

Abstract
Acute alcohol drinking induces steatosis, and effective prevention of steatosis can protect liver from progressive damage caused by alcohol. Increased oxidative stress has been reported as one mechanism underlying alcohol-induced steatosis. We evaluated whether cannabidiol, which has been reported to function as an antioxidant, can protect the liver from alcohol-generated oxidative stress-induced steatosis. Cannabidiol can prevent acute alcohol-induced liver steatosis in mice, possibly by preventing the increase in oxidative stress and the activation of the JNK MAPK pathway. Cannabidiol per se can increase autophagy both in CYP2E1-expressing HepG2 cells and in mouse liver. Importantly, cannabidiol can prevent the decrease in autophagy induced by alcohol. In conclusion, these results show that cannabidiol protects mouse liver from acute alcohol-induced steatosis through multiple mechanisms including attenuation of alcohol-mediated oxidative stress, prevention of JNK MAPK activation, and increasing autophagy.
Keywords: Alcohol, Steatosis, Cannabidiol, Oxidative stress, Autophagy, Free radicals
Both acute and chronic alcohol drinking induces hepatosteatosis [1,2]. Although steatosis is a reversible injury to liver, its progression can develop into more severe liver problems such as hepatitis and cirrhosis. Recent research showed that oxidative stress is an important factor contributing to mechanisms that induce steatosis [2–4]. Although exact pathways for this have not been clearly identified or studied in detail, possible mechanisms may include reactive oxygen activation of lipogenic transcription factors, e.g., SREBP, or decline in lipolytic factors, e.g., PPARα, or mitochondrial dysfunction and decline in fatty acid oxidation, or activation of the unfolded protein response and endoplasmic reticulum stress. CYP2E1, a cytochrome P450 enzyme, has been shown to play a major role in alcohol-induced oxidative stress and fat accumulation as well as non-alcohol-induced steatosis [1–4]. Studies with CYP2E1-knockout mice showed little or no oxidative stress produced by alcohol compared to CYP2E1-overexpressing mice [1]. Increased oxidative stress was also found in CYP2E1-overexpressing HepG2 cells compared to HepG2 cells lacking CYP2E1 expression [4,5]. These data indicate that oxidative stress is important for the induction of steatosis by alcohol and suggest that compounds that could effectively prevent the induction of oxidative stress represent potential drugs for the prevention of liver steatosis caused by alcohol drinking.
Cannabidiol (CBD) is a nonpsychoactive molecule from the cannabis plant that exhibits unique pharmacological and biological activities. In recent years, CBD has been shown to act as an anti-inflammatory molecule, to be able to block the progression of arthritis [6] and type 1 diabetes [7], to have antioxidant actions [8], and to act as an antiproliferative compound inducing a loss in viability in various tumor cell lines [9–11]. However, the molecular mechanisms mediating the biological effects of CBD are not well understood.
Given the antioxidant properties of CBD, we hypothesized that this compound could exhibit therapeutic properties in the context of alcohol-induced liver injury, by alleviating oxidative stress-induced hepatocellular injury and fat accumulation.
Cannabinoid compounds have also been shown to induce autophagy [12,13]. This is of particular interest in the context of liver injury, as autophagy has recently emerged as a major regulator of lipid homeostasis in the liver [14–18]. Autophagy, especially macroautophagy, is a mechanism by which hepatocytes break down lipids (a mechanism called lipophagy). Inhibition of macroautophagy causes lipid droplets to increase in size and number and to accumulate in hepatocytes. A decreased rate of lipolysis leads to an increase in cellular triglyceride (TG) content, a feature observed in steatosis. In some studies, acute alcohol was shown to decrease autophagy in the liver, leading to steatosis [2,19]. However, other studies showed that acute and chronic alcohol increased autophagy, possibly to protect against alcohol-induced fat accumulation [20,22]. In line with these observations and the proautophagic properties of cannabinoids, we determined whether CBD can protect against alcohol-induced liver steatosis through induction of autophagy.
Materials and methods
Materials
CBD was obtained from Tocris Bioscience (Ellisville, MO, USA). Reactive oxygen species (ROS) detection kit was purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA). ATP determination kit was from Molecular Probes (Eugene, OR, USA). Antibodies against phospho-c-Jun N-terminal kinase (pJNK), JNK, microtubule-associated protein 1A/1B light-chain 3 (LC3), pP38 mitogen-activated protein kinase (MAPK), and P38 MAPK were obtained from Cell Signaling (Danvers). Antibody against CYP2E1 was a generous gift from Dr. Jerry Lasker.
