

Abstract
Calcineurin is an evolutionarily conserved, ubiquitously expressed protein phosphatase that serves as a major effector of Ca2+ signals, regulating diverse biological processes such as gene expression, tissue differentiation, immune responses, and neural plasticity. The following method describes how to monitor real-time calcineurin activity in cultured mammalian cells using a fluorescence resonance energy transfer (FRET)-based activity reporter.
Keywords: Calcineurin, Phosphatase activity, Biosensor, Calmodulin, NFAT, FRET, Live-cell imaging
1 Introduction
1.1 Calcineurin
The Ca2+- and calmodulin (CaM)-dependent, serine/threonine protein phosphatase calcineurin (aka PP2B/PP3) is an evolutionarily conserved signaling enzyme found in virtually all eukaryotes [1]. Calcineurin is comprised of a stable heterodimer between a catalytic A subunit (CNA) and a regulatory B subunit (CNB). The CNA subunit includes an N-terminal catalytic domain, which shares homology with the conserved PPP family of serine/threonine phosphatases, as well as a C-terminal regulatory arm containing a CNB-binding domain, a CaM-binding domain, and an autoinhibitory domain. Calcineurin activation occurs in response to elevated Ca2+ levels; Ca2+ binds to CNB and Ca2+/CaM binds to CNA, leading to conformational rearrangements that release autoinhibition [2]. A major effector of Ca2+ signaling, calcineurin regulates a number of critical biological processes including tissue differentiation, cardiac development, neuronal function, and immune activation [2]. The importance of calcineurin is further underscored by the involvement of dysregulated calcineurin signaling in a number of human pathologies, such as heart disease, diabetes, Alzheimer’s disease, and Down syndrome [3–6].
1.2 A FRET-Based Reporter for Calcineurin Activity
The introduction of genetically encoded fluorescence resonance energy transfer (FRET)-based enzyme activity reporters markedly enhanced our ability to directly visualize the native, real-time dynamics of signaling enzymes with high spatiotemporal resolution in living cells [7, 8]. Typically, these reporters utilize a molecular switch, capable of undergoing a conformational change in response to a specific enzymatic activity, sandwiched between a pair of fluorescent proteins, such that the conformational change results in a change in FRET between the fluorescent protein pair. FRET is a photophysical process in which excitation of a donor fluorophore (e.g., a cyan fluorescent protein, CFP) results in non-radiative energy transfer to an acceptor fluorophore (e.g., a yellow fluorescent protein, YFP), followed by acceptor emission. Efficient transfer depends on several factors, such as spectral overlap between donor and acceptor. Given the significant overlap between the CFP emission spectrum and YFP excitation spectrum, and the minimal overlap between CFP excitation and YFP excitation, CFP and YFP are well suited to this purpose and are a commonly used FRET pair.
Energy transfer also depends on the relative proximity (e.g., <10 nm) and orientation of the fluorophores, rendering FRET highly sensitive to the conformation of the molecular switch. In the case of protein kinase activity reporters, this switch is typically comprised of a kinase-specific consensus phosphorylation motif fused to a phosphoamino acid-binding domain (PAABD). Phosphorylation of the substrate motif promotes binding by the PAABD, inducing a conformational change in the reporter. This modular design has been widely used to generate activity reporters specific to a variety of different protein kinases [9–15]. However, whereas kinase activity reporters have gained widespread use, the development of similar reporters for protein phosphatase activity has lagged. Unlike a kinase activity reporter, which responds to the addition of a phosphate group, a phosphatase activity reporter must already be phosphorylated in its basal state, so that it will respond to the phosphate group’s removal. Therefore, one reason for the comparatively slow development of phosphatase activity reporters is the challenge of designing just such a “dephosphorylation-competent” molecular switch.
In designing a FRET-based calcineurin activity reporter (CaNAR), we chose to address this problem by utilizing a well-studied substrate of calcineurin, nuclear factor of activated T-cells (NFAT) [16]. NFAT proteins are a family of transcription factors that are ubiquitously co-expressed alongside calcineurin in vertebrates, serving as the main molecular target of calcineurin signaling in a multitude of cellular processes [17–19]. In CaNAR, the N-terminal 297 amino acids from NFAT1 are sandwiched between CFP and YPF (Fig. 1a). The N-terminal portion of each NFAT isoform functions as a regulatory domain that is constitutively phosphorylated at multiple serine-rich sites by cytosolic kinases [18, 20], making CaNAR “competent” for dephosphorylation without additional manipulation. The phosphorylated residues are thought to interact with positive charges found in the nuclear localization signal (NLS) within the NFAT regulatory domain, with dephosphorylation by calcineurin inducing a conformational change that exposes the NLS and promotes transit of NFAT into the nucleus [20, 21]. This conformational change forms the basis of the calcineurin-dependent molecular switch found in CaNAR, resulting in a FRET increase in response to calcineurin activity (Fig. 1b).
