patients [12] or chronic lymphocytic leukemia patients [6]) are at 5- to 50-fold increased risk of developing MCC. MCC tumors sometimes regress following improvement in immune function [13, 14] underscoring the importance of immune surveillance in the development of MCC. Additionally, there are several reported cases of complete spontaneous regression in the MCC literature (a far greater number than expected for its rarity) that suggest a sudden recognition by the immune system leading to the clearance of MCC [15-20]. These epidemiologic data raised the possibility of an infectious etiology for MCC. Indeed, the recent discovery of the Merkel Cell polyomavirus (MCV or MCPyV) has provided the missing link between MCC and its association with immune suppression [21].
The Merkel cell polyomavirus was discovered in 2008 [21]. Yuan Chang, Patrick Moore and their colleagues created cDNA libraries from MCC tumor mRNA and used the Digital Transcriptome Subtraction method to identify a novel transcript with high homology to the African green monkey lymphotropic polyomavirus (AGM LPyV). The circular genome of MCPyV (∼5200 base pairs) has an early gene expression region containing the oncoprotein tumor (T) antigen locus with large-T (LT) and small-T (ST) open reading frames. A late gene region contains the viral structural proteins that encode capsid proteins. MCpyV was found to have the highest homology with the murine polyomavirus subgroup (includes AGM LPyV) and lesser homology to the known human polyomaviruses (BK or JC viruses) or to simian virus 40 (SV40). PCR-Southern hybridization revealed MCPyV sequences to be present in 8 of 10 (80%) MCC tumors, but uncommon in non-MCC tissues (8%) and normal skin or non-MCC skin tumor tissues (16%), suggesting strong association between MCPyV infection and MCC. The monoclonal pattern of integration of the viral genome into the tumor genome was suggestive of MCPyV infection and genomic integration prior to or very early in tumorigenesis. Since the original description of the virus in 2008, several groups around the world have independently verified the association between MCPyV and MCC [22-28].
Epidemiology of MCPyV Infection
Similar to the other known human polyomaviruses (BK, JC, KI and WU viruses) [29], exposure to MCPyV as measured by serum antibodies to viral capsid proteins appears to be widely prevalent among healthy subjects [30-32]. In one study, the prevalence of MCPyV seropositivity was 0% in infants, 43% among children aged 2-5 years old, and increased to 80% among adults older than 50 years [30]. A similar trend of increasing seroprevalence with age was seen in another study suggesting that primary exposure to MCPyV occurs during childhood [29]. Consistent with the serologic data, MCPyV DNA was detected in cutaneous swabs from clinically healthy subjects with a prevalence of 40-100% in 3 independent studies [33-35]; it appears that the virus is being shed chronically from clinically normal skin in the form of assembled virions [33]. Besides the skin, viral DNA has been detected in lower frequencies among respiratory secretions, on oral and anogenital mucosa, and in the digestive tract [36-41]. The exact mode of transmission remains to be elucidated and could involve cutaneous, fecal-oral, mucosal or respiratory routes. Importantly, although widely prevalent, active MCPyV infection appears to be asymptomatic and with the exception of MCC, this virus has not yet been convincingly associated with any other human disease.
Role of MCPyV in pathogenesis of MCC
Cancer-associated viruses may contribute to carcinogenesis directly via expression of viral oncogenes that promote cell transformation or indirectly via chronic infection and inflammation, which may predispose host cells to acquire carcinogenic mutations [42]. Polyomaviruses are a genus of non-enveloped viruses with a circular double-stranded DNA genome of approximately 5000 base pairs. The ability of certain polyomaviruses to transform mammalian cells is well known. The best studied example is the SV40 polyomavirus that was originally discovered in the primary monkey kidney cells used to prepare polio vaccines. Alarmingly, SV40 was found to induce multiple tumors in newborn hamsters [43]). Fortunately, despite their prevalence, the known polyomaviruses other than MCPyV have not been associated with formation of any human tumors. Typically, human polyomavirus infection is asymptomatic except in immunosuppressed individuals who can develop nephropathy (BK virus) or progressive multifocal leukoencephalopathy (JC virus). In humans, MCPyV is the first polyomavirus with demonstrated integration into genomic DNA. Several significant observations suggest that MCPyV contributes to the pathogenesis of MCC (Figure 1): (1) it is present in a substantial portion of MCC tumors [21]; (2) monoclonality of MCPyV integration in MCC tumor cells suggests viral integration is an early event in tumorigenesis [21]; and (3) LT antigen transcript and protein is expressed in most MCC tumors, (4) the MCPyV LT antigen expressed in MCC tumors is truncated due to mutations that preserve critical cell-cycle progression functions, but eliminate cell-lethal virus-replication activities [44] and (5) persistent expression of these MCPyV proteins is required for continued growth of MCC cell lines in vitro [26]. These findings strongly suggest that MCPyV plays a key role in MCC carcinogenesis rather than merely being a passenger virus that secondarily infects tumor cells.
Figure 1. Although infection with MCPyV is common, a progression of several rare mutagenic events and escape from immune surveillance likely precede the development of Merkel cell carcinoma.