Mouse acute alcohol model
Animal experiments were performed with the approval of the Mount Sinai Animal Care and Use Committee. C57Bl/6 mice weighing 25–30 g, at 8–10 weeks of age (purchased from The Jackson Laboratory), were gavaged with ethanol (30% v/v in saline, 4 g/kg, every 12 h) for 5 days. CBD (5 mg/kg, every 12 h) or vehicle (Tween 80 2%–saline) was injected intraperitoneally (ip) 30 min before each ethanol gavage. After the stated treatment period, mice were anesthetized by ip administration of 50 mg/kg pentobarbital, and serum and liver were collected. Hematoxylin and eosin (H&E) and oil red O staining of liver slices was performed as previously described [2,4,5]. Paraffin slides were deparaffinized and blocked as previously reported [20]. Antibodies against 4-hydroxynonenal (4-HNE), 3-nitrotyrosine (3-NT), and pJNK were added at 1:200 dilution and incubated at 4°C overnight. Slides were washed with phosphate-buffered saline and stained with the Histostain Plus Broad Spectrum (DAB) kit. Images were acquired under the light microscope at 200 × magnitude. The oil red O staining was quantified by ImageJ software from the U.S. National Institutes of Health.
ATP assay
ATP content was determined using an ATP Determination Kit (A22066, Molecular Probes). Each reaction contained 1.25 μg/ml firefly luciferase, 50 μM D-luciferin, and 1 mM dithiothreitol in 1 × reaction buffer. Cells were treated with vehicle or CBD or ethanol or CBD plus ethanol. After a 15-min incubation with 1 mg liver protein, luminescence was measured in a luminometer. Results are expressed as arbitrary units of luminescence compared to that of the vehicle-treated control cells, which was taken as 100 arbitrary units.
Triglyceride analysis
TG levels in the liver lysates were analyzed following the instructions in the TG analysis kit from Pointe Scientific (Canton, MI, USA). TGs in the sample are hydrolyzed by lipase to glycerol and fatty acids. The glycerol is then phosphorylated by ATP to glycerol 3-phosphate. Glycerol 3-phosphate is then converted to dihydroxyacetone phosphate and hydrogen peroxide by glycerophosphate oxidase. The hydrogen peroxide then reacts with 4-aminoantipyrine and 3-hydroxy-2,4,6-tribomobenzoic acid in a reaction catalyzed by peroxidase to yield a red quinoneimine dye. The intensity of the color produced is directly proportional to the concentration of triglycerides in the sample when measured at 540 nm.
Oil red O and ROS assays
E47 cells, which express CYP2E1 [21], were incubated with 50 or 100 mM ethanol for 5 days (fresh medium with ethanol was added every 24 h) in the absence or presence of CBD (2 and 5 μM). Oil red O staining and ROS detection by flow cytometry of E47 cells were performed as previously described [2,4,5,19]. Total ROS detection was assayed using the Total ROS Detection Kit (Enzo Life Sciences) which contains a nonfluorescent, cell-permeative ROS detection dye. The dye can react directly with a wide range of reactive species, such as hydrogen peroxide, peroxynitrite, and hydroxyl radicals, to yield a green fluorescent product indicative of cellular production of various reactive oxygen or nitrogen species. Upon staining, the fluorescent product generated can be visualized using a flow cytometer equipped with a blue (488 nm) laser. The shift of fluorescence peaks to the right is indicative of increased ROS production. Flow cytometric analyses were performed on a three-laser LSRII with DiVa software (BD Biosciences). Data were analyzed with FlowJo software. Activity of CYP2E1 was assayed by the oxidation of p-nitrophenol [2,5].
Western blot analysis
Levels of CYP2E1, pJNK, JNK, LC3, pP38 MAPK, and P38 MAPK in 20–100 μg of protein samples from freshly prepared liver homogenate fractions were determined using Western blot analysis. Blots were scanned using an Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE, USA). All specific bands were quantified with the Automated Digitizing System (ImageJ programs, version 1.34 S, National Institutes of Health). Results are shown for one experiment but quantification is from three separate experiments.
LC3 and autophagy assay
LC3 is microtubule-associated protein 1 A/1B light-chain 3, a soluble protein with a molecular mass of approximately 17 kDa. It is distributed ubiquitously in mammalian tissues and cultured cells. During autophagy, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine (lipidation) to form an LC3–phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. The lipidated form of LC3 is referred to LC3-II, whereas LC3-I is not lipidated. The LC3-II/LC3-I ratio is often used to assay autophagy and, when assayed in the presence of lysosomal inhibitors such as chloroquine, it represents autophagic flux.
Statistical analysis
Values reflect means ± SD. One-way ANOVA with subsequent post hoc comparisons by Tukey HSD were performed by SPSS analysis software (version 10.0). A p value of less than 0.05 was considered statistically significant and results are from experiments using four to six mice or six replicates of each group of cells.