Fig. 1.
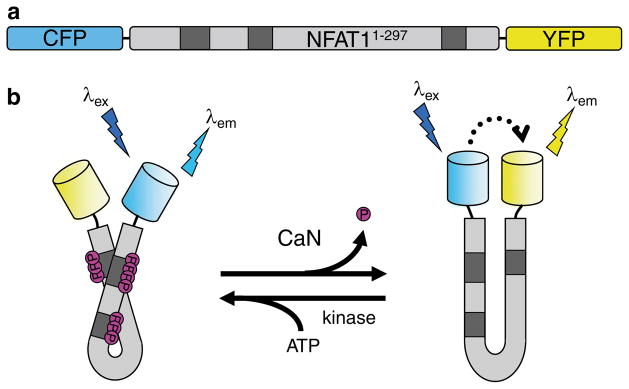
The design of CaNAR. (a) CaNAR features the N-terminal 297 amino acids of NFAT1 sandwiched between the FRET donor CFP and the FRET acceptor YFP. Shaded boxes indicate the sites where NFAT is constitutively phosphorylated. (b) Dephosphorylation of the NFAT N-terminal domain by CaN results in a conformational change that exposes an NLS. In CaNAR, this serves as a molecular switch that induces a change in the proximity and relative orientation of CFP and YFP, leading to a FRET change (dotted arrow). This is reversed upon rephosphorylation of the reporter by cellular kinases
CaNAR can be used to directly visualize calcineurin activity dynamics in live cells by monitoring FRET changes in response to various stimuli capable of eliciting increases in the cytosolic Ca2+ concentration (see Note 1). Using epifluorescence microscopy, FRET can be observed as a decrease in donor (CFP) fluorescence intensity combined with an increase in acceptor (YFP) fluorescence intensity, often expressed numerically as an acceptor-to-donor emission ratio. Since CaNAR is a unimolecular reporter (i.e., donor and acceptor are present in fixed amounts), the value of the emission ratio serves as a convenient readout of FRET [22]. It is also possible to quantitatively determine the FRET efficiency by bleaching the acceptor fluorophore, thereby abolishing FRET and dequenching donor fluorescence [22, 23].
Below, we describe a general method for using CaNAR to measure live-cell calcineurin activity, including specific, detailed procedures for the maintenance of Human Embryonic Kidney (HEK) 293T and HeLa cells, transfection of cells with CaNAR plasmid DNA, preparation of cells for imaging experiments, preparation of the imaging equipment, imaging of calcineurin activity in live cells, and analysis of the acquired imaging data to quantify any observed changes in FRET.
2 Materials
2.1 Cell Culture and Transfection
Cell lines: Human Embryonic Kidney—SV40 T Antigen (HEK 293T) and HeLa (American Type Culture Collection, Manassas, VA).
Dulbecco’s Phosphate Buffered Saline—without Mg2+ and Ca2+ (DPBS, Gibco).
35-mm glass-bottom imaging dishes (MatTEK, Ashland, MA).
Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco/BRL, Bethesda, MD) supplemented with 10 % fetal bovine serum (FBS, Sigma) and 1 % penicillin-streptomycin (Sigma-Aldrich) (DMEM-HEK 293T) for use with both HEK 293T and HeLa cells (see Note 2).
Solution of trypsin (0.05 % for HEK 293T, 0.25 % for HeLa) and ethylenediamine tetraacetic acid (EDTA, 0.53 mM) (Invitrogen, Carlsbad, CA).
Lipofectamine 2000 (Invitrogen).
OPTI-MEM I Reduced Serum Medium (Opti-MEM, Gibco).
CaNAR plasmid DNA.
2.2 Preparing Epifluorescence Microscope
All of the experiments described below are performed on an Axiovert 200 M inverted microscope using a 40×/1.3NA oil-immersion objective lens equipped with an Aqua Stop to prevent liquid from running down the objective (Zeiss, Thornwood, NY). Images are captured using a MicroMAX BFT512 cooled charge-coupled device camera (Roper Scientific, Trenton, NJ).
Xenon lamp: XBO 75 W (Zeiss).
Neutral density filters 0.6 and 0.3 (Chroma Technology, Bellows Falls, VT).