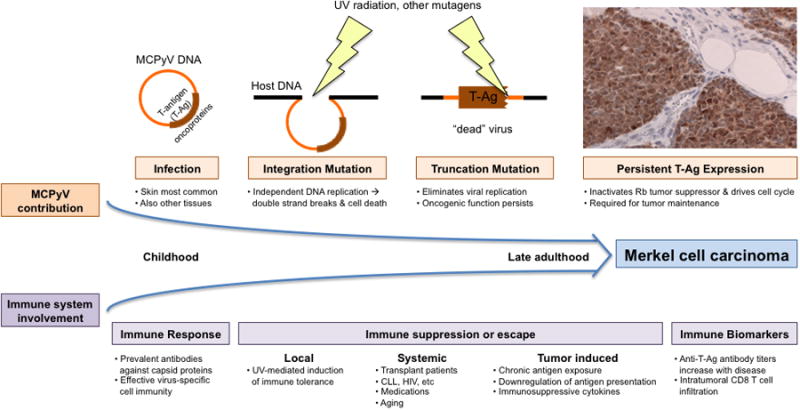
Infection with MCPyV occurs early in childhood [30], is clinically asymptomatic and likely induces an appropriate humoral and cellular immune response. Ultraviolet (UV) radiation or other environmental mutagens may mediate virus integration into the host genome and Large T (LT)-antigen truncation mutations [44]. These sequential mutational events result in persistent T-Ag expression (brown stain with IHC anti-LT antibody, CM2B4) that plays a key role in MCC pathogenesis [26, 42, 46]. Importantly, in parallel, local, systemic or tumor induced loss of immune surveillance may allow for an unsupervised increase in both wild-type virus burden and T-Ag dependent MCC disease. Oftentimes, disease progression can be monitored via immune biomarkers such as anti-T-Ag antibody levels [60] and disease outcome can be predicted by levels of CD8 T cell infiltration [63].
The MCPyV LT antigen appears to retain the major conserved features of other polyomavirus LT antigens, including the DnaJ motif (binds to heat-shock proteins) and the LxCxE motif (inactivates retinoblastoma family proteins), and the origin-binding and helicase/ATPase domains (promote viral replication) [44]. These various domains allow the polyomaviruses to use host cell machinery for viral genome replication, but can also target tumor suppressor proteins resulting in cellular transformation [45]. The LT antigen transcripts are commonly expressed in MCC tumors [44]. However, tumor-specific truncating mutations retain LT-antigen DnaJ and LxCxE motifs that promote cellular growth, but eliminate origin-binding and helicase domains that are essential for production of progeny virions [44]. This acquired inability of tumor-derived LT antigen to initiate constitutive viral genome replication protects virus-infected tumor cells from apoptosis triggered by DNA-damage response mechanisms.
The mechanisms by which MCPyV may contribute to MCC carcinogenesis continue to be elucidated. MCPyV T antigen appears to be essential for cell survival among tumors infected with the virus. In MCPyV-infected MCC cell lines and xenograft models, the expression of T-antigen appears to be essential for sustained proliferation; knockdown of this viral protein leads to growth arrest and/or cell death while restoration of T-antigen expression rescues cell growth [26, 46]. Furthermore, interaction with the retinoblastoma (Rb) tumor suppressor protein appears to be critical to the observed growth-promoting effects of LT antigen [46]. Immunohistochemistry (IHC) data from human MCC tumors shows strong positive association between tumor Rb expression and MCPyV LT-antigen expression with LT-antigen positive MCC tumors also expressing Rb and 87% of LT-antigen negative tumors being Rb-negative as well [47, 48]. Similar to the well-characterized interactions between SV40 LT-antigen and the Rb family of proteins (Rb, p107, p130), the MCPyV LT-antigen is likely to sequester hypophosphorylated Rb that usually binds to E2F transcription factors. This sequestration of Rb allows E2F-mediated transcription that leads to the entry of the cell into S-phase. The integrity of the DnaJ- and the LxCxE-motifs is required for this mechanism in SV40, and the retention of these domains (with intact Rb-binding ability) in the truncated MCPyV LT-antigen is consistent with this mechanism being relevant to MCC pathogenesis.
The other putative mechanism by which polyomaviruses contribute to transformation is interference with the p53 tumor suppressor pathway. The usual functions of p53 are not conducive to viral replication as p53 transactivates genes that lead to cell cycle arrest, which could deprive the virus of essential replication factors. Additionally, active p53 could lead to cellular apoptosis in response to the presence of viral or cellular oncoproteins. In order to complete their normal infectious cycles, the polyomaviruses have developed the ability to block p53 function through several mechanisms. The bipartite domain of the SV40 LT-antigen can bind directly to the specific DNA binding domain of p53, hence interfering with p53-dependent gene transcription [49, 50] (this binding has also been shown to increase the half-life and steady-state levels of p53 in cells [51]. As the MCPyV LT-antigen seems to be prematurely truncated in the MCC tumor cells lacking the helicase domain and the supposed p53-binding sites [44], the significance of the p53 pathway in pathogenesis of MCPyV-associated MCC is unclear. However, even if the truncated T-antigen does not bind to p53, MCPyV may play a role in suppressing p53 function in MCC tumors via other mechanisms. For example, there is evidence that the binding of T-antigen to p53 in SV40 may not be sufficient to block p53 function and that other indirect mechanisms (involving small T-antigen and/or the J- and Rb-binding domains of the LT-antigen) are also important in functional suppression of p53 [52, 53]. Consistent with MCPyV somehow disabling p53 function in MCC tumors, inactivating mutations in TP53 gene and/or overexpression of p53 have been seen only in a small subset of MCC tumors [54, 55]. Moreover, recent studies have indicated an inverse relationship between p53 expression and MCPyV viral abundance in MCC tumors as well as p53 overexpression potentially being associated with poor outcome [56, 57].
In addition to the processes described above, there are likely additional mechanisms by which MCPyV contributes to the development/maintenance of MCC tumors. While some of these pathways may also be relevant to MCPyV-negative MCC tumors (albeit via non-viral mechanisms), the virus-associated MCC subgroup is likely to have important biological distinctions from the virus-negative subgroup. Understanding the molecular mechanisms that contribute to disease progression in various MCC subgroups will be crucial to the development of mechanism-based targeted therapies for this disease.