Results
CBD decreases ethanol-induced liver injury
To assess the effect of CBD on ethanol-induced hepatotoxicity, we induced liver injury by treating mice with ethanol (30% v/v in saline, 4 g/kg, every 12 h for 5 days). CBD (5 mg/kg, every 12 h) or vehicle was injected ip 30 min before ethanol gavage. Ethanol gavage led to an increase in serum aspartate aminotransferase (AST) that was prevented by CBD (Fig. 1A). Ethanol gavage produced a 60% decrease in hepatic ATP levels (Fig. 1C), suggestive of hepatic bioenergetic injury. This decline in ATP was completely reversed by CBD (Fig. 1C). Ethanol gavage led to a 60% elevation in hepatic triglycerides. This effect was significantly attenuated by CBD treatment (Fig., 1B). The CBD treatment also lowered basal TG levels. These effects of CBD were accompanied by a prevention of ethanol-induced steatosis as demonstrated by H&E (Fig. 2A) and oil red O staining of liver sections (Fig. 2B).
Fig. 1.
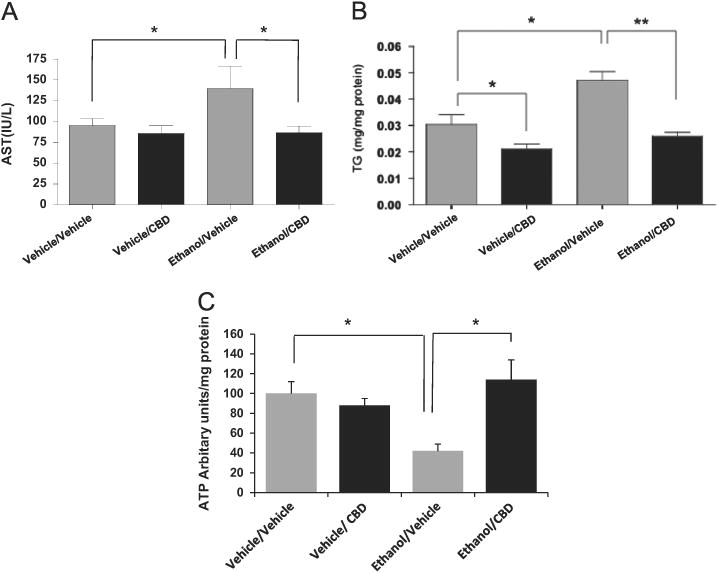
Binge ethanol induces liver injury and accumulation of lipids in mice, and CBD reverses these effects. (A) Serum AST levels; (B) TG levels in liver; (C) ATP levels in liver. Results are from four to six mice in each group. *p < 0.05 and **p < 0.01.
Fig. 2.
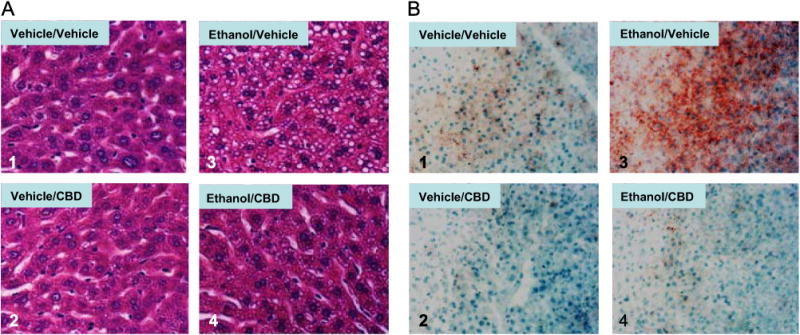
(A) H&E staining and (B) oil red O staining showing increased lipid accumulation in mouse liver after binge alcohol treatment. CBD decreases this lipid accumulation (image 4 compared to image 3 in (A) and (B)).
CBD reduces ethanol-induced oxidative stress in livers of ethanol-treated mice
We then examined if CBD was attenuating liver injury by acting as an antioxidant molecule. Liver sections were treated with anti-4-HNE and anti-3-NT IgG to detect 4-HNE or 3-NT adducts (Figs. 3A and B). Ethanol led to an increase in 4-HNE staining. Interestingly, this effect was attenuated in livers from mice treated with CBD (Fig. 3A, images 3 and 4). There was, however, no change in 3-NT protein adducts by ethanol, or CBD, or ethanol/CBD (Fig. 3B).
Fig. 3.
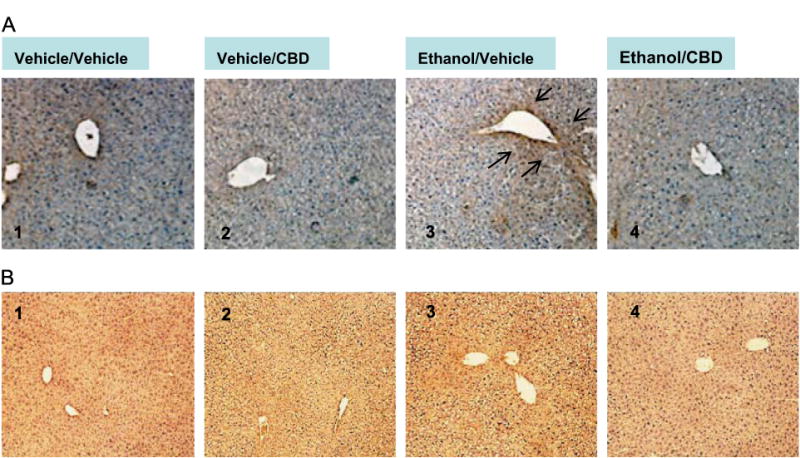
(A) Binge alcohol drinking increases 4-HNE staining in mouse liver (image 3 compared to image 1), and CBD reverses this increase (image 4 compared to image 3). Black arrows point to areas of positive staining of 4-HNE. (B) Binge alcohol, CBD, or ethanol/CBD do not change 3-NT staining in mouse liver (image 2, 3, or 4 compared to image 1).
