

Abstract
Pseudomonas aeruginosa (PA) forms biofilms in lungs of cystic fibrosis CF) patients, a process regulated by quorum sensing molecules including N-(3-oxododecanoyl)-L-homoserine lactone, C12. C12 (10–100 μM) rapidly triggered events commonly associated with the intrinsic apoptotic pathway in JME (CFΔF508CFTR, nasal surface) epithelial cells: depolarization of mitochondrial (mito) membrane potential (Δψmito) and release of cytochrome C (cytoC) from mitos into cytosol and activation of caspases 3/7, 8 and 9. C12 also had novel effects on the endoplasmic reticulum (release of both Ca2+ and ER-targeted GFP and oxidized contents into the cytosol). Effects began within 5 minutes and were complete in 1–2 hrs. C12 caused similar activation of caspases and release of cytoC from mitos in Calu-3 (wtCFTR, bronchial gland) cells, showing that C12-triggered responses occurred similarly in different airway epithelial types. C12 had nearly identical effects on three key aspects of the apoptosis response (caspase 3/7, depolarization of Δψmito and reduction of redox potential in the ER) in JME and CFTR-corrected JME cells (adenoviral expression), showing that CFTR was likely not an important regulator of C12-triggered apoptosis in airway epithelia. Exposure of airway cultures to biofilms from PAO1wt caused depolarization of Δψmito and increases in Cacyto like 10–50 μM C12. In contrast, biofilms from PAO1ΔlasI (C12 deficient) had no effect, suggesting that C12 from P. aeruginosa biofilms may contribute to accumulation of apoptotic cells that cannot be cleared from CF lungs. A model to explain the effects of C12 is proposed.
INTRODUCTION
The gram negative bacterium Pseudomonas aeruginosa uses N-(3-oxododecanolyl)-L-homoserine lactone (C12), the product of the lasI gene, as a quorum-sensing molecule (Pearson et al, 1995; Wagner and Iglewski, 2008). When C12 accumulates in the P. aeruginosa-infected lungs, it permeates into neighboring bacteria, binds to lasR and contributes to the two-component lasI/lasR signaling-regulated gene expression pathways involved in bacterial adaptation to the cystic fibrosis (CF) lungs (Christensen et al, 2011). A critical effect of C12 is the activation of genes involved in the production of P. aeruginosa biofilms, communities of sessile cells encased in exopolysaccharide that provides protection against environmental insults, including antimicrobials and host immune responses (Kirisits and Parsek, 2006).
In addition to its gene regulatory effects in P. aeruginosa, C12 also affects multiple functions of airway and other host cells, a process termed inter-kingdom signaling (Pacheko and Sperandio, 2009; Davis et al, 2010). The C12 molecule stimulates airway epithelial cells to produce IL8 and other proinflammatory mediators (DiMango et al, 1995, Smith et al, 2001, Jahoor et al, 2008), but also inhibits LPS-stimulated NF-κB and proinflammatory cytokine responses in macrophages and airway epithelial cells (Kravchenko et al, 2006, 2008). C12 can also stimulate CFTR-dependent Cl− and fluid secretion in airway epithelia (Schwarzer et al, 2010), and it also damages or kills many host cells. C12 causes apparent degradation of tight junctions and loss of barrier function in the intestinal cell line CaCo-2 (Vikstrom et al, 2006, 2010) and triggers apoptosis in mast cells, fibroblasts, endothelia, macrophages and neutrophils (Shiner et al, 2006, Li et al, 2009, Tateda et al, 2003). In contrast, the ability of C12 to induce apoptosis in epithelia appears to depend on the particular cell type. C12 triggers apoptosis in mammary epithelial cells (Li et al, 2004), but not in the liver cell line Hep2 or in the lung epithelial cell line CCL185 (Tateda et al, 2003). C12 also caused apparently damaging changes in ultrastructure of mitochondria (mitos) and endoplasmic reticulum (ER) and killed primary cultures of airway epithelial cells (Kravchenko et al, 2006), but there were no tests of the role of apoptosis as opposed to some other type of cell death.
The first goal of the present studies was to determine whether synthetic C12 triggers apoptosis in the CF airway epithelial cell line (JME, homozygous for mutant ΔF508CFTR, nasal-derived). Previous data showed that C12 triggered changes in Ca2+ and cAMP signaling that began within a few mins, so we tested for C12 triggered apoptosis-related events (activation of caspases and changes of structure and function of both mitos and ER) over the course of a few mins up to four hrs. We performed a subset of these experiments on Calu-3 (wtCFTR, gland-like) cells to test whether the effects of C12 were applicable to other airway epithelia.