Immunology of Merkel Cell Cancer
The discovery of MCPyV and its role in MCC pathogenesis raise several interesting questions about interactions between the host immune system and MCC tumor cells. The sero-epidemiologic data (discussed above) suggests that exposure to MCPyV is widely prevalent and that viral capsid proteins are recognized by the human immune system in infected individuals [30, 31]. Also, as discussed above, MCC tumor cells commonly express the MCPyV LT antigen [44, 58] and the LT antigen is essential for continued growth of cells infected with the virus [26, 46]. Despite this persistent expression of viral proteins, however, MCC tumor cells are somehow able to evade the immune system. While this can be explained by the presence of generalized T-cell dysfunction in a small subset of MCC patients with comorbidities such as HIV-infection, immunosuppressive medications or concurrent hematologic malignancies, the vast majority (>90%) of MCC patients have no clinically apparent immune dysfunction [6]. Our understanding of host-virus immune interactions in MCC pathogenesis is increasing rapidly with new insights into the humoral and cellular immunity in MCC patients (Figure 1).
Humoral immune response
Although the prevalence of antibodies to viral capsid proteins (VP) in the general population is high, all studies have found that IgG antibodies to MCPyV VP1 and VP2 are even more prevalent in MCC patients [27, 30, 31, 59]. Interestingly, the titer of antibodies to viral capsid proteins is typically higher in MCC patients than in control populations [30-32]. This finding is not attributable to increased viral capsid antigen production by tumor cells because MCC tumor cells do not express viral capsid proteins [31, 32]. One possible explanation for higher antibody titers in MCC patients could be exposure to a greater virus burden in MCC patients. Supporting this hypothesis, the MCPyV DNA levels in cutaneous swabs from MCC patients were found to be significantly higher than levels in control population [34] and another study reported a positive correlation between serum MCPyV antibody titers and MCpyV DNA levels in skin biopsies [59]). The apparently higher virus burden in MCC patients could possibly be a risk factor that predisposes to subsequent development of MCC in these patients; alternatively, the development of MCC could somehow have resulted in a MCPyV-specific immunodeficiency that leads to the higher virus levels on the skin of MCC patients (further discussed below). Interestingly, higher anti-MCPyV capsid antibody titers have also been associated with better progression-free survival in MCC patients [32]; whether this indicates the presence of a more robust host immune system remains unclear.
The limited serologic data from patients with MCPyV-negative MCC tumors suggests that the majority of these patients have been exposed to MCPyV [27], and in many patients, antibody titers can be very high, similar to patients with MCPyV-positive MCC [30]. This raises the fascinating possibility of MCPyV infection possibly playing a role in tumor initiation with subsequent selection for less immunogenic, MCPyV-negative MCC tumor subclones in these patients. Indeed, the heterogeneity of MCPyV DNA or T-antigen expression levels in MCC tumors supports immune selection within the tumors and is consistent with the ‘hit and run’ hypothesis for tumorigenesis in MCPyV-negative MCC tumors.
As compared to antibodies to viral capsid proteins, antibodies to MCPyV T-Ag oncoproteins are more specifically associated with MCC; these antibodies are rarely detected in the general population (<1%) but appear to be present in a substantial proportion (∼40%) of patients with active MCC [60]. Importantly, the titer of antibodies to T-antigen oncoproteins correlates strongly with the presence of MCPyV DNA and the expression of T-antigens in MCC tumor cells [60]. Moreover, the antibody titer to T-Ag oncoproteins can potentially serve as a biomarker of MCC disease burden; the antibody titer drops rapidly after successful treatment of MCC tumors and a rising titer in a previously treated patient has been shown to herald disease progression prior to development of symptoms [60]. This apparent correlation between the humoral response to T-antigens and MCC disease burden is not completely unexpected because T antigen expression is selectively linked to MCC tumors. Specifically, in contrast to viral capsid proteins that are readily visible to the host humoral immune system, T-antigens are not present in viral particles, are only expressed after viral entry into host cells, are located in the nucleus [61], and are thus less likely to trigger an antibody response except in the setting of dying or diseased tissue (such as a tumor that persistently expresses T-antigens).
Cellular immune response
The presence of MCPyV T-antigen specific antibodies that appear to correlate with tumor burden in MCC patients [60] suggests ongoing expression of viral proteins in tumor cells and their recognition by the adaptive arm of the immune system. Histologic analyses have revealed the presence of variable numbers of tumor-infiltrating lymphocytes (TILs) in the MCC tumors with possible prognostic significance [62]. Our group has recently documented that intratumoral (but not peritumoral) infiltration of CD8+ lymphocytes is an independent predictor of improved survival among MCC patients. In this study, unbiased gene expression analyses revealed overexpression of immune response genes in tumors with favorable prognoses. These immune response genes included genes that encode components of cytotoxic granules (granzymes), chemokines (CCL19), lymphocyte-activation molecules and CD8 receptor molecules [63]. Importantly, in an independent cohort of 156 cases, patients with robust CD8+ intratumoral infiltration had 100% MCC-specific survival as compared to 60% survival among patients with sparse or no CD8+ intratumoral infiltration [63]. This evidence highlights the important role of cellular immune responses in the natural history of MCC and further explains the increased incidence of MCC in patients with cellular immune suppression. While the antigen specificity and differentiation phenotype of infiltrating CD8+ T cells in MCC tumors are yet to be defined, the MCPyV-specific T cell response could presumably be an excellent target for therapeutic manipulation in MCC patients.