CBD treatment prevents JNK activation
Binge ethanol has been shown to activate the JNK and P38 MAPK pathways [2,22–24]. Inhibition of MAPK partially prevents alcohol- and other xenobiotic-mediated liver injury [2,25,26]. We examined the effect of CBD on ethanol-induced JNK and P38 MAPK phosphorylation. Staining of liver sections with anti-pJNK IgG indicated that CBD prevented JNK activation by binge ethanol (Fig. 4A). Western blot analyses of liver homogenates confirmed that ethanol induced the phosphorylation of JNK1 about fourfold and that CBD treatment partially blocked this increase in JNK1 phosphorylation (Fig. 4B). The binge ethanol treatment had no effect on activation of P38 MAPK (Fig. 4C).
Fig. 4.
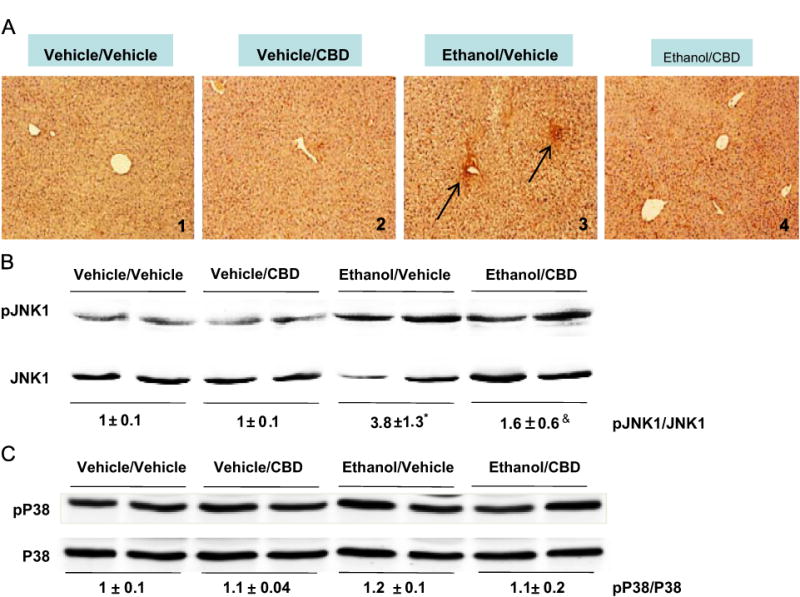
Binge alcohol drinking increases phosphorylation of c-Jun N-terminal kinase (JNK) as shown by (A) immunohistochemistry (image 3 compared to image 1) and (B) Western blot. Livers were collected 18 h after the last ethanol gavage. CBD partially blocked the activation of JNK by binge alcohol. *p < 0.05 comparing ethanol/vehicle to vehicle/vehicle. &p < 0.05 comparing ethanol/CBD to ethanol/vehicle. (C) P38 MAPK phosphorylation is not increased by binge alcohol consumption.
CBD reduces ethanol-induced oxidative stress in CYP2E1-expressing HepG2 cells
To confirm the antioxidant action of CBD as an explanation for its effect on ethanol-induced liver injury, we examined the effect of CBD on ethanol-induced oxidative stress in vitro. HepG2 cells stably expressing CYP2E1 (E47 cells) were used for this purpose. We have previously demonstrated the requirement of CYP2E1 activity to facilitate ethanol toxicity in these cells [4,5]. E47 cells were incubated with 50 or 100 mM ethanol for 5 days, in the absence or presence of CBD (5 μM). Ethanol treatment led to increases in ROS levels that were decreased by cotreatment with CBD (Fig. 5A). Quantification of the areas in Fig. 5A indicated that there were 2.5- and 3.5-fold increases produced by 50 and 100 mM ethanol, respectively, and that CBD lowered these to increases of about 10–20% (data not shown). In addition we observed that treatment with CBD also reduced the basal levels of ROS produced by CYP2E1 in the E47 cells (Fig. 5A, graph 1). CBD treatment also decreased the ethanol-induced increase in the TG content in these cells (Fig. 5B) and decreased lipid droplet accumulation induced by ethanol as shown by oil red O staining (Figs. 5C and D). CBD effects were not due to changes in CYP2E1 protein levels or activity in the E47 cells (Figs. 5E and F). These results in a well-established model to study the effects of alcohol metabolism on cellular injury are consistent with the ability of CBD to mitigate ethanol-induced liver injury in vivo.
