

Abstract
Constituting the largest group of membrane proteins identified in the human genome, G protein-coupled receptors (GPCRs) help control many physiological processes by responding to various stimuli. As targets for more than 40% of all prescribed pharmaceuticals, detailed understanding of GPCR structures is vital for the design and development of more specific medications and improved patient therapies. But structural information for membrane proteins and GPCRs, in particular, is limited despite considerable interest. The major impediment to obtaining sufficient quantities of highly purified GPCRs in their native form for crystallization lies in their low tissue levels, poor yields, and stability. The only exception is rhodopsin, which is abundantly expressed in the eye and stabilized by its covalently bound chromophore, 11-cis-retinal. Expression systems and purification protocols have yet to be developed for all other GPCRs. Here, we present a novel expression system for human GPCRs in Caenorhabditis elegans that produces sufficient amounts of recombinant proteins to allow their biochemical and structural characterization.
1. Introduction
Although several expression systems such as cell-free (Klammt, Schwarz, Dotsch, & Bernhard, 2007), Escherichia coli-based (Dodevski & Pluckthun, 2011), yeast-based (Shiroishi et al., 2011), mammalian cell-based (Standfuss et al., 2011), and animal-based (Panneels, Kock, Krijnse-Locker, Rezgaoui, & Sinning, 2011; Salom et al., 2008; Zhang et al., 2005) systems have been developed that yield GPCRs in quantities suitable for crystallization, no GPCR other than rhodopsin (Palczewski et al., 2000) has yet been crystallized in its native form. Because production of recombinant functional receptors is difficult, due in part to GPCR conformational flexibility, different strategies have been used to stabilize GPCR structures. Examples include formation of complexes between GPCRs and specific antibody fragments, deglycosylation, introduction of multiple point mutations, or replacing the deleted third cytoplasmic flexible loop with T4-lysozyme. Though all those strategies allowed the determination of a few GPCR crystal structures (reviewed in Katritch, Cherezov, & Stevens, 2012), they also altered the pharmacological and functional properties of these receptors. Therefore, developing new approaches for expressing stable unmodified GPCRs remains a high priority. In this chapter, we describe a novel expression system in Caenorhabditis elegans that allows production of milligram quantities of functional heterologous GPCRs in their native form suitable for structural studies. C. elegans can meet this challenge because it contains the machinery to express over 1000 endogenous GPCRs.
2. Expression of Transgenic GPCRs in C. Elegans
Equipment
Leica DMI 3000B microscope, micromanipulator, and MZ16F fluorescence stereomicroscope (Leica Microsystems, Bannockburn, IL, USA); FemtoJet microinjector (Eppendorf, Hauppauge, NY, USA); and SpectroLinker XL-1500 UV crosslinker (Spectronics Corporation, Westbury, NY, USA).
Materials
pBluescript KS(+) vector, mammalian GPCR cDNA, and C. elegans Bristol N2 strain from the Caenorhabditis Genetic Center (CGC; University of Minnesota, Minneapolis, MN, USA), nematode growth medium (NGM: 0.25% peptone, 51 mM NaCl, 25 mM potassium phosphate buffer, pH 6.0, 5 μg/ml cholesterol, 1 mM CaCl2, and 1 mM MgCl2) plates (LabExpress, Ann Arbor, MI, USA) seeded with OP50 bacteria (CGC), 2% agarose pad, and halocarbon oil (Halocarbon Oil Series HC-700, Halocarbon Product Corporation, River Edge, NY, USA).
Solutions
1 M stock potassium phosphate buffer, pH 6.0 (108.3 gKH2PO4 and 35.6 g K2HPO4 dissolved in 1 l H2O), M9 buffer, pH 7.5 (3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl and 1 ml of 1 M MgSO4 dissolved in 1 l H2O).
C. elegans was chosen as the host organism for expression of heterologous GPCRs because this worm expresses more than 1000 GPCRs in its neurons along with their cognate heterotrimeric G proteins. Thus, they contain all molecular machinery needed to produce properly folded and functional receptors in response to extracellular stimuli.
2.1. Worm maintenance
Bristol N2 strain worms should be cultured on NGM plates seeded with OP50 bacteria at 16–25 °C. (But remember that worms grow 2.1 times faster at 25 °C than at 16 °C.) Cryostorage and recovery from frozen stocks should be carried out by previously described protocols (Stiernagle, 2006).
2.2. Preparation of GPCR constructs
To generate a mammalian GPCR expression construct, insert either the promotor of myo-3 (Okkema, Harrison, Plunger, Aryana, & Fire, 1993) that drives strong gene expression in worm body wall muscles or H20 (Yabe, Suzuki, Furukawa, Ishihara, & Katsura, 2005) which drives gene expression in the nervous system into pBluescript KS(+) vector at HindIII/Xbal or Pstl, respectively. Then, insert the cDNA of a specific transgenic GPCR (which can be synthesized by Genescript, Piscataway, NJ, USA) followed by a tobacco etch virus (TEV) protease cleavage site T7 tag (a sequence encoding the initial 11 amino acids of the leader sequence of T7 bacteriophage gene 10 that comprises an epitope for T7 monoclonal antibody [mAb]) and either a Rho9 tag (the C-terminal 9 amino acids of bovine Rho, an epitope for 1D4 mAb) (Oprian, Molday, Kaufman, & Khorana, 1987) or a His tag, followed by the polyadenine [poly] tail of unc-54 (Fire & Waterston, 1989) between Notl and XhoI. Both T7 and Rho9 tags can be removed from the GPCR construct by TEV protease treatment. Only the opsin construct can be inserted without additional tags because it already contains a C-terminal Rho9 tag. The entire GPCR fusion protein construct should be sequenced after the construct is generated to confirm the presence of the transgenic GPCR and absence of random mutations.