Immune evasion mechanisms in MCC
Despite the expression of immunogenic virus-encoded oncoproteins in the majority of tumors [44, 60], MCCs that became clinically evident were significantly able to evade host immune responses. According to the cancer immunoediting hypothesis [64], development of tumors generally requires cancer cells to navigate successfully through three distinct (and usually sequential) phases of the interaction between the cancer and the host immune system: (1) Elimination phase, an immunosurveillance phase in which the innate and adaptive immune systems work together to detect the presence of nascently transformed cells and destroy them before a tumor becomes clinically apparent; (2) Equilibrium phase, a tumor dormancy phase in which the adaptive immune system restrains the outgrowth of tumors and sculpts the immunogenicity of the tumor cells; (3) Escape phase, a tumor progression phase in which the tumor cells are able to circumvent the host immune response manifesting as clinically progressing tumors. The lack of a good animal model for MCC pathogenesis and the inherent challenges of conducting longitudinal studies in at-risk individuals for a rare cancer render it difficult to study the precise events during the elimination and equilibrium phases of MCC tumorigenesis. However, the potential mechanisms of immune escape by MCC tumors are becoming increasingly apparent (Figure 1).
The progression from equilibrium to the escape phase may occur due to changes in tumor cell population that may acquire new immune evasive characteristics or due to changes in the host immune system that may get suppressed either generally or more selectively toward the tumor cells. Both of these broad mechanistic categories appear relevant to MCC:
Tumor cell changes: Under the pressures of immune selection, MCC tumor cells may acquire new features to become either “less visible” to the immune system or “more resistant” to the effects of the cytotoxic immune cells. The former may occur via loss of tumor antigen expression. Cell surface major histocompatibility complex class I (MHC-I) serves to present intracellular peptides to CD8+ T lymphocytes; specifically, viral oncoproteins expressed in MCC tumor cells would be presented to T cells via MHC-I. Indeed, multiple viruses (eg. adenovirus and HSV) and virus-associated cancers (e.g. Kaposi's sarcoma, cervical cancer) are known to directly or indirectly down-regulate the expression of MHC-I as a key mechanism of immune escape [65-70]. Besides MHC-I loss, dysregulation of other components of cellular antigen presenting machinery such as the transporter associated with antigen processing (TAP) [71] or downregulation of appropriate tissue-specific T cell homing signals may also preclude the presentation of persistently expressed tumor antigens to T cells and need to be investigated further in MCC. Indeed, our laboratory findings suggest that 46% of MCC tumors exhibit a 'stalled phenotype' of lymphocytic infiltration where CD8+ cells accumulated near the tumor-stroma border but were unable to infiltrate into the tumors [63]. Such ‘peritumoral’ T cells were not associated with significantly improved survival. These features together likely lead to poor visibility of the MCC tumor cells to the immune system and may explain the sparse infiltrates of T cells in most MCC tumors that are associated with poor outcomes [63]. Another important adaptation at the tumor cell level that can result in immune escape is increased resistance of the tumor cell to immune control mechanisms. Innate immune signaling networks and tumor suppressor pathways share some key proteins such as p53 [72] and cyclin-dependent kinase inhibitor p21 [73]. Due to this functional overlap, the targeting of tumor-suppressor pathways by MCC oncoproteins may also serve as an immune evasion mechanism for MCC. In addition, tumor cells may secrete proteins that interfere with the functioning of the immune cells (discussed below).
Immune system changes: Immunosuppression resulting in T-cell dysfunction may predispose to the immune escape of transformed cancer cells; however, clinically evident systemic immunosuppression due to comorbidities such as post-transplant status, concurrent hematological malignancy, HIV infection etc. is present only in fewer than 10% of MCC patients. What may be of even greater relevance to the pathogenesis of MCC, a disease of the elderly population, could be the altered phenotype and functional incapacity of an aging immune system that allows the development and progression of the disease (Figure 1). This phenomenon of immunosenescence, an erosion of the immune response with aging, is associated with phenotypic and functional changes in both innate and adaptive arms of the immune system, including a contracted repertoire of naïve and cytotoxic T-cells and impaired function of effector T cells [74]. Ultraviolet radiation (UVR), another risk factor for MCC, may not only promote critical LT-antigen mutations and ST-antigen upregulation [75], but may also play a key role in cutaneous immune system inhibition and tolerance [76]. Specifically, UVR has been implicated in recruitment of regulatory T cells and in inhibition of antigen presentation via direct damage to antigen presentation cells (APCs) or via functional inhibition of APCs by cytokines (interleukin 10, tumor necrosis factor-alpha) released by keratinocytes and mast cells [77, 78]. In addition to systemic immune dysfunction contributing to immune escape, it is likely that MCC tumor cells establish a local immune suppressive microenvironment in order to thrive. In this scenario, immunologically sculpted tumor cell subclones may overproduce immunosuppressive cytokines, such as TGF-β [79], Fas-L [80], IL-10 [81] or inhibitors of T cell responses such as galectin-1 [82] and indoleamine 2,3-dioxygenase (IDO) [83]. Tumors could also suppress proinflammatory danger signals through pathways involving activated STAT3, leading to impaired dendritic cell maturation [84] or could downregulate the NKG2D receptor on immune effector cells by secretion of soluble forms of the MIC NKG2D ligands thereby attenuating lymphocyte-mediated cytotoxicity [85]. Tumor cells may also facilitate the generation, activation, or function of immunosuppressive cells [86], such as CD4+CD25+ regulatory T cells (T-regs) [87] or myeloid-derived suppressor cells [88]. T-cell exhaustion, originally described in the context of chronic viral infection in mice [89, 90], is being found to be increasingly relevant to human cancers. In response to chronic antigen exposure, antigen-specific CD8+ T-cells often develop an exhausted phenotype with poor effector function, sustained expression of inhibitory receptors, and a transcriptional state distinct from that of functional effector or memory T cells. The final stage of exhaustion may involve physical deletion of antigen-specific T cells [89, 91]. In the context of viral infection, more severe CD8+ T cell exhaustion has been correlated with higher viral load. Moreover, in the setting of the same viral load, epitopes that were present in larger amounts led to more extreme exhaustion and/or deletion than epitopes present in smaller amounts [91]. This phenomenon may possibly be relevant in MCC as well and could explain the observed higher MCPyV viral load on the skin of MCC patients as compared to the general population (discussed above) if MCPyV-specific T cells are exhausted by chronic antigen exposure in the tumors and hence fail to suppress MCPyV colonization [34]. The interaction of programmed death (PD)-1 expressed on T-cells with its ligand B7H1 or PDL-1 is an important mechanism of T-cell exhaustion [92] that could be harnessed for therapeutic purposes.