Fig. 5.
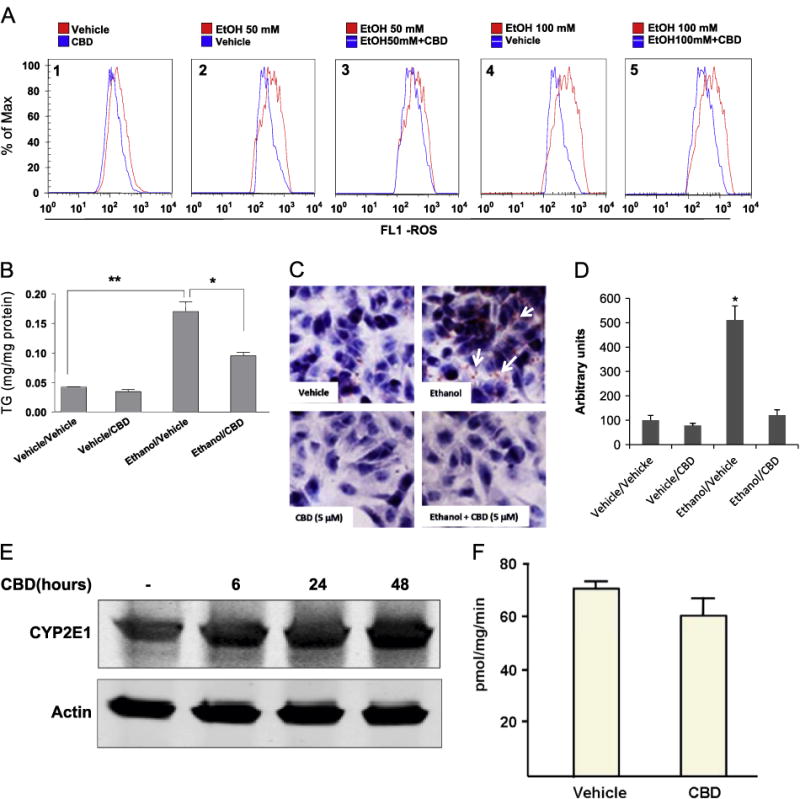
(A) In cytochrome P450 2E1 (CYP2E1)-expressing HepG2 cells, either 50 or 100 mM ethanol treatment increased ROS production (graphs 2 and 4), and CBD partially prevented the increase in ROS (graphs 3 and 5 compared to 2 and 4). ROS were assayed using the Total ROS Detection Kit. The distribution of fluorescence intensity is shown as a histogram, with the y axis indicating the percentage of cells in the total population displaying the specific fluorescence intensity shown on the x axis. (B and C) In vitro 100 mM ethanol treatment increased lipid accumulation in CYP2E1-expressing HepG2 cells. (B) TG levels, expressed as mg/mg protein; *p < 0.05, **p < 0.01. (C) Oil red O staining showing that CBD prevents this lipid accumulation. (D) Quantification of oil red O staining was carried out using the ImageJ program. Staining in the vehicle/vehicle group was taken as 100, and staining of all other groups was expressed relative to the vehicle/vehicle group. *p < 0.05 compared to vehicle/vehicle group. (E and F) CYP2E1 protein expression and catalytic activity were assayed 48 h after the last treatment with CBD. CBD itself does not have any effect on either CYP2E1 protein expression or CYP2E1 activity.
CBD stimulates autophagy in vitro and in vivo
Several recent studies have demonstrated the importance of autophagy in hepatocyte homeostasis. In particular, growing evidence indicates that stimulation of autophagy decreases hepatocyte oxidative stress and fat accumulation by clearing damaged mitochondria and lipid droplets from hepatocytes [27–29]. We examined the possibility that one mechanism by which CBD exerted its protective effects against ethanol toxicity was by stimulating autophagy. During autophagy, LC3-I (cytosolic form of LC3) is conjugated to phosphatidylethanolamine (lipidation) to form LC3-II (LC3–phosphatidylethanolamine conjugate), which is recruited to autophagosomal membranes. The lipidated form of LC3 is referred to as LC3-II, whereas LC3-I is not lipidated. The LC3-II/LC3-I ratio is often used to assay autophagy and when assayed in the presence of lysosomal inhibitors such as chloroquine, it assays autophagic flux. LC3 lipidation was evaluated as an index of autophagy by Western blot analysis. Treatment of E47 cells with CBD led to an increase in levels of LC3-II, indicating an increase in autophagosome formation. This effect was further increased upon cotreatment with chloroquine, confirming an induction of autophagy by CBD (Fig. 6A). We next examined autophagy in livers from the ethanol-treated mice. Ethanol decreased autophagy, as indicated by a decrease in LC3-II levels and in the LC3-II/LC3-I ratio (Fig. 6B). This inhibition of autophagy by ethanol was not seen in livers from mice that received CBD plus ethanol (Fig. 6B), indicating that CBD prevents alcohol-mediated inhibition of autophagy in vivo. Both in vitro and in vivo experiments showed that CBD promotes autophagy, which could play a role in the mechanisms by which CBD protects liver from acute alcohol-induced steatosis.