2.3. Generation of transgenic worm lines
To obtain a transgenic worm line, microinject the GPCR construct (10 ng/μl) mixed with marker DNA (3 ng/μl) encoding the coral-derived red fluorescent protein (DsRed) into the syncytial gonad of a healthy, non-starved, young adult day 1 (d1) worm according to the protocol described in Stiernagle (2006). Both DNA constructs should be under control of the same promoter (either Pmyo-3 or PH20). First, to help immobilize a worm that is going to be injected, transfer it to the center of a 2% agarose injection pad (a coverslip with a layer of 2% agarose in water) and add a drop of halocarbon oil to avoid its rapid dehydration. Fill the injection needle with the DNA mixture and inject it into the worm gonad. Remove the injected animal from the agarose pad, wash it with M9 buffer to remove excess oil and transfer it onto a new plate with food and incubate at 20 °C. Up to five worms can be placed onto one plate. After 3 or 4 days, score the DsRed expression levels of the first generation (F1). Expression of DsRed can be detected by visualizing its fluorescence through a dissecting microscope. (Worms manifesting strong DsRed expression are selected based on the assumption that the GPCR expression level positively correlates with this independently expressed marker protein.) Then, place selected single transgenic F1 worms on individual plates and culture them for three to four generations to determine which ones produce stable transgenic lines. To integrate GPCR cDNA into the worm genome, select larva stage 4 (L4) worms from the identified stable transgenic worm lines and expose them to 350 × 100 μJ/cm2 ultraviolet (UV) light. Then culture them for four generations (Fig. 11.1). Select F4 (fourth generation) progeny of integrated transgenic line worms and back-cross them with wild-type worms to clarify the genomic background because exposure to UV light can cause nonspecific mutations.
Figure 11.1.
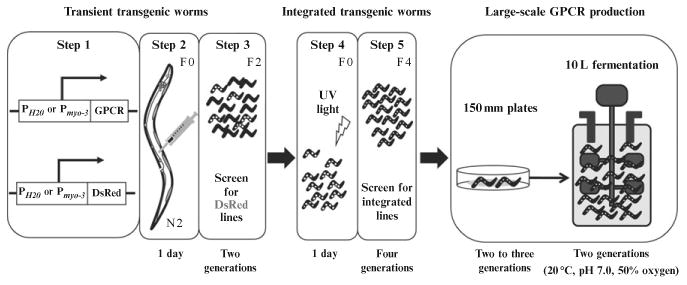
Flowchart for expression of vertebrate GPCRs in C. elegans. Generation of transient transgenic worms is carried out in three steps (Steps 1–3). After DNA constructs, expressing GPCR and DsRed are mixed (Step 1) and injected into a worm's gonad (Step 2), worms are cultured for two generations and then screened for those expressing the DsRed marker (Step 3). (White dots indicate worms expressing DsRed.) Selected transient transgenic worm lines are then exposed to UV light to generate integrated transgenic worm lines (Step 4). UV-exposed worms are cultured for four generations to establish stable integrated transgenic worm lines (Step 5).
3. Detection of Heterologous GPCR Gene Expression
3.1. Immunohistochemistry
Equipment
Leica TCS SP2 confocal microscope (Leica Microsystems).
Materials
2% Agarose pads, NGM plates, Alexa-488-conjugated 1D4 antibody (conjugate Alexa-488 (Molecular Probes, Eugene, OR, USA) to 1D4 antibody by using the Alexa Fluor 488 monoclonal antibody labeling kit (Molecular Probes)), and Triton X-100.
Solutions
Injection buffer (20 mM K3PO4, 3 mM potassium citrate, and 2% polyethylene glycol, pH 7.5), M9 buffer, methanol, acetone, and PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.76 mM KH2PO4, pH 7.4).
3.1.1 Staining of expressed transgenic GPCRs in live C. elegans
The protocol for immunohistochemical staining of live animals is adopted from Gottschalk and Schafer (2006). Mount d1 adult worms from transient or integrated transgenic worm lines on a 2% agarose pad with halocarbon oil. Resuspend Alexa-488-conjugated 1D4 antibody in the injection buffer containing 0.4–0.6% Triton X-100 at a final concentration 33 μg/ml. Inject the diluted antibody into the pseudocoelom, transfer animals from the agarose pad to an NGM plate containing M9 buffer and allow them to recover for about 6 h at 20 °C. Check live animals for Rho9 immunoreactivity under a confocal microscope (λex= 488 nm, λem = 510–530 nm). Under these conditions, 1D4 antibody should enter cells and bind to the expressed transgenic GPCRs localized in membranes because the Rho9 tag is located intracellularly.
3.1.2 Staining of expressed transgenic GPCRs in fixed C. elegans
Place L4 or d1 animals from integrated transgenic worm lines between two coverslips and bury them in dry ice for 30 min. Then fix worms in 100% methanol for 10 min, followed by 100% acetone for 10 min. Wash worms with PBS for 30 min and then incubate them overnight at 4°C with Alexa-488-conjugated 1D4 antibody diluted with PBS and 0.1% Triton X-100 to a final concentration 33 μg/ml. Next day, wash the stained worms 3 × with PBS and examine them by confocal microscopy (λex = 488 nm, λem = 510–530 nm).