Moving towards biology-driven immunotherapy
The discovery of the MCPyV and the increasing recognition of the importance of the immune system in MCC pathogenesis suggest several new targets for therapeutic exploration; rational immunotherapeutic approaches can possibly advance outcomes for this aggressive disease. The critical role of viral oncoproteins in tumorigenesis of MCPyV-positive MCC tumors and the resultant cellular expression of viral peptides could not only be exploited to develop virus-targeting therapies interfering with the function of the oncoproteins, but also be harnessed to stimulate immune responses against virus-infected tumor cells. As an example, the T-antigen specific antibody response is confined to a 78 amino acid N-terminus domain shared by the small- and large- T-antigens [60], which could provide a suitable vaccine or adoptive T-cell therapy target. Similarly, other non-viral tumor associated-antigens such as survivin [93] or the oncoprotein HIP1 that interacts with c-KIT [94] may also be suitable immunotherapy targets. Immunostimulatory cytokines, such as Interferons, interleukin(IL)-2, IL-12, IL-15, or IL-21 could be delivered systemically or intratumorally to counteract immune evasion mechanisms employed by MCC tumors. A phase II trial using intratumoral delivery of IL-12 followed by in vivo electroporation of MCC tumors will be opening to accrual soon. Other therapeutic agents that look appealing to investigate for MCC treatment include CTLA-4 receptor blocking agents such as Ipilimumab (recently approved by FDA for metastatic melanoma), drugs targeting the PD-1/PDL-1 pathway to reverse immune exhaustion of infiltrating lymphocytes or drugs targeting the costimulatory 4-1BB pathways that could promote T cell infiltration, proliferation and cytokine production [95, 96].
Given the heterogeneity of MCC tumors and individual variations in host immune systems, it is unlikely that one single approach will be effective in all patients. Rather, a combination of various strategies and personalization to the unique biologic characteristics of MCC tumors in individual patients will be required. Nevertheless, it is an exciting time for investigation of novel targeted and/or immune therapies in this fascinating malignancy.
Conclusion
The discovery of Merkel cell polyomavirus has revolutionized our understanding of MCC pathogenesis. The immune system appears to be playing a major role in MCC biology with increasing evidence of virus-specific cellular and humoral immune responses that influence the prognosis of MCC patients. MCC tumors are able to evade the immune system by establishing a local immunosuppressive microenvironment. Understanding the mechanisms of immune evasion by MCC tumors will offer opportunities for development of biologically driven therapies to improve patient outcomes from this often-lethal virus-associated cancer.
References
* Of importance
** Of outstanding importance
- 1*.Lemos BD, Storer BE, Iyer JG, et al. Pathologic nodal evaluation improves prognostic accuracy in Merkel cell carcinoma: Analysis of 5,823 cases as the basis of the first consensus staging system for this cancer. J Am Acad Dermatol. 2010 doi: 10.1016/j.jaad.2010.02.056. A publication especially relevant to the clinician, as it determines the prognostic significance of tumor size, clinical vs pathologic nodal evaluation, and extent of disease at presentation and thereby derives the first consensus staging/prognostic system for MCC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemos B, Nghiem P. Merkel cell carcinoma: more deaths but still no pathway to blame. J Invest Dermatol. 2007;127:2100–2103. doi: 10.1038/sj.jid.5700925. [DOI] [PubMed] [Google Scholar]
- 3.Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009 doi: 10.1111/j.1600-0560.2009.01370.x. [DOI] [PubMed] [Google Scholar]
- 4.Hodgson NC. Merkel cell carcinoma: changing incidence trends. Journal of surgical oncology. 2005;89:1–4. doi: 10.1002/jso.20167. [DOI] [PubMed] [Google Scholar]
- 5.Moll R, Löwe A, Laufer J, Franke WW. Cytokeratin 20 in human carcinomas. A new histodiagnostic marker detected by monoclonal antibodies. Am J Pathol. 1992;140:427–447. [PMC free article] [PubMed] [Google Scholar]
- 6.Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375–381. doi: 10.1016/j.jaad.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinstock MA, Gardstein B. Twenty-year trends in the reported incidence of mycosis fungoides and associated mortality. Am J Public Health. 1999;89:1240–1244. doi: 10.2105/ajph.89.8.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.American Cancer Society. Cancer Facts & Figures 2006. Atlanta: American Cancer Society; 2006. [Google Scholar]
- 9.Tai PT, Yu E, Winquist E, et al. Chemotherapy in neuroendocrine/Merkel cell carcinoma of the skin: case series and review of 204 cases. J Clin Oncol. 2000;18:2493–2499. doi: 10.1200/JCO.2000.18.12.2493. [DOI] [PubMed] [Google Scholar]
- 10.Penn I. Posttransplant malignancies. Transplant Proc. 1999;31:1260–1262. doi: 10.1016/s0041-1345(98)01987-3. [DOI] [PubMed] [Google Scholar]
- 11.Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49:832–841. doi: 10.1016/s0190-9622(03)02108-x. [DOI] [PubMed] [Google Scholar]
- 12.Engels EA, Frisch M, Goedert JJ, et al. Merkel cell carcinoma and HIV infection. Lancet. 2002;359:497–498. doi: 10.1016/S0140-6736(02)07668-7. [DOI] [PubMed] [Google Scholar]