Fig. 6.
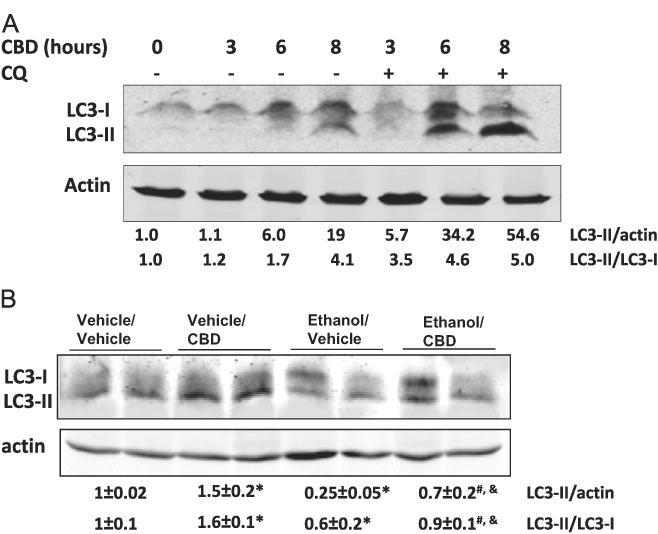
Effects of CBD and CBD + ethanol on autophagy. LC3 is microtubule-associated protein 1 A/1B light-chain 3. During autophagy, a cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine (lipidation) to form LC3–phosphatidylethanolamine (LC3-II), which is recruited to autophagosomal membranes. The LC3-II/LC3-I ratio is often used to assay autophagy and, when assayed in the presence of lysosomal inhibitors such as chloroquine, it indicates autophagic flux. (A) E47 HepG2 cells were incubated with 5 μM CBD in the absence or presence of 10 mM chloroquine (CQ) for up to 8 h and immunoblots were carried out to assay for LC3-II, LC3-I, and actin. (B) Binge alcohol drinking decreases autophagy in mouse liver. Pretreatment of mice with 5 mg/kg CBD prevents the decrease in autophagy by alcohol. *p < 0.05 compared to vehicle/vehicle group. #p < 0.05 compared to ethanol/vehicle group. &p < 0.05 compared to vehicle/CBD group.
Discussion
Acute alcohol drinking induces liver steatosis [2,19,27,29]. The induction of liver steatosis is promoted by CYP2E1, as wild-type mice with CYP2E1 expression displayed steatosis, but CYP2E1-knockout mice exhibit strongly decreased steatosis after acute alcohol treatment [1,2,19]. In the current study, lipid accumulation was found in CYP2E1-expressing HepG2 cells after ethanol treatment, and liver steatosis was observed in mice treated with acute alcohol. Mechanisms by which CYP2E1 promotes ethanol induction of steatosis include elevated ROS, activation of MAPKs such as JNK, inhibition of autophagy, and decreases in levels of PPARα, a major regulator of fatty acid oxidation [1,2]. Oxidative stress has been reported to be one major cause of liver steatosis by alcohol. Antioxidants such as N-acetylcysteine attenuate acute alcohol-induced steatosis [2,30]. We previously found that a JNK inhibitor partially blocked acute ethanol-induced steatosis [2]. CBD is a nonpsychotic cannabinoid, which has been shown to have anti-inflammatory and antioxidant properties. CBD has also been reported to function as an antioxidant in preventing glutamate toxicity and preventing neurotoxicity by acute alcohol [31]. Consistent with reports that CBD has antioxidant activity, we found that CBD can reduce ROS production in HepG2 cells expressing CYP2E1 and that CBD inhibited the increase in ROS induced by alcohol treatment. CBD protected liver from acute alcohol-induced steatosis and lowered 4-HNE levels via its antioxidant property. CBD did not alter CYP2E1 levels or catalytic activity; therefore the CBD prevention of alcohol-induced steatosis and oxidant stress is not due to effects on CYP2E1.
How activation of CYP2E1 by alcohol consumption, in addition to oxidative stress, promotes steatosis requires further mechanistic evaluation. The JNK MAPK pathway is a major signaling pathway, which initiates apoptosis and cell death [32,33]. Recent reports also showed that activation of the JNK MAPK pathway by alcohol may be a key factor regulating the increase in liver steatosis, because inhibition of JNK activation prevented acute alcohol-induced steatosis [2]. We found that CBD can inhibit the increase in JNK MAPK phosphorylation by acute alcohol, which may be due to its antioxidant actions. Inhibition of JNK activation may contribute to CBD's anti-steatosis effects.