3.2. SDS-PAGE and immunoblotting
To confirm either transient or integrated GPCR expression in transgenic worms, use either a worm membrane fraction (see Section 5), intact worm pellets or worms sonicated for ∼20 s. To the resulting pellet add electrophoresis loading buffer and either sonicate briefly with a water bath sonicator or vortex vigorously. Then, centrifuge for 5 min at 13,000 rpm in an Eppendorf bench top centrifuge at 15 °C to avoid overheating or cold SDS precipitation. Load the solubilized fraction on a 4–12% SDS-PAGE gel. Transfer separated proteins from the gel to PVDF transfer membranes. Before immunoblotting, block PVDF membrane-containing proteins with blocking solution for 1 h at room temperature or overnight at 4 °C. Discard the blocking buffer and incubate membranes with primary antibody (alkaline phosphatase-conjugated 1D4 mAb) in blocking solution for 1 h. Wash membranes with TBST (0.1% Tween in 50 mM Tris and 150 mM NaCl, pH 7.4) 3 × for 10 min. To detect proteins of interest, incubate membranes with Western Blue stabilized substrate for alkaline phosphatase for several minutes until protein bands become visible. Stop the reaction by washing membranes with ddH2O (Fig. 11.2A).
Figure 11.2.

Expression and purification of heterologous GPCRs expressed in C. elegans. (A) Expression of (h)A2AR in a lysate prepared from a transgenic worm line expressing (h)A2AR in muscle and detected by immunoblotting with alkaline phosphatase-conjugated 1D4 mAb. The two monomeric bands shown correspond to glycosylated (top) and nonglycosylated (bottom) forms of this receptor. Figure adapted with permission from Salom et al. (2012), Federation of American Societies for Experimental Biology. (B) Representative silver-stained SDS-PAGE of (b)isoRho purified from transgenic worms expressing opsin in muscles by 1D4 immunoaffinity chromatography and compared with 500 ng of (b)Rho purified from bovine retina. F1–F4 fractions were eluted from the 1D4 column.
Purified GPCRs can be visualized directly on SDS-PAGE gel after either Coomassie Blue or silver staining, depending on the purification scale. For Coomassie Blue staining, incubate the gel with 1% Coomassie Blue R-250 in 50% methanol and 10% acetic acid for 30 min, then destain the gel with 10% methanol and 10% acetic acid for 2 h. For silver staining, fix the gel with 30% ethanol and 10% acetic acid for 1 h and then wash with 30% ethanol 3 × for 15 min each. Next, wash the gel with ddH2O 3 × for 10 min each, followed by soaking in 2 mM sodium thiosulfate for 1 min. Wash the gel with ddH2O twice for 1 min and then stain with 50 ml of 12 mM silver nitrate containing 4 μl of 37% formaldehyde for 5–20 min. Rinse stained gels with ddH2O twice for 30 s and then develop with 0.4M sodium carbonate, 80 μM sodium thiosulfate, and 0.02% formaldehyde. To terminate the developing reaction, soak the gel in 5% acetic acid for 5 min, followed by extensive washing with ddH2O.
4. Large-Scale Expression of Heterologous GPCRs
Equipment
Fermentation system (BioFlo/CelliGen 115; New Brunswick Scientific, Edison, NY, USA) and centrifuges (JA-10 Beckman, Brea, CA, USA; Allegra 6KR Beckman).
Materials
Falcon tubes (50 ml), high growth medium (HGM; 2% peptone, 51 mM NaCl, 25 mM potassium phosphate buffer, pH 6.0, 5 μg/ml cholesterol, 1 mM CaCl2, 1 mM MgCl2, 2.5% agar) plates (150 mm) seeded with HB101 bacteria, S medium (for growing worms in solution; detailed protocol in Stiernagle, 2006), and protease inhibitor cocktail (Complete Mini, EDTA-free, Roche Branchburg, IN, USA).
Solutions
Sucrose (35% in M9 buffer), M9 buffer, resuspension buffer (20 mM HEPES, pH 7.4, 2 mM EDTA, supplemented with protease inhibitor cocktail), 1 M stock potassium phosphate buffer, pH 6.0 (108.3 g KH2PO4 and 35.6 g K2HPO4 dissolved in 1 l H2O).
The protocol for worm fermentation is described in Fabian and Johnson (1994). First, culture a selected transgenic integrated worm line on 25 150-mm HGM plates seeded with HB101 bacteria for two to three generations over 1–2 weeks and then transfer them into a fermenter in S medium at a final volume of 10 l. Culture worms for two generations over about 1 week in the fermenter (pH 7.0, 20 °C, 50% dissolved oxygen, 300 rpm agitation with a low shear pitched blade impeller) until most reach the young adult stage (Fig. 11.1). To harvest worms, centrifuge the liquid culture at 6000 × g in a JA-10 Beckman centrifuge for 15 min. Suspend the worm pellet in a minimum volume of S medium by vigorous shaking. Carefully load the resulting worm suspension (∼3 ml at a time) onto 30 ml of ice-cold 35% sucrose-containing M9 buffer in a 50-ml Falcon tube and centrifuge at 1000 × g in an Allegra 6KR Beckman centrifuge for 10 min at 4 °C. Carefully collect the top and interface layers containing live worms and dilute with an equal volume of ice-cold M9 buffer, followed by a 2500 × g centrifugation for 10 min (Allegra 6KR) to remove the sucrose. Wash worms again in M9 buffer and then resuspend the pellet in an equal volume of resuspending buffer. This worm preparation, defined as “wet worms,” can be used immediately for GPCR purification or frozen at −80 °C until needed.