- 13.Burack J, Altschuler EL. Sustained remission of metastatic Merkel cell carcinoma with treatment of HIV infection. J R Soc Med. 2003;96:238–239. doi: 10.1258/jrsm.96.5.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muirhead R, Ritchie DM. Partial regression of Merkel cell carcinoma in response to withdrawal of azathioprine in an immunosuppression-induced case of metastatic Merkel cell carcinoma. Clin Oncol (R Coll Radiol) 2007;19:96. doi: 10.1016/j.clon.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Miller RW, Rabkin CS. Merkel cell carcinoma and melanoma: etiological similarities and differences. Cancer Epidemiol Biomarkers Prev. 1999;8:153–158. [PubMed] [Google Scholar]
- 16.Pan D, Narayan D, Ariyan S. Merkel cell carcinoma: five case reports using sentinel lymph node biopsy and a review of 110 new cases. Plast Reconstr Surg. 2002;110:1259–1265. doi: 10.1097/01.PRS.0000025287.96915.88. [DOI] [PubMed] [Google Scholar]
- 17.Karkos PD, Sastry A, Hampal S, Al-Jafari M. Spontaneous regression of Merkel cell carcinoma of the nose. Head Neck. 2010;32:411–414. doi: 10.1002/hed.21095. [DOI] [PubMed] [Google Scholar]
- 18.Kubo H, Matsushita S, Fukushige T, et al. Spontaneous regression of recurrent and metastatic Merkel cell carcinoma. J Dermatol. 2007;34:773–777. doi: 10.1111/j.1346-8138.2007.00382.x. [DOI] [PubMed] [Google Scholar]
- 19.Wooff JC, Trites JR, Walsh NMG, Bullock MJ. Complete Spontaneous Regression of Metastatic Merkel Cell Carcinoma: A Case Report and Review of the Literature. Am J Dermatopathol. 2010 doi: 10.1097/DAD.0b013e3181cd3158. [DOI] [PubMed] [Google Scholar]
- 20.Ciudad C, Avilés JA, Alfageme F, et al. Spontaneous regression in merkel cell carcinoma: report of two cases with a description of dermoscopic features and review of the literature. Dermatol Surg. 2010;36:687–693. doi: 10.1111/j.1524-4725.2010.01531.x. [DOI] [PubMed] [Google Scholar]
- 21**.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. The original publication identifying the novel Merkel cell polyomavirus and its association with MCC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garneski KM, Warcola AH, Feng Q, et al. Merkel Cell Polyomavirus Is More Frequently Present in North American than Australian Merkel Cell Carcinoma Tumors. Journal of Investigative Dermatology. 2009;129:246–248. doi: 10.1038/jid.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becker JC, Houben R, Ugurel S, et al. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol. 2009;129:248–250. doi: 10.1038/jid.2008.198. [DOI] [PubMed] [Google Scholar]
- 24.Kassem A, Schöpflin A, Diaz C, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68:5009–5013. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- 25.Katano H, Ito H, Suzuki Y, et al. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi's sarcoma. J Med Virol. 2009;81:1951–1958. doi: 10.1002/jmv.21608. [DOI] [PubMed] [Google Scholar]
- 26*.Houben R, Shuda M, Weinkam R, et al. Merkel Cell Polyomavirus-Infected Merkel Cell Carcinoma Cells Require Expression of Viral T Antigens. Journal of Virology. 2010;84:7064–7072. doi: 10.1128/JVI.02400-09. This article reports that MCC cells depend on persistent expression of MCPyV oncoproteins and supports causative role of MCPyV and therapeutic targeting of viral oncoproteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter JJ, Paulson KG, Wipf GC, et al. Association of Merkel cell polyomavirus-specific antibodies with Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:1510–1522. doi: 10.1093/jnci/djp332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulitzer MP, Amin BD, Busam KJ. Merkel cell carcinoma: review. Adv Anat Pathol. 2009;16:135–144. doi: 10.1097/PAP.0b013e3181a12f5a. [DOI] [PubMed] [Google Scholar]
- 29.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolstov YL, Pastrana DV, Feng H, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250–1256. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastrana DV, Tolstov YL, Becker JC, et al. Quantitation of Human Seroresponsiveness to Merkel Cell Polyomavirus. PLoS Pathog. 2009:1–20. doi: 10.1371/journal.ppat.1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Touze A, Le Bidre E, Laude H, et al. High Levels of Antibodies Against Merkel Cell Polyomavirus Identify a Subset of Patients With Merkel Cell Carcinoma With Better Clinical Outcome. Journal of Clinical Oncology. 2011:1–9. doi: 10.1200/JCO.2010.31.1704. [DOI] [PubMed] [Google Scholar]
- 33.Schowalter RM, Pastrana DV, Pumphrey KA, et al. Merkel Cell Polyomavirus and Two Previously Unknown Polyomaviruses Are Chronically Shed from Human Skin. Cell Host and Microbe. 2011;7:509–515. doi: 10.1016/j.chom.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foulongne V, Kluger N, Dereure O, et al. Merkel cell polyomavirus in cutaneous swabs. Emerging Infect Dis. 2010;16:685–687. doi: 10.3201/eid1604.091278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wieland U, Silling S, Scola N, et al. Merkel Cell Polyomavirus Infection in HIV-Positive Men. Arch Dermatol. 2011;147:401–406. doi: 10.1001/archdermatol.2011.42. [DOI] [PubMed] [Google Scholar]
- 36.Dworkin AM, Tseng SY, Allain DC, et al. Merkel cell polyomavirus in cutaneous squamous cell carcinoma of immunocompetent individuals. J Invest Dermatol. 2009;129:2868–2874. doi: 10.1038/jid.2009.183. [DOI] [PubMed] [Google Scholar]
- 37.Loyo M, Guerrero-Preston R, Brait M, et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int J Cancer. 2010:NA–NA. doi: 10.1002/ijc.24737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kantola K, Sadeghi M, Lahtinen A, et al. Merkel cell polyomavirus DNA in tumor-free tonsillar tissues and upper respiratory tract samples: implications for respiratory transmission and latency. J Clin Virol. 2009;45:292–295. doi: 10.1016/j.jcv.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babakir-Mina M, Ciccozzi M, Lo Presti A, et al. Identification of Merkel cell polyomavirus in the lower respiratory tract of Italian patients. J Med Virol. 2010;82:505–509. doi: 10.1002/jmv.21711. [DOI] [PubMed] [Google Scholar]