Second, we tested the role of CFTR in C12-triggered apoptosis by comparing time-course responses to C12 in JME cells and CFTR-corrected (adenoviral expression) JME cells. There have been a number of previous studies showing that apoptosis, triggered either by cycloheximide (Gottlieb and Dosanjh 1996), oxidative stress (Jungas et al, 2002, Boncoeur et al, 2006) or thapsigargin (Keribiriou et al, 2009), occurred to a greater extent in cells expressing wtCFTR compared to cells expressing ΔF508CFTR. These activators triggered apoptosis after 24–48 hrs. Another reason for testing CF vs. CFTR in the C12-triggered apoptosis arises from the finding that C12 increases Ca2+ responses to C12 in CF vs. CFTR-corrected cells (Mayer et al, 2011), and increases in cytosolic Ca2+ may trigger apoptosis.
A final goal of these studies was to determine whether effects observed with synthetic C12 were recapitulated upon exposure to intact P. aeruginosa biofilms. Although PA biofilms are likely to be important in CF pathophysiology, and C12 is produced by these biofilms (Williams and Camara, 2009; Charlton et al, 2000), it remains controversial whether C12 concentrations produced by biofilms are large enough to trigger the various inflammatory and apoptotic events observed using synthetic C12. For example, activation of inflammatory and apoptotic events by C12 requires 10 – 1000 μM, but CF sputum has been reported to contain C12 only in the low nM range of concentrations (Erickson et al, 2002, Chambers et al, 2005). In addition, there have been only a few tests of biofilms on airway cells. One approach has been to grow P. aeruginosa biofilms on coverglasses, and to then add macrophages or neutrophils to the biofilms (Van Gennip et al, 2009; Bjarnsholt et al, 2010; Christensen et al, 2011). In another approach, P. aeruginosa biofilms were grown on airway epithelial cell lines, and the biofilms grew better on CF than on CFTR-expressing airway epithelia (Anderson et al, 2008); this difference resulted from CF cells releasing more iron into the media (to support the bacterial biofilm) than the CFTR-expressing cells (Moreau-Marquis et al, 2008, 2009). These investigators also found that P aeruginosa biofilms killed CF airway epithelia, though the role of apoptosis in this cell killing was not tested (Anderson et al, 2008). Here we describe a different approach involving culture of P. aeruginosa biofilms on sterile semi-permeable nylon membranes on LB medium for 48 hours, prior to application to JME cells that had been grown separately. Responses to biofilms were compared to responses to synthetic C12. Comparisons were also made between PAO1wt and PAO1ΔlasI (C12 deficient) biofilms to test the role of C12 in PAO1 biofilm-activation of apoptosis in airway epithelia.
RESULTS
C12 activates caspases 3/7, 8 and 9 in airway epithelial cells
Caspases 3/7, 8 and 9 are commonly activated during apoptosis (Brenner and Mak, 2009; Mace and Riedl, 2010). We determined the time course of activation of caspases 3/7 in JME (nasal, ΔF508CFTR) cells in response to C12. As summarized in Fig. 1A, 50 μM C12 activated caspases 3/7, 8 and 9 in JME cells beginning within 20 mins, reaching a maximum and remaining constant from 60 mins up to 4 hrs. Control experiments were also performed to test DMSO and another P. aeruginosa quorum-sensing molecule butyryl homoserine lactone (C4). Neither DMSO (added in amounts equivalent to those in experiments with 50 μM C12) nor C4 (50 μM) activated caspase 3/7 compared to untreated controls after one or two hrs treatment. After one hr treatment, relative caspase activities were DMSO = 0.99 +/− 0.08 (p > 0.9), C4 = 0.95 +/− 0.06 (p > 0.5) and C12 = 1.61 +/− 0.03 (p < 0.05) (n = 3 expts); after two hrs treatment, relative caspase activities were DMSO = 1.04 +/− 0.07 (p > 0.6), C4 = 1.02 +/− 0.02 (p > 0.3) and C12 = 1.54 +/− 0.11 (p < 0.05) (n = 3 expts). Thus, there was no increase in caspase 3/7 activity with either DMSO or C4 at either one or two hrs, while C12 increased caspase 3/7 equivalently at these times. For comparison, we also tested staurosporine, a commonly used activator of apoptosis (Eckenrode et al, 2010). Staurosporine (50 μM) caused a similar activation of the caspases in JME cells (Fig. 1B). Similar experiments were performed on Calu-3 (gland, wtCFTR) cells to test for cell specificity of responses. C12 (50 μM) increased activities of caspases 3/7, 8 and 9 in Calu-3 cells after 2 hr (Fig. 2).
Fig. 1. Time dependent increase of caspases 3/7, 8 and 9 activities by C12.