5. Purification of Heterologous GPCRs
5.1. Membrane preparation and solubilization
Equipment
Microfluidizer (M-110Y microfluidizer processor; Microfluidics, Newton, MA, USA) and ultracentrifuge (Optima L-90K Ultracentrifuge Beckman).
Materials
Syringe filter (0.8 μm; Sterlitech, Kent, WA, USA), rotating platform, porcine pancreas phospholipase PLA2 (Sigma, St. Louis, MO, USA), n-dodecyl-β-D-maltopyranoside (DDM; Affymetrix Inc., Santa Clara, CA, USA), ligands 9-cis-retinal agonist for bovine opsin ((b)opsin; Toronto Research Chemicals, Toronto, ON, Canada) and ZM241385 antagonist for human β-adrenergic receptor ((h)A2AR; Tocris Bioscience, Ellisville, MO, USA), and protease inhibitor cocktail (Complete Mini, EDTA-free).
Solutions
Buffer A: 50 mM bis-tris-propane (BTP) buffer, pH 7.0, supplemented with protease inhibitor cocktail and buffer B: 50 mM BTP, pH 7.0, and 250 mM NaCl supplemented with protease inhibitor cocktail.
Thaw “wet worms” from the −80 °C stock and resuspend them in 1 volume of buffer A. Homogenize worms with microfluidizer (120 psi, four cycles). Centrifuge the homogenate at 100,000 × g for 1 h at 4 °C. Resuspend the resulting membrane pellet in buffer B to achieve the same volume as the original homogenate. Then incubate GPCR-containing membranes with a specific ligand for 1 h at 4 °C. Thus for the opsin preparation, incubate membranes in the dark with an excess of 9-cis-retinal agonist to obtain ground state recombinant bovine isorhodopsin (b)isoRho; for (h)A2AR, incubate membranes with an excess of the (h)A2AR antagonist, ZM241385. PNGase-F can be added at this point or later if degly-cosylation is required.
To solubilize the membrane pellet, incubate the membrane suspension with porcine pancreas phospholipase PLA2 (50 U 1 ml−1 of wet worms) and 1 mM CaCl2 for 30 min at 4 °C and then add DDM to a final concentration of 20 mM and rotate for at least 1–2 h at 4 °C. Separate unsolubilized material by centrifugation at 48,400 × g in an optima L-90K ultracentrifuge for 30 min at 4 °C. Clarify the resulting supernatant by passing it through a 0.8-μm filter.
5.2. Transgenic GPCR purification by 1D4-affinity chromatography
Equipment
Peristaltic pump, fraction collector, 1.5-ml capless graduated tubes (Fisher Scientific, Pittsburgh, PA, USA), chromatographic column ∼1 cm wide, and 50 or 100 kDa molecular weight cut-off AmiconUltra (Millipore, Billerica, MA, USA).
Materials
1D4 competing peptide (TETSQVAPA, ∼90% purity) and DDM.
Solutions
Equilibration buffer: 50 mM BTP, pH 7.0, 250 mM NaCl, and 1 mM DDM; elution buffer: 50 mM BTP, pH 7.0, 250 mM NaCl, and 1 mM DDM supplemented with 1 mg/ml of 1D4 peptide.
Equilibrate 1D4 resin bearing agarose-immobilized anti-rhodopsin 1D4 antibody prepared as described in Salom et al. (2012) with at least 5 column volumes of equilibration buffer. Incubate the solubilized protein extract with equilibrated 1D4 resin (5–10 μl of settled gel per milligram of starting “wet worms”) on a rotating platform for 1 h at 4 °C. Load the resulting gel onto a column and wash with at least 20 column volumes of equilibration buffer. Elute bound GPCR with elution buffer, collecting 200–300 μl fractions.
Tip
For more effective GPCR elution, load with 0.9 column volumes of the above buffer, close the column, and incubate for 1 h at 4°C, then elute protein from the resin. Check each fraction by SDS-PAGE gel electrophoresis, followed by either Coomassie Blue or silver staining (Fig. 11.2B) immunoblotting, and absorbance determination. Pool fractions containing purified GPCR and concentrate to ∼5–10 mg/ml with 50 or 100 kDa molecular weight cut-off AmiconUltra and subject the resulting protein to crystallization trials.
6. Determination of Transgenic GPCR Activity and Function
6.1. Light-dependent assay
Light response assays only test the functionality of photoreceptive heterologous GPCRs expressed in worms and regenerated with their agonist retinals, so these procedures are not applicable to other transgenic GPCRs. Because Rho is abundantly expressed and can be efficiently purified from native retina, heterologous expression creates the opportunity to produce and determine the structure of opsin mutants that cause visual disorders.
Because Rho and isoRho are sensitive to light, experiments must be performed in a dark room under dim light (Kodak two-way Safe-lamp with Kodak 1A Safelight Filter and 15 W bulb).