-
Filter sets for individual channels:
FRET—420DF20 excitation filter, 450DRLP dichroic mirror, 535DF25 emission filter.
CFP—420DF20 excitation filter, 450DRLP dichroic mirror, 475DF40 emission filter.
YFP—495DF10 excitation filter, 515DRLP dichroic mirror, 535DF25 emission filter.
YFP photobleaching—525DF40 excitation filter, 560DRLP dichroic mirror (All from Chroma Technology).
A Lambda 10-2 filter changer (Sutter Instruments, Novato, CA) alternates the filters being used.
Immersion oil “Immersol” 518F fluorescence free (Zeiss).
METAFLUOR 7.7 software (Molecular Devices, Sunnvale, CA) (see Note 3).
2.3 Prepare Cells for Imaging
Hanks’ Balanced Salt Solution for Imaging (HBSS*): 10× Hanks’ Balanced Salt Solution (Gibco), 20 mM HEPES (Invitrogen), 2.0 g/L D-glucose (Sigma). Adjust pH to 7.4, then filter sterilize using a 0.22 μm filter (see Note 4). Store a 50 mL aliquot at room temperature in the microscope room and store the remainder at 4 °C (see Note 5).
2.4 Acquiring Images and Data
Ionomycin (iono, Calbiochem) and thapsigargin (TG, Sigma) are both dissolved to 1 mM in dimethyl sulfoxide (DMSO) and stored at −20 °C.
2.5 Analyzing Images and Data
Spreadsheet application (e.g., Microsoft Office Excel).
3 Methods
3.1 Cell Culture and Transfection
The cells are maintained in T-25 cm2 flasks in a humidified 37 °C incubator with a 5 % CO2 atmosphere. Cells should be passaged whenever they reach 70–80 % confluency (every 2–3 days) into flasks for maintenance or 35-mm dishes for imaging.
To passage cells, aspirate culture media from the flask and wash cells gently with 2 mL DPBS. Add 300 μL of trypsin/EDTA, gently rocking the flask from side to side to disperse the solution, and let sit 2–5 min (see Note 6). Add 4.7 mL of fresh media into the flask and mix (see Note 7). For imaging, perform a 1:10 split of cells into the 35 mm glass-bottom dishes. Cell should reach 60–70 % confluence after approximately 24 h (see Note 8). Transfect the cells at this confluence.
For each 35-mm dish being transfected, prepare 2 separate microcentrifuge tubes. Tube 1 contains 1 μg CaNAR plasmid DNA and 50 μL Opti-MEM. Tube 2 contains 2 μL Lipofectamine 2000 and 50 μL Opti-MEM. Allow tubes to incubate at room temperature for 5 min, then add contents from Tube 1 drop-wise into Tube 2 and mix well with pipet (see Note 9). Incubate this solution at room temperature for 20 min.
Gently add the CaNAR transfection solution to the cells drop-wise, and carefully rock the dish back and forth to evenly disperse the solution. Incubate at 37 °C with 5 % CO2 for 48 h (see Note 10).
3.2 Preparing Epifluorescence Microscope
Turn on lamp, microscope, filter changer, camera, and computer. Load the METAFLUOR 7.7 application plus an appropriate protocol for acquiring a time series of sets of images for the FRET, CFP, and YFP channels (see Note 11). Confirm that all of the appropriate filters are in place.
Set the excitation exposure times for the FRET, CFP, and YFP channels to 500, 500, and 50 ms, respectively (see Note 12). In addition, select the time interval between each set of acquisitions. This is typically 30 s, but may be any number between 10 and 120 s.
Apply a small drop of immersion oil directly onto the objective. Make sure not to use an excess amount (i.e., 1 drop from the attached applicator). Avoid touching the objective with the applicator tip.
3.3 Preparing Cells for Imaging
Aspirate off media from transfected cells in imaging dish and wash twice with 1 mL HBSS*.
Gently add 1–2 mL HBSS* to the imaging dish, while holding the dish on a slight angle. Slowly return the dish to a level position and place securely on microscope stage (see Note 13).
Raise the objective until the immersion oil comes fully into contact with the glass cover-slip and then bring the cells into focus while viewing through the eyepiece.
In the dark, use either the FRET or CFP channel to select cells with good morphology and good CaNAR expression, meaning intermediate emission intensity and uniformly distributed fluorescence (see Note 14).
3.4 Acquiring Images and Data
Select several regions of interest to follow during the course of the experiment (see Note 15). A background region consisting of an untransfected cell must also be selected to correct for cell autofluorescence and other background fluorescence.