While a basal level of autophagy is required for the survival of cells or organisms, prolonged activation of autophagy may have adverse effects. In mammalian systems, autophagy is stimulated by nutrient starvation or deprivation of growth factors [34–38]. Although it is not clear yet whether alcohol increases or decreases autophagy under different conditions, it is consistent that autophagy is a protective mechanism against alcohol-induced liver injury [2,19,27,29]. Increasing autophagy can protect cells from injury by various stimuli, as inhibition of autophagy increases toxicity to cells. Genetically enhancing autophagy by overexpressing Atg7 could alleviate hepatic steatosis induced by a high-fat diet [39]. Carbamazepine, an FDA-approved antiepileptic drug, can alleviate fatty liver by inducing autophagy [40]. In contrast, loss of autophagy in vitro or in vivo increases lipid accumulation in cells and in liver [2,4,19]. We found that CBD can stimulate autophagy in HepG2 cells that express CYP2E1. The increased flux from LC3-I to LC3-II was confirmed by treating cells with a lysosome inhibitor. Because the LC3-II/LC3-I ratio is used to assay autophagy, the increased flux from LC3-I to LC3-II indicates increases in autophagy. CBD-mediated increase in autophagy may be important for the prevention of alcohol-induced steatosis by CBD.
In conclusion, this is the first report demonstrating that CBD can alleviate lipid accumulation in both an in vitro HepG2 cell model and an in vivo binge alcohol treatment model by multiple mechanisms. These mechanisms may involve activation of autophagy, inhibition of the JNK MAPK pathway, and inhibition of oxidative stress, and all three mechanisms could be important for alleviating steatosis.
Acknowledgments
This work was supported by Grants AA-018790 and AA-021362 from the NIAAA (to A.I.C.) and NIH Grant DA 008863 (to L.A.D.).
Contributor Information
Lili Yang, Email: yangll7@mail.sysu.edu.cn.
Zhenfeng Zhang, Email: zhangzhf@sysucc.org.cn.
Arthur Cederbaum, Email: arthur.cederbaum@mssm.edu.
References
- 1.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radic Biol Med. 2010;49:1406–1416. doi: 10.1016/j.freeradbiomed.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Wu D, Wang X, Cederbaum AI. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic Biol Med. 2012;53:1170–1180. doi: 10.1016/j.freeradbiomed.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdelmegeed MA, Banerjee A, Yoo SH, Jang S, Gonzalez FJ, Song BJ. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J Hepatol. 2012;57:860–866. doi: 10.1016/j.jhep.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu D, Wang X, Zhou R, Cederbaum A. CYP2E1 enhances ethanol-induced lipid accumulation but impairs autophagy in HepG2 E47 cells. Biochem Biophys Res Commun. 2010;402:116–122. doi: 10.1016/j.bbrc.2010.09.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Wu D, Wang X, Cederbaum AI. Depletion of cytosolic or mitochondrial thioredoxin increases CYP2E1-induced oxidative stress via an ASK-1-JNK1 pathway in HepG2 cells. Free Radic Biol Med. 2011;51:185–196. doi: 10.1016/j.freeradbiomed.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malfait AM, Gallily R, Sumariwalla PF, Malik AS, Andreakos E, Mechoulam R, Feldmann M. The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc Natl Acad Sci USA. 2000;97:9561–9566. doi: 10.1073/pnas.160105897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss L, Zeira M, Reich S, Har-Noy M, Mechoulam R, Slavin S, Gallily R. Cannabidiol lowers incidence of diabetes in non-obese diabetic mice. Autoimmunity. 2006;39:143–151. doi: 10.1080/08916930500356674. [DOI] [PubMed] [Google Scholar]
- 8.Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (–) Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998;95:8268–8273. doi: 10.1073/pnas.95.14.8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ligresti A, Moriello AS, Starowicz K, Matias I, Pisanti S, De Petrocellis L, Laezza C, Portella G, Bifulco M, Di Marzo V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther. 2006;318:1375–1387. doi: 10.1124/jpet.106.105247. [DOI] [PubMed] [Google Scholar]
- 10.McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther. 2007;6:2921–2927. doi: 10.1158/1535-7163.MCT-07-0371. [DOI] [PubMed] [Google Scholar]
- 11.Vaccani A, Massi P, Colombo A, Rubino T, Parolaro D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br J Pharmacol. 2005;144:1032–1036. doi: 10.1038/sj.bjp.0706134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrivastava A, Kuzontkoski PM, Groopman JE, Prasad A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol Cancer Ther. 2011;10:1161–1172. doi: 10.1158/1535-7163.MCT-10-1100. [DOI] [PubMed] [Google Scholar]