6.1.1 In vivo light response assay
Equipment
Macam L202 photometer (MacamPhotometers, Livingstone, UK) and a modified automated and quantitative analysis of behavior of nematode (AQUABN) system that includes: a Zeiss Stemi SV11-Apo microscope (Carl Zeiss, Oberkochen, Germany) mounted with a Kramer Universal Stereo Fluorescence Attachment and Cubes (USFAC) unit (Kramer Scientific, Amesbury, MA, USA), an Andor iXon DV897 electron multiplying charge-coupled device (EMCDD) camera (Andor, South Windsor, CT, USA), a ProScan II H117 motorized stage (Prior Scientific, Rockland, MA, USA), and an EXFO X-Cite 120PC-Q unit housing a metal halide short arc bulb (Lumen Dynamics, Mississauga, ON, Canada).
Materials
NGM plates seeded with OP50 bacteria, DMSO stock solutions of both 9-cis-retinal (10 mM) and all-trans-retinal (10 mM; Toronto Research Chemicals), and aluminum foil.
One day before each experiment, transfer L4 worms expressing (b)opsin to NGM plates seeded with OP50 bacteria that contain either 10 μM 9-cis-retinal or all-trans-retinal. Wrap these plates with aluminum foil and incubate them overnight at 20 °C. (After overnight culture, L4 worms become d1 young adults.) Perform light exposure/response experiments in a dark room at room temperature. A Zeiss Stemi SV11-Apo microscope mounted with a Kramer USFAC unit, an Andor iXon DV897 EMCDD camera, and ProScan II H117 motorized stage can be used together with a 1.6 × objective lens in combination with a 2.5 × magnifying lens to track worm motor behavior visualized under 7 lux of transmitted white light. For light response experiments, blue light (488 ± 20 nm) should be chosen as the stimulus because (b)isoRho has a maximum absorption at ∼485 nm and is sensitive to this wavelength. Moreover, blue light per se does not modify worm behavior (Fabian & Johnson, 1994). To start an experiment, transfer d1 worms with embedded platinum wires from NGM plates seeded with OP50 bacteria supplemented with 9-cis-retinal or all-trans-retinal to a tracking plate (unseeded NGM plate) and place the plate on the microscope stage. Upon changing conditions, worms crawl vigorously and then slow down. After 5 s of imaging, deliver 1000 lux of blue light (488±20 nm) to animals expressing (b)opsin in neurons from a metal halide short arc bulb housed in an EXFO X-Cite 120PC-Q unit through a Kramer USFAC for 1 s and image them continuously for 6 min. For tracking the behavior of worms expressing (b)opsin in muscles, image them for 2 min after transfer to the tracking plates, deliver 1500 lux of blue light (488±20 nm) and then image them continuously for 4 min. Worm locomotion, before and after illumination, should be recorded in AVI movies at 30 Hz. We developed a software package to capture images, control the onset and duration of illumination, and integrate this information. A previously published algorithm can be used to compute worm locomotion velocity (Zhang et al., 2011). The light intensity output of the EXFO unit must be calibrated to reach a targeted intensity (±5%) at the microscopic field as measured with a Macam L202 photometer. To score the extent of motor activity change, captured images can be analyzed with frame-by-frame, home-made software that constitutes part of AQUABN (Zhang et al., 2011). (Figure 11.3 shows the locomotion response of worms expressing (b)opsin in neurons under various conditions.)
Figure 11.3.
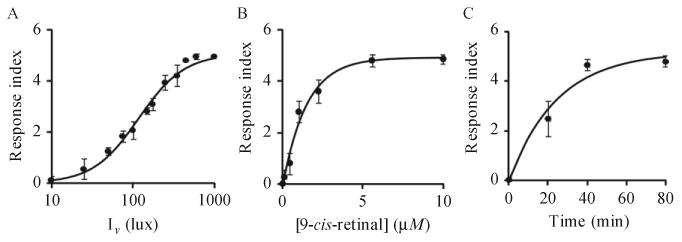
Light-associated motor responses of worms expressing (b)opsin in neurons. (A) Vigorously crawling d1 transgenic animals preincubated with 10 μM 9-cis-retinal and expressing (b)opsin in neurons were exposed to blue light (488±20 nm) for 1 s at indicated intensities and their motor responses were recorded and scored. (B) L4 transgenic animals expressing (b)opsin in neurons were preincubated overnight with 9-cis-retinal at indicated concentrations and then transferred onto OP50 bacteria-seeded plates. These vigorously crawling integrated transgenic animals then were exposed to blue light (1000 lux, 488±20 nm) (hv) for 1 s and their motor responses were scored. (C) Day 1 transgenic animals expressing (b)opsin in neurons were pretreated with 10 μM 9-cis-retinal for 20, 40, or 80 min. Their motor responses to blue light (1000 lux, 488±20 nm, 1 s) (hv) are shown. Figure adapted with permission from Cao et al. (2012), The Federation of American Societies for Experimental Biology.
6.1.2 UV–vis spectroscopy
Concentrations of purified (b)Rho or (b)isoRho can be quantified from knowledge of their absorption maximum and absorption coefficient values. The absorption coefficient for (b)Rho is 40.600 M−1 cm−1 (Matthews, Hubbard, Brown, &Wald, 1963) and for (b)isoRho regenerated with 9-cis retinal it is 43.000 M−1 cm−1 (Spalink, Reynolds, Rentzepis, pening, & Applebury, 1983). The A280/A500 absorbance ratio reflects Rho purity and a value 1.56 is the theoretical maximum for pure Rho. Samples with absorbance ratios up to 1.8 are generally suitable for structural studies. A detailed protocol for measuring Rho concentrations can be found in Matthews et al. (1963). Similarly, the A280/A485 theoretical maximum for (b)isoRho is 1.47, but samples with values up to 1.7 are usually suitable for structural studies.