Record a baseline by acquiring 3–5 min of data (all three channels) from unstimulated cells. Remove ~ 300 μL of HBSS* from the imaging dish and mix with a 1–2 μL aliquot of 1 mM iono or TG in a 1.5 mL tube, then gently pipet this solution back into the imaging dish. The final concentration of drug should be 1 μM. Be sure to record the time of drug addition. The yellow-to-cyan emission ratio (FRET channel intensity/CFP channel intensity) should increase, indicating a change in calcineurin activity (see Note 16).
- At the end of the experiment, remove all neutral density filters, use the YFP photobleaching excitation filter, and then excite for 5 min. This should sufficiently photobleach YFP, although it is important to verify this by acquiring the YFP channel. The acquired data can be used to calculate absolute FRET efficiency with the following formula:
3.5 Analyzing Images and Data
Use METAFLUOR 7.7 to generate pseudo-colored images for each acquisition, where pseudo-coloring is used to indicate the yellow-to-cyan emission ratio (FRET channel intensity/ CFP channel intensity) (see Note 17). These images can be strung together as a movie, or a subset can be used to illustrate the observed real-time FRET changes. An example is shown in Fig. 2.
- In a spreadsheet application, calculate emission ratio at each time point using the logged emission intensity data and the following formula:
Plot the ratio time course (yellow-to-cyan emission ratio versus time).
Fig. 2.

CaNAR response in live cells. HeLa cells were transiently transfected with CaNAR plasmid DNA and imaged after 48 h. The cells were treated with (a, b) 1 μM ionomycin, a Ca2+ ionophore, or (c, d) 1 μM thapsigargin, a SERCA pump inhibitor. (a, c) Plotting the yellow-to-cyan emission ratio versus time shows the change in CaN activity upon stimulation. (b, d) Pseudo-colored images showing the CaNAR response
Footnotes
It is important to verify effective drug concentrations and other conditions affecting drug function. For example, when using receptor agonists to elevate cytosolic Ca2+, be sure to confirm receptor expression in the cell line being used. Western blots using antibodies against the receptor of interest are helpful in this regard. Additionally, effective drug concentrations can be verified by monitoring the phosphorylation of CaNAR via gel mobility shift on western blots employing an anti-GFP antibody (eBioscience, San Diego, CA) [16].
All solutions should be made under sterile conditions in a tissue culture hood and cell culture media should be warmed to 37 °C before using with cells.
Metafluor requires a PC running Microsoft Windows XP, Windows Vista, or Windows 7. Other imaging software with similar or equivalent functionality is also suitable for these experiments.
All solutions should be prepared with water that has an 18.2 MΩ-cm resistivity unless otherwise noted.
Imaging is typically performed at room temperature. However, FRET responses in certain cells lines are enhanced by imaging at 37 °C using an optional Heatable Insert P for Scanning Stage and Mechanical Stage (Zeiss). When using this setup, an aliquot of HBSS* should be preheated to 37 °C prior to imaging.
Be sure the cells are fully detached before continuing. Gently swaying the flask side to side should help.
Gently pipet cells up and down to break up cell clumps, while being careful not to over-pipet and damage the cells.
This protocol can be adapted for other cell lines by following appropriate cell culture and transfection guidelines for the cell line of choice. Additionally, splitting times may vary, so it is important to verify doubling times for each cell line used.
Be sure to mix gently. Do not vortex the solution.
CaNAR generally requires longer expression times compared to other FRET-based reporters [24], possibly owing to its size or need to be pre-phosphorylated by cells. Optimal CaNAR expression time should be determined empirically for each cell type used.
The FRET channel logs sensitized YFP emission intensity upon CFP excitation, the CFP channel logs direct CFP emission intensity upon CFP excitation, and the YFP channel logs direct YFP emission intensity upon YFP excitation. The YFP channel serves to control for YFP photobleaching and does not factor into emission ratio calculations.
These exposure times are a good starting point for most experiments. Strict adherence to these numbers is not required, and exposure times can be adjusted as needed, based on the observed brightness of the reporter.
Securing the dish to the stage is essential to minimize slight movement of the dish that may occur while imaging.