- 13.Wu HY, Chu RM, Wang CC, Lee CY, Lin SH, Jan TR. Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8. Toxicol Appl Pharmacol. 2008;226:260–270. doi: 10.1016/j.taap.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Christian P, Sacco J, Adeli K. Autophagy: emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta. 2013;1831:819–824. doi: 10.1016/j.bbalip.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 15.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stankov MV, Panayotova-Dimitrova D, Leverkus M, Vondran FW, Bauerfeind R, Binz A, Behrens GM. Autophagy inhibition due to thymidine analogues as novel mechanism leading to hepatocyte dysfunction and lipid accumulation. AIDS. 2012;26:1995–2006. doi: 10.1097/QAD.0b013e32835804f9. [DOI] [PubMed] [Google Scholar]
- 18.Younce C, Kolattukudy P. MCP-1 induced protein promotes adipogenesis via oxidative stress, endoplasmic reticulum stress and autophagy. Cell Physiol Biochem. 2012;30:307–320. doi: 10.1159/000339066. [DOI] [PubMed] [Google Scholar]
- 19.Wu D, Wang X, Zhou R, Yang L, Cederbaum AI. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radic Biol Med. 2012;53:1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Wu D, Yang L, Cederbaum AI. Hepatotoxicity mediated by pyrazole (cytochrome P450 2E1) plus tumor necrosis factor alpha treatment occurs in c-Jun N-terminal kinase 2 −/− but not in c-Jun N-terminal kinase 1 −/− mice. Hepatology. 2011;54:1753–1766. doi: 10.1002/hep.24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu D, Cederbaum AI. Development and properties of HepG2 cells that constitutively express CYP2E1. Methods Mol Biol. 2008;447:137–150. doi: 10.1007/978-1-59745-242-7_11. [DOI] [PubMed] [Google Scholar]
- 22.Gu L, Fink AM, Chowdhury SA, Geenen DL, Piano MR. Cardiovascular responses and differential changes in mitogen-activated protein kinases following repeated episodes of binge drinking. Alcohol Alcohol. 2012;48:131–137. doi: 10.1093/alcalc/ags090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishitani Y, Matsumoto H. Ethanol rapidly causes activation of JNK associated with ER stress under inhibition of ADH. FEBS Lett. 2006;580:9–14. doi: 10.1016/j.febslet.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 24.Aroor AR, James TT, Jackson DE, Shukla SD. Differential changes in MAP kinases, histone modifications, and liver injury in rats acutely treated with ethanol. Alcohol Clin Exp Res. 2010;34:1543–1551. doi: 10.1111/j.1530-0277.2010.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu D, Cederbaum AI. Role of p38 MAPK in CYP2E1-dependent arachidonic acid toxicity. J Biol Chem. 2003;278:1115–1124. doi: 10.1074/jbc.M207856200. [DOI] [PubMed] [Google Scholar]
- 26.Wu D, Cederbaum A. Glutathione depletion in CYP2E1-expressing liver cells induces toxicity due to the activation of p38 mitogen-activated protein kinase and reduction of nuclear factor-kappaB DNA binding activity. Mol Pharmacol. 2004;66:749–760. doi: 10.1124/mol.104.002048. [DOI] [PubMed] [Google Scholar]
- 27.Ding WX, Li M, Yin XM. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy. 2010;7:248–249. doi: 10.4161/auto.7.2.14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol. 2013;44:311–326. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- 29.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y, Zhang XH, Cederbaum AI. Ethanol induction of CYP2A5: role of CYP2E1–ROS-Nrf2 pathway. Toxicol Sci. 2012;128:427–438. doi: 10.1093/toxsci/kfs164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamelink C, Hampson A, Wink DA, Eiden LE, Eskay RL. Comparison of cannabidiol, antioxidants, and diuretics in reversing binge ethanol-induced neurotoxicity. J Pharmacol Exp Ther. 2005;314:780–788. doi: 10.1124/jpet.105.085779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guegan JP, Ezan F, Theret N, Langouet S, Baffet G. MAPK signaling in cisplatin-induced death: predominant role of ERK1 over ERK2 in human hepatocellular carcinoma cells. Carcinogenesis. 2012;34:38–47. doi: 10.1093/carcin/bgs317. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Chen GG, Zhang Z, Chun S, Leung BC, Lai PB. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis. 2011;17:325–334. doi: 10.1007/s10495-011-0685-y. [DOI] [PubMed] [Google Scholar]
- 34.Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP–LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol. 2010;300:C308–317. doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dall'Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol. 2013;23:R33–45. doi: 10.1016/j.cub.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen M, Flatt T, Aguilaniu H. Reproduction, fat metabolism, and life span: what is the connection? Cell Metab. 2013;17:10–19. doi: 10.1016/j.cmet.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puls F, Goldschmidt I, Bantel H, Agne C, Brocker V, Dammrich M, Lehmann U, Berrang J, Pfister ED, Kreipe HH, Baumann U. Autophagy-enhancing drug carbamazepine diminishes hepatocellular death in fibrinogen storage disease. J Hepatol. 2013;59:626–630. doi: 10.1016/j.jhep.2013.05.018. [DOI] [PubMed] [Google Scholar]