6.1.3 G protein binding
Equipment
Perkin Elmer L55 luminescence spectrophotometer (Perkin Elmer, Wellesley, MA, USA) and a fiber optics light (Dolan Jenner Industries, Boxborough, MA, USA).
Materials
Band-pass 480–520 nm filter (Chroma Technology, Rockingham, VT, USA), quartz cuvette with stirring bar, and guanosine 5′-[γ-thio] triphosphate tetralithium salt (GTPγS) (Sigma–Aldrich).
Solutions
Gt binding buffer: 20 mM BTP, pH 7.0, 120 mM NaCl, 2 mM MgCl2, and 1 mM DDM.
The function of recombinant (b)isoRho purified from worms can be evaluated by the Gt (transducin) activation fluorescence assay (Farrens, Altenbach, Yang, Hubbell, & Khorana, 1996). Mix Gt purified by the protocol described in Goc et al. (2008) with purified (b)isoRho at concentrations of 25 nM and 250 nm in the Gt binding buffer. Load this mixture into the quartz cuvettes and expose them to light for 15 s from a 150 W fiber light covered with a band-pass filter (480–520 nm). Follow the reactions in a continuously stirred cuvette located in the spectrofluorometer at 20 °C. After 300 s of incubation, add GTPγS (5 μM final concentration) and record the fluorescence for the next 2100 s. The intrinsic tryptophan fluorescence increase emanating from Gtα can be quantified by using the excitation and emission wavelengths of 300 and 345 nm, respectively (Fig. 11.4). No change in tryptophan fluorescence should be detected in the control experiment without added GTPγS. Pseudo-first order kinetic rates (k) can be readily derived from the function A(t) = Amax(1 − exp−kt), where Amax is the maximal Gt fluorescence change and A(t) is the relative fluorescence change at time t.
Figure 11.4.
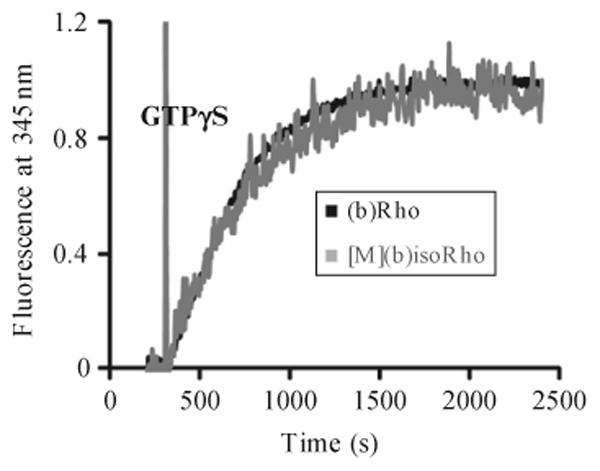
Gt activation by (b)Rho. Fluorescent assay of Gt activation by (b)Rho purified from bovine retina and (b)isoRho purified from transgenic worms expressing (b)opsin in muscles. Activation of Gt was monitored by the increase in intrinsic tryptophan fluorescence (λex = 300 nm, λem = 345 nm) due to nucleotide exchange catalyzed by Rho. GTPγS (5 μM) was added to initiate the reaction after the signal was recorded for 300 s. Figure adapted with permission from Salom et al. (2012), The Federation of American Societies for Experimental Biology.
6.2. Ligand-binding assays
A ligand-binding assay must be performed to determine if the GPCR heterologously expressed in worms is functional. This assay can be carried out either in live animals or in vitro, with crude membranes isolated from transgenic animals. For example, we expressed native human (h)A2AR in either C. elegans muscles or neurons because of its biomedical importance and available functional and structural data (Cao et al., 2012). Several transgenic worm lines expressing native (h)A2AR in muscles or neurons were generated and selected for further studies.
Equipment
Centrifuge (Allegra 6KR Beckman), 150 T ultrasonic dismembrator (Fisher Scientific), LS-6500 Beckman scintillation counter and automated and quantitative analysis of behavior of nematode (AQUABN) system to quantify motor behavior of C. elegans.
Materials
NGM plates (100 mm), GF/B glass microfiber filters (Whatman, Piscataway, NJ, USA), [3H]-CGS21680 (Perkin Elmer, Waltham, MA, USA), CGS21680 (Acros Organics, Morris Plains, NJ, USA), adenosine (Acros Organics), caffeine (Sigma–Aldrich), scintillation cocktail (Perkin Elmer), liquid NGM, and OP50 bacteria-seeded NGM plates.
Solutions
Buffer A: 25 mM HEPES, pH 7.4, 1 mM EDTA, and 2 mM MgCl2; buffer B: 25 mM HEPES, pH 7.4, 1 mM EDTA, 2 mM MgCl2, and 0.5% BSA.