JME and CalLu-3 cells were grown on 12-well plates and exposed to C12 or staurosporine (positive control), then processed for activities of caspase 3/7, 8 and 9. JME cells were left untreated or exposed to C12 (50 μM) (A) or staurosporine (50 μM) (B), and activities of caspases 3/7, 8 and 9 were measured after 0.5, 1, 2, and 4 hrs. C. Similar experiments were performed on Calu-3 cells exposed to C12 (50 μM) for 2 hrs. Activities of caspases 3/7, 8 and 9 were expressed relative to untreated controls. Avg +/− SD (n = 2–9 expts for all averages).
Fig. 2. C12 depolarizes Δψmito.

A. Confocal image (ex: 488 nm, em: 520–560 nm) of JC-1-stained JME cells shows punctate staining in cytosol characteristic of mitos in control, untreated cells. Nuclei appear as dark, nonfluorescent areas. B. Image of the same field after 10 mins with 50 μM C12. Punctate staining was replaced by diffuse staining, including in the nuclei. C. Time course of JC-1 fluorescence in the nucleus, which tracks loss of JC-1 from the mitos resulting from depolarization of Δψmito. JC-1 signal was normalized by setting initial value in the nucleus as zero and value after FCCP (10 μM) as 100%. D. Summary of effects of C12 (50 μM, 20 min) and FCCP (10 μM, 2 min) on change in nuclear JC-1 fluorescence (ΔJC-1) in JME cells. Avg +/−SD (n = 6 expts, >10 cells in each experiment)..
C12 causes rapid depolarization of mitochondrial membrane potential and slower release of cytochrome C in JME cells
Some proapoptotic drugs cause rapid depolarization of mito membrane potential (Δψmito) as a critical step in triggering apoptosis (Wang, et al, 2000; Degterev et al, 2001; Milanesi et al, 2006). When JME cells were loaded with JC-1 (a cationic dye that selectively accumulates in mitos and indicates Δψmito) and examined by confocal microscopopy, the dye was localized to puncta throughout the cytosol, but not in the nucleus (Fig. 2A). When these cells were then treated with C12 (50 μM), the punctate appearance gradually disappeared as the dye was lost from the mitos and diffused into the cytosol and nucleus (Fig. 2B). Quantitation of the time course of changes in JC-1 fluorescence in the nucleus showed that 50 μM C12 caused a rapid, small increase in JC-1 signal followed by a slower, larger increase during the next 20 mins. The rate and magnitude of this depolarization response depended on cell density, with faster and larger responses occurring in sparsely grown cells and slower and smaller responses in densely grown cells (as in Fig. 2C). These quantitiative differences likely reflected the reduced accessibility of C12 to key cellular sites in densely grown cells. Treatment with the protonophore FCCP (10 μM) to totally depolarize Δψmito caused a further, rapid increase in JC-1 signal (Fig. 2C). In this set of experiments C12 caused JC-1 signal to increase over the course of 20 mins to about 40% of maximal (Fig 2D). There were no effects of adding either DMSO or 50 μM C4 on JC-1 fluorescence (data not shown). These results indicated that C12 caused selective effects to depolarize Δψmito.
Release of cytochrome C (cytoC) from mitos into the cytosol is common to both intrinsic and extrinsic pathways to apoptosis (Ow et al, 2008). This possibility was tested using immunofluorescence in JME cells, which were left untreated or treated with 50 μM C12 for one hr followed by staining. Control cells showed punctate staining characteristic of mitos and distinct nuclei that lacked staining (Fig. 3A), while cells treated with C12 (50 μM) for 1 hr showed diffuse cytoC staining along with marked decreases in size of cells and nuclei and increases in cytoC-filled plasma membrane blebs (Fig. 3B). Similar experiments were performed on Calu-3 cells mounted in Ussing chambers so that C12 (50 μM) could be added selectively to the apical surface, as would occur in vivo. Control cells showed punctate cytoC staining (Fig. 3C). After one hr of C12 (50 μM), many cells, usually in groups, lost puncta, and there was an increase in diffuse staining throughout the cytosol and nucleus, consistent with release of cytoC from mitos into the cytosol and nuclei (Fig. 3D). These results showed that C12 caused cytoC to be released from mitos into the cytosol in both JME and Calu-3 cells within 1 hr.
Fig. 3. C12 causes mitos to release cytoC into cytosol.

Immunofluorescence of cytoC of JME (A, B) and Calu-3 (C, D) cells under control conditions (A, C) and after treatment with 50 μM C12 for 1 hr (B, D). Results typical of six similar experiments for each.