- 40.Bialasiewicz S, Lambert SB, Whiley DM, et al. Merkel cell polyomavirus DNA in respiratory specimens from children and adults. Emerging Infect Dis. 2009;15:492–494. doi: 10.3201/eid1503.081067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goh S, Lindau C, Tiveljung-Lindell A, Allander T. Merkel cell polyomavirus in respiratory tract secretions. Emerging Infect Dis. 2009;15:489–491. doi: 10.3201/eid1503.081206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat Rev Cancer. 2010;10:878–889. doi: 10.1038/nrc2961. An excellent review of viral carcinogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eddy BE, Borman GS, Grubbs GE, Young RD. Identification of the oncogenic substance in rhesus monkey kidney cell culture as simian virus 40. Virology. 1962;17:65–75. doi: 10.1016/0042-6822(62)90082-x. [DOI] [PubMed] [Google Scholar]
- 44**.Shuda M, Feng H, Kwun HJ, et al. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc Natl Acad Sci USA. 2008;105:16272–16277. doi: 10.1073/pnas.0806526105. The first of several publications that identified tumor-specific truncating mutations in MCPyV T-antigen DNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol. 2001;11:15–23. doi: 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- 46.Houben R, Adam C, Baeurle A, et al. An intact retinoblastoma protein binding site in merkel cell polyomavirus large T antigen is required for promoting growth of merkel cell carcinoma cells. International journal of cancer Journal international du cancer. 2011 doi: 10.1002/ijc.26076. [DOI] [PubMed] [Google Scholar]
- 47.Sihto H, Kukko HM, Koljonen VS, et al. Merkel Cell Polyomavirus Infection, Large T Antigen, Retinoblastoma Protein and Outcome in Merkel Cell Carcinoma. Clinical Cancer Research. 2011:1–9. doi: 10.1158/1078-0432.CCR-10-3363. [DOI] [PubMed] [Google Scholar]
- 48.Bhatia K, Goedert JJ, Modali R, et al. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. International journal of cancer Journal international du cancer. 2010;126:2240–2246. doi: 10.1002/ijc.24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Segawa K, Minowa A, Sugasawa K, et al. Abrogation of p53-mediated transactivation by SV40 large T antigen. Oncogene. 1993;8:543–548. [PubMed] [Google Scholar]
- 50.Jiang D, Srinivasan A, Lozano G, Robbins PD. SV40 T antigen abrogates p53-mediated transcriptional activity. Oncogene. 1993;8:2805–2812. [PubMed] [Google Scholar]
- 51.Oren M, Maltzman W, Levine AJ. Post-translational regulation of the 54K cellular tumor antigen in normal and transformed cells. Mol Cell Biol. 1981;1:101–110. doi: 10.1128/mcb.1.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pipas JM, Levine AJ. Role of T antigen interactions with p53 in tumorigenesis. Semin Cancer Biol. 2001;11:23–30. doi: 10.1006/scbi.2000.0343. [DOI] [PubMed] [Google Scholar]
- 53.Ahuja D, Sáenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 54.Schmid M, Janssen K, Dockhorn-Dworniczak B, et al. p53 abnormalities are rare events in neuroendocrine (Merkel cell) carcinoma of the skin. An immunohistochemical and SSCP analysis. Virchows Arch. 1997;430:233–237. doi: 10.1007/BF01324807. [DOI] [PubMed] [Google Scholar]
- 55.Van Gele M, Kaghad M, Leonard JH, et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. British Journal of Cancer. 2000;82:823–826. doi: 10.1054/bjoc.1999.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhatia K, Goedert JJ, Modali R, et al. Immunological detection of viral large T antigen identifies a subset of merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. International journal of cancer Journal international du cancer. 2009 doi: 10.1002/ijc.25136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waltari M, Sihto H, Kukko H, et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. International journal of cancer Journal international du cancer. 2010 doi: 10.1002/ijc.25720. [DOI] [PubMed] [Google Scholar]
- 58.Shuda M, Arora R, Kwun HJ, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125:1243–1249. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faust H, Pastrana DV, Buck CB, et al. Antibodies to Merkel Cell Polyomavirus Correlate to Presence of Viral DNA in the Skin. Journal of Infectious Diseases. 2011;203:1096–1100. doi: 10.1093/infdis/jiq173. [DOI] [PubMed] [Google Scholar]
- 60*.Paulson KG, Carter JJ, Johnson LG, et al. Antibodies to Merkel Cell Polyomavirus T Antigen Oncoproteins Reflect Tumor Burden in Merkel Cell Carcinoma Patients. Cancer research. 2010 doi: 10.1158/0008-5472.CAN-10-2128. This report identifies a potential biomarker to track MCC burden and predict relapse prior to conventional clinical approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakamura T, Sato Y, Watanabe D, et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology. 2010;398:273–279. doi: 10.1016/j.virol.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 62.Andea AA, Coit DG, Amin B, Busam KJ. Merkel cell carcinoma: histologic features and prognosis. Cancer. 2008;113:2549–2558. doi: 10.1002/cncr.23874. [DOI] [PubMed] [Google Scholar]