6.2.1 In vivo ligand response assay
Transfer d1 adult worms from stock plates to OP50-seeded NGM tracking plates with or without ligands. Quantify locomotion speed of these animals for 10 min at 30 Hz with the AQUBAN system (Feng et al., 2006). Under these conditions, worms initially move vigorously and then enter a steady locomotion state after several minutes. To eliminate the acclimation phase, ignore the first 6 min of locomotion data and calculate the average speed from between 7 and 10 min as the worm locomotion velocity. Use worms that only express DsRed as controls. To prepare tracking plates containing ligand, dissolve adenosine (10 mM) or CGS21680 (27 μM) in NGM medium and add to NGM plates containing OP50 bacteria. Adenosine and CGS21680 enhance locomotion velocity in a dose-dependent manner in worms expressing human (h)A2AR in either muscles or neurons (Fig. 11.5 shows data for CGS21680).
Figure 11.5.
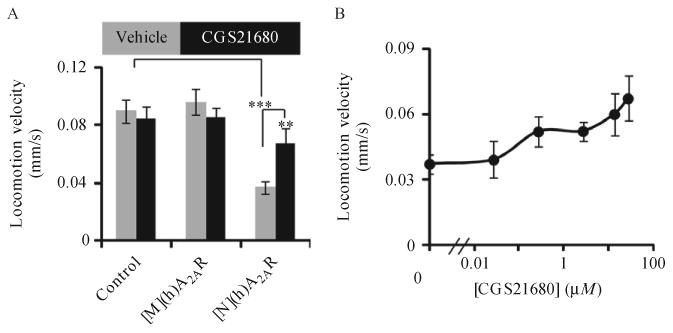
In vivo functional analysis of recombinant (h)A2AR. (A) Day 1 transgenic worms expressing DsRed only (control) or (h)A2AR in either neurons ([N](h)A2AR) or muscles ([M](h)A2AR) were transferred onto OP50-seeded NGM plates containing either the A2AR agonist CGS21680 or the control vehicle (Vehicle). Though the locomotion velocity of these animals was quantified for 10 min at 30 Hz, data from the first 6 min were discounted to eliminate the acclimation phase. Locomotion velocities, representing the average speed of animals from 7 to 10 min then were computed. (A) Bar graph showing locomotion velocities in response to 27 μM CGS21680. Error bars indicate S.E.M. **p<0.01, ***p<0.001, t-test, n=12 for the number of animals tested. (B) Locomotion velocities of transgenic worms expressing (h)A2AR in neurons in response to different concentrations of CGS21680. Figure adapted with permission from Salom et al. (2012), The Federation of American Societies for Experimental Biology.
6.2.2 Radioligand-binding assay
Collect d1 worms expressing a transgenic GPCR, for example, (h)A2AR, from eight 100 mm NGM plates. Wash worms with M9 buffer, followed by a 200 × g centrifugation for 5 min (Allegra 6KR Beckman). Resuspend the worm pellet (∼0.5 ml) in 20 ml of buffer A and sonicate on ice with a 150 T ultrasonic dismembrator (70% amplitude, six times for 30 s). Briefly centrifuge sonicated extract at 500 × g for 5 min to remove worm debris. Then centrifuge the supernatant at 48,000 × g for 20 min to pellet membranes. Discard the supernatant and resuspend membrane pellet in 500 μl of buffer A to obtain worm crude membranes.
Add 20 μl of (h)A2AR containing crude membranes to buffer B containing various concentrations of the isotopic exogenous agonist, [3H]-CGS21680, with or without competing ligand to reach a total volume 50 μl. Incubate the mixture for 1 h at room temperature. In a control experiment to check ligand-binding specificity, include 10 mM nonradioactive caffeine (a nonspecific ligand). Filter reaction mixtures through GF/B glass microfiber filters in a vacuum manifold, followed by multiple washes of the filters with 15 ml of cold buffer A. Next, soak the washed filters in 5 ml of scintillator cocktail and measure their radioactivity in a LS-6500 Beckman scintillation counter. Use obtained results to generate a 1-site-saturation curve with SigmaPlot 11 (SySat Software, Inc., San Jose, CA, USA) to determine Kd values.
7. Concluding Remarks
C. elegans is one of the best-studied multicellular organisms by state-of-the-art genetic methods. This nematode expresses 1100 GPCRs in chemosensory neurons to detect environmental stimuli. Yet C. elegans is one of the simplest organisms, with 302 neurons out of just 959 total somatic cells. Protein trafficking, folding, and degradation of misfolded proteins are essential for its survival. The power of this system can be harnessed for the production of membrane proteins.
Methods outlined in this chapter describe experiments that foster generation of recombinant GPCRs in C. elegans that serves as an excellent host for these important membrane proteins. C. elegans has a short reproductive life cycle (3.5 days at 20 °C), relatively simple genetics and survives cryostorage. Thus, it takes relatively little time to generate worm lines stably expressing heterologous GPCRs and only about 1 month starting from plate culturing of integrated transgenic worms to obtain “wet worms” from a fermenter. Such GPCR production is easily scalable, which is still not true of other systems. Moreover, transgenic GPCRs expressed in C. elegans exhibit limited N-glycosylation, which makes it easier to identify these proteins on immunoblots without the need for deglycosylation. Therefore, we believe this expression system can be expanded to other membrane proteins that are difficult to express in other available systems. This robust expression system is applicable not only to GPCRs but also likely to other eukaryotic membrane proteins as well.
Acknowledgments
We would like to thank Dr. L.T. Webster, Jr., and members of Palczewski's laboratory for critical comments on the manuscript.
We thank Dr. I. Katsura (The Graduate University for Advanced Studies, Mishima, Japan) for PH20 DNA and Mr. M.H. Zheng for technical assistance.