This step focuses on the key criteria for proper cell selection. First, cell morphology should be verified before starting an experiment, as healthy cells are required for successful imaging experiments. For instance, when imaging HEK 293 cells, select cells that are spread out and lying flat rather than balled-up and rounded, as the latter could indicate unhealthy cells. Second, the fluorescence intensity of CaNAR should be closely monitored, though a recommended range cannot be given as the intensity values will vary between microscope setups. However, cells with a moderate intensity level are typically used. Cells with very dim fluorescence intensities will have a low signal-to-noise ratio, rendering changes in FRET difficult to visualize, whereas cells with very high fluorescence intensities may have perturbed endogenous signaling pathways because of excessive expression of CaNAR.
The selected regions of interest will need to remain in the same cellular region throughout the time series, and may be manually readjusted should the cells move. Alternatively, cell tracking software (e.g., Imaris Track) can be used to overcome this problem.
It is important to confirm that changes in FRET from CaNAR are specifically due to calcineurin activity. However, as CaNAR is highly phosphorylated, generating a CaNAR variant that cannot be dephosphorylated is impractical. Rather, specificity can be determined by pretreating cells with a specific calcineurin inhibitor, such as cyclosporin A, prior to stimulation [16].
If the software being used lacks this feature, an image processing application (e.g., ImageJ) can be used to create pseudo-colored ratiometric images from the raw emission intensity images from the individual channels.
References
- 1.Hilioti Z, Cunningham KW. Calcineurin: roles of the Ca2+/calmodulin-dependent protein phosphatase in diverse eukaryotes. Top Curr Genet. 2004;5:73–90. [Google Scholar]
- 2.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80(4):1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 3.Harris CD, Ermak G, Davies KJ. Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product calcipressin 1 (or RCAN1) in disease. Cell Mol Life Sci. 2005;62(21):2477–2486. doi: 10.1007/s00018-005-5085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 5.Heit JJ. Calcineurin/NFAT signaling in the beta-cell: from diabetes to new therapeutics. Bioessays. 2007;29(10):1011–1021. doi: 10.1002/bies.20644. [DOI] [PubMed] [Google Scholar]
- 6.Xie CW. Calcium-regulated signaling pathways: role in amyloid beta-induced synaptic dysfunction. Neuromolecular Med. 2004;6(1):53–64. doi: 10.1385/NMM:6:1:053. [DOI] [PubMed] [Google Scholar]
- 7.Mehta S, Zhang J. Reporting from the field: genetically encoded fluorescent reporters uncover signaling dynamics in living biological systems. Annu Rev Biochem. 2011;80:375–401. doi: 10.1146/annurev-biochem-060409-093259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newman RH, Fosbrink MD, Zhang J. Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem Rev. 2011;111(5):3614–3666. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fosbrink M, Aye-Han NN, Cheong R, Levchenko A, Zhang J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc Natl Acad Sci U S A. 2010;107(12):5459–5464. doi: 10.1073/pnas.0909671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453(7198):1132–1136. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao X, Zhang J. Spatiotemporal analysis of differential Akt regulation in plasma membrane microdomains. Mol Biol Cell. 2008;19(10):4366–4373. doi: 10.1091/mbc.E08-05-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunkel MT, Toker A, Tsien RY, Newton AC. Calcium-dependent regulation of protein kinase D revealed by a genetically encoded kinase activity reporter. J Biol Chem. 2007;282(9):6733–6742. doi: 10.1074/jbc.M608086200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161(5):899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434(7036):1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437(7058):569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- 16.Newman RH, Zhang J. Visualization of phosphatase activity in living cells with a FRET-based calcineurin activity sensor. Mol Biosyst. 2008;4(6):496–501. doi: 10.1039/b720034j. [DOI] [PubMed] [Google Scholar]
- 17.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 18.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17(18):2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 19.Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol. 2002;156(5):771–774. doi: 10.1083/jcb.200111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JP, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell. 2000;6(3):539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 21.Porter CM, Havens MA, Clipstone NA. Identification of amino acid residues and protein kinases involved in the regulation of NFATc subcellular localization. J Biol Chem. 2000;275(5):3543–3551. doi: 10.1074/jbc.275.5.3543. [DOI] [PubMed] [Google Scholar]
- 22.Ananthanarayanan B, Ni Q, Zhang J. Chapter 2: molecular sensors based on fluorescence resonance energy transfer to visualize cellular dynamics. Methods Cell Biol. 2008;89:37–57. doi: 10.1016/S0091-679X(08)00602-X. [DOI] [PubMed] [Google Scholar]
- 23.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 24.Depry C, Zhang J. Using FRET-based reporters to visualize subcellular dynamics of protein kinase A activity. Methods Mol Biol. 2011;756:285–294. doi: 10.1007/978-1-61779-160-4_16. [DOI] [PMC free article] [PubMed] [Google Scholar]