- 63*.Paulson KG, Iyer JG, Tegeder AR, et al. Transcriptome-Wide Studies of Merkel Cell Carcinoma and Validation of Intratumoral CD8+ Lymphocyte Invasion As an Independent Predictor of Survival. J Clin Oncol. 2011 doi: 10.1200/JCO.2010.30.6308. This report underscores the importance of immune cell infiltration in MCC tumors and its association with improved prognosis, hence supporting the use of immunotherapy in MCC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64**.Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. This is an excellent summary of key steps in tumor immune evasion. [DOI] [PubMed] [Google Scholar]
- 65.Hansen TH, Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 66.Haque M, Ueda K, Nakano K, et al. Major histocompatibility complex class I molecules are down-regulated at the cell surface by the K5 protein encoded by Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. J Gen Virol. 2001;82:1175–1180. doi: 10.1099/0022-1317-82-5-1175. [DOI] [PubMed] [Google Scholar]
- 67.Hayashi H, Tanaka K, Jay F, et al. Modulation of the tumorigenicity of human adenovirus-12-transformed cells by interferon. Cell. 1985;43:263–267. doi: 10.1016/0092-8674(85)90031-5. [DOI] [PubMed] [Google Scholar]
- 68.Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 69.Koopman LA, van Der Slik AR, Giphart MJ, Fleuren GJ. Human leukocyte antigen class I gene mutations in cervical cancer. J Natl Cancer Inst. 1999;91:1669–1677. doi: 10.1093/jnci/91.19.1669. [DOI] [PubMed] [Google Scholar]
- 70.Cromme FV, van Bommel PF, Walboomers JM, et al. Differences in MHC and TAP-1 expression in cervical cancer lymph node metastases as compared with the primary tumours. Br J Cancer. 1994;69:1176–1181. doi: 10.1038/bjc.1994.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seliger B, Maeurer MJ, Ferrone S. TAP off--tumors on. Immunol Today. 1997;18:292–299. doi: 10.1016/s0167-5699(97)01052-9. [DOI] [PubMed] [Google Scholar]
- 72.Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424:516–523. doi: 10.1038/nature01850. [DOI] [PubMed] [Google Scholar]
- 73.Chin YE, Kitagawa M, Su WC, et al. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science. 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 74.Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol. 2007;42:400–406. doi: 10.1016/j.exger.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mogha A, Fautrel A, Mouchet N, et al. Merkel Cell Polyomavirus Small T Antigen mRNA Level Is Increased following In Vivo UV-Radiation. PLoS ONE. 2010;5:e11423. doi: 10.1371/journal.pone.0011423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res. 2005;571:185–205. doi: 10.1016/j.mrfmmm.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 77.Granstein RD, Matsui MS. UV radiation-induced immunosuppression and skin cancer. Cutis; cutaneous medicine for the practitioner. 2004;74:4–9. [PubMed] [Google Scholar]
- 78.Halliday GM, Bestak R, Yuen KS, et al. UVA-induced immunosuppression. Mutat Res. 1998;422:139–145. doi: 10.1016/s0027-5107(98)00185-7. [DOI] [PubMed] [Google Scholar]
- 79.Teicher BA. Transforming growth factor-beta and the immune response to malignant disease. Clin Cancer Res. 2007;13:6247–6251. doi: 10.1158/1078-0432.CCR-07-1654. [DOI] [PubMed] [Google Scholar]
- 80.Houston A, Bennett MW, O'Sullivan GC, et al. Fas ligand mediates immune privilege and not inflammation in human colon cancer, irrespective of TGF-beta expression. British Journal of Cancer. 2003;89:1345–1351. doi: 10.1038/sj.bjc.6601240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rubinstein N, Ilarregui JM, Toscano MA, Rabinovich GA. The role of galectins in the initiation, amplification and resolution of the inflammatory response. Tissue Antigens. 2004;64:1–12. doi: 10.1111/j.0001-2815.2004.00278.x. [DOI] [PubMed] [Google Scholar]
- 83.Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 84.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 85.Groh V, Rhinehart R, Randolph-Habecker J, et al. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. 2001;2:255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 86.Terabe M, Berzofsky JA. Immunoregulatory T cells in tumor immunity. Current Opinion in Immunology. 2004;16:157–162. doi: 10.1016/j.coi.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 87.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 88.Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol. 2005;175:4583–4592. doi: 10.4049/jimmunol.175.7.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zajac AJ, Blattman JN, Murali-Krishna K, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gallimore A, Glithero A, Godkin A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–1393. doi: 10.1084/jem.187.9.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wherry EJ, Blattman JN, Murali-Krishna K, et al. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of Virology. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 93.Kim J, Mcniff JM. Nuclear expression of survivin portends a poor prognosis in Merkel cell carcinoma. Mod Pathol. 2008;21:764–769. doi: 10.1038/modpathol.2008.61. [DOI] [PubMed] [Google Scholar]
- 94.Ames HM, Bichakjian CK, Liu GY, et al. Huntingtin-Interacting Protein 1: A Merkel Cell Carcinoma Marker that Interacts with c- Kit J Invest Dermatol. 2011:1–8. doi: 10.1038/jid.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Curran MA, Kim M, Montalvo W, et al. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE. 2011;6:e19499. doi: 10.1371/journal.pone.0019499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palazon A, Teijeira A, Martinez-Forero I, et al. Agonist Anti-CD137 mAb Act on Tumor Endothelial Cells to Enhance Recruitment of Activated T Lymphocytes. Cancer Research. 2011;71:801–811. doi: 10.1158/0008-5472.CAN-10-1733. [DOI] [PubMed] [Google Scholar]