This research was supported in part by grants EY008061, EY009339, and P30 EY11373 (to K. P.) from the National Institutes of Health and Mt. Sinai Health Care Foundation Scholars Program in the Basic Science (to Z. F.). The work also received funding from a U54 award to the New York SGX Research Center for Structural Genomics (NYSGXRC) by the National Institute of General Medical Sciences (GM074945; PI: Stephen K. Burley) under a contract to Polgenix, Inc.. K. P. is John H. Hord Professor of Pharmacology.
References
- Cao P, Sun W, Kramp K, Zheng M, Salom D, Jastrzebska B, et al. Light-sensitive coupling of rhodopsin and melanopsin to Gi/o and Gq signal transduction in Caenorhabditis elegans. The FASEB Journal. 2012;26:480–491. doi: 10.1096/fj.11-197798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodevski I, Pluckthun A. Evolution of three human GPCRs for higher expression and stability. Journal of Molecular Biology. 2011;408:599–615. doi: 10.1016/j.jmb.2011.02.051. [DOI] [PubMed] [Google Scholar]
- Fabian TJ, Johnson TE. Production of age-synchronous mass cultures of Caenorhabditis elegans. Journal of Gerontology. 1994;49:B145–B156. doi: 10.1093/geronj/49.4.b145. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Feng Z, Li W, Ward A, Piggott BJ, Larkspur ER, Sternberg PW, et al. A C. elegans model of nicotine-dependent behavior: Regulation by TRP-family channels. Cell. 2006;127:621–633. doi: 10.1016/j.cell.2006.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Waterston RH. Proper expression of myosin genes in transgenic nematodes. The EMBO Journal. 1989;8:3419–3428. doi: 10.1002/j.1460-2075.1989.tb08506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goc A, Angel TE, Jastrzebska B, Wang B, Wintrode PL, Palczewski K. Different properties of the native and reconstituted heterotrimeric G protein transducin. Biochemistry. 2008;47:12409–12419. doi: 10.1021/bi8015444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk A, Schafer WR. Visualization of integral and peripherial cell surface proteins in live Caenorhabditis elegans. J Neurosci Methods. 2006;154(1–2):68–79. doi: 10.1016/j.jneumeth.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends in Pharmacological Sciences. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klammt C, Schwarz D, Dotsch V, Bernhard F. Cell-free production of integral membrane proteins on a preparative scale. Methods in Molecular Biology. 2007;375:57–78. doi: 10.1007/978-1-59745-388-2_3. [DOI] [PubMed] [Google Scholar]
- Matthews RG, Hubbard R, Brown PK, Wald G. Tautomeric forms of metarhodopsin. The Journal of General Physiology. 1963;47:215–240. doi: 10.1085/jgp.47.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Panneels V, Kock I, Krijnse-Locker J, Rezgaoui M, Sinning I. Drosophila photoreceptor cells exploited for the production of eukaryotic membrane proteins: Receptors, transporters and channels. PLoS One. 2011;6:e18478. doi: 10.1371/journal.pone.0018478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom D, Cao P, Sun W, Kramp K, Jastrzebska B, Jin H, et al. Heterologous expression of functional G-protein-coupled receptors in Caenorhabditis elegans. The FASEB Journal. 2012;26:492–502. doi: 10.1096/fj.11-197780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom D, Wu N, Sun W, Dong Z, Palczewski K, Jordan S, et al. Heterologous expression and purification of the serotonin type 4 receptor from transgenic mouse retina. Biochemistry. 2008;47:13296–13307. doi: 10.1021/bi8018527. [DOI] [PubMed] [Google Scholar]
- Shiroishi M, Kobayashi T, Ogasawara S, Tsujimoto H, Ikeda-Suno C, Iwata S, et al. Production of the stable human histamine H(1) receptor in Pichia pastoris for structural determination. Methods. 2011;55:281–286. doi: 10.1016/j.ymeth.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Spalink JD, Reynolds AH, Rentzepis PM, Sperling W, Applebury ML. Bathorhodopsin intermediates from 11-cis-rhodopsin and 9-cis-rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:1887–1891. doi: 10.1073/pnas.80.7.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D'Antona A, Fransen M, Xie G, Oprian DD, et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 2011;471:656–660. doi: 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. Maintenance of C. elegans. WormBook. 2006 Feb 11; doi: 10.1895/wormbook.1.101.1. ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe T, Suzuki N, Furukawa T, Ishihara T, Katsura I. Multidrug resistance-associated protein MRP-1 regulates dauer diapause by its export activity in Caenorhabditis elegans. Development (Cambridge, England) 2005;132:3197–3207. doi: 10.1242/dev.01909. [DOI] [PubMed] [Google Scholar]
- Zhang S, Jin W, Huang Y, Su W, Yang J, Feng Z. Profiling a Caenorhabditis elegans behavioral parametric dataset with a supervised K-means clustering algorithm identifies genetic networks regulating locomotion. Journal of Neuroscience Methods. 2011;197:315–323. doi: 10.1016/j.jneumeth.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Salom D, He J, Okun A, Ballesteros J, Palczewski K, et al. Expression of functional G protein-coupled receptors in photoreceptors of transgenic Xenopus laevis. Biochemistry. 2005;44:14509–14518. doi: 10.1021/bi051386z. [DOI] [PubMed] [Google Scholar]