Effects of C12 on ER: rapid release of Ca2+ and slower release of roGFP and oxidized contents into the cytosol of JME cells
Previous experiments on Calu-3 cells showed that C12 caused slow increases in cytosolic [Ca2+] (Cacyto) by activating the IP3 receptor to release Ca2+ from the thapsigargin-sensitive store in the ER (Schwarzer et al, 2010). Similar experiments were performed on JME cells for comparison to these previous data and also to compare to later experiments with biofilms. As shown in Fig. 4A, 1 μM C12 did not affect Cacyto, while 10 μM C12 caused a small increase, and 100 μM C12 caused a large, transient increase in Cacyto; following 50 or 100 μM C12 there was no effect of thapsigargin (Fig. 4A). In contrast, when cells were treated first with thapsigargin, there was a larger, initial increase in Cacyto, and no further effect of C12 (Fig. 4B). These results were similar to those reported previously for Calu-3 cells (Schwarzer et al, 2010) and indicated that 50–100 μM C12 emptied the ER store of Ca2+.
Fig. 4. C12 increases Cacyto.

Fura-2-loaded JME cells were treated with C12 1, 10 and 100 μM followed by thapsigargin (10 μM, positive control for release from ER) (A) or with thapsigargin followed by 1, 10 and 100 μM C12 (B). Traces show average Cacyto for 10–20 cells, and are typical of four similar experiments.
Kravchenko et al (2006) previously showed that C12 caused the ER of human bronchial epithelial cells to become dilated. Recent experiments on mouse embryonic fibroblasts showed that ER stress activated by thapsigargin or tunicamycin increased the permeability, leading to the loss of GFP and other large proteins (60 kDa) from the ER into the cytosol (Wang et al, 2011). This thapsigargin-triggered ER leakage of proteins began after about 10 hrs, but once it began it was completed within about 30 mins. Since C12 triggered both loss of Ca2+ from the ER (Schwarzer et al, 2010) and also ER stress (Kravchenko et al, 2008; C. Schwarzer and T.E. Machen, unpublished data) in airway epithelia, it seemed possible that C12 might cause similar loss of ER proteins into the cytosol. We used ER-targeted redox-sensitive roGFP to test this possibility. JME cells expressing ER-targeted roGFP were grown on coverslips and mounted in either the confocal or widefield imaging microscopes. Under control conditions, ER-targeted roGFP exhibited a typical reticular appearance, and there was little or no ER-localized roGFP apparent in the nucleus (Fig. 5A). When these cells were exposed to C12, the ER began to condense and fragment after 30–45 mins, similar to the effects of thapsigargin (Ribeiro et al, 2000, Subramanian et al, 1997, Wang et al, 2010). C12 also caused release of ER-targeted roGFP, which was seen most easily as an increase in fluorescence in confocal sections through the middle of the nucleus. Beginning after 30–45 mins, ER-targeted roGFP started to appear in the nucleus of some cells (Fig. 5B). The apparent release of roGFP from the ER and diffusion into the cytosol and nucleus continued in those cells and became apparent in most cells after 60–90 mins (Fig. 5C).
Fig. 5. C12 causes release of ER-targeted roGFP into the cytosol.
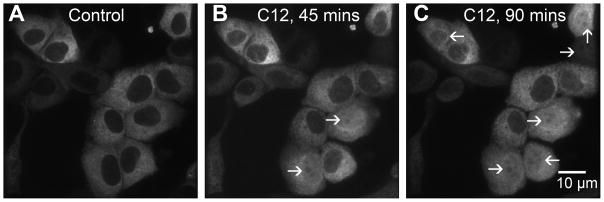
Cells were transfected with ER-targeted roGFP and observed in the confocal microscope under control conditions (A) and after treatment with C12 (50 μM) for 45 (B) and 90 mins (C). Cells in which roGFP had leaked from the ER and diffused into the cytosol and nucleus are shown by arrows. Results typical of >10 similar.
We used the redox-sensitivity of roGFP and the oxidized vs. reduced properties of the ER and cytosol, respectively, to quantitate leakage of ER contents into the cytosol. Results from a typical experiment are shown in Fig. 6A. In the control condition, the ER exhibited a characteristically oxidized redox potential (redoxer = −230 mV), while the cytosol was reduced (redoxcyto = −310 mV), similar to values reported previously (Schwarzer et al, 2007). C12 caused, after a delay, ER-roGFP to become more reduced while cytosol-roGFP became more oxidized. These changes in redoxer and redoxcyto occasionally began before there was noticeable leakage of ER-targeted roGFP into the cytosol and nucleus. A summary of these experiments is shown in Fig. 6B. C12 caused redox potential measured with the ER-targeted probe to become reduced and cytosol-targeted probe to become oxidized, and the two probes measured nearly equal redox potentials (approx. −290 mV). These data were consistent with equilibration of redox of the two compartments.
Fig. 6. C12 causes reduction of redoxer and oxidation of redoxcyto.

JME cells were transfected separately with ER-targeted and cytosolic roGFP, and cells were plated onto one coverslip for measurement of redoxer and redoxcyto, as described previously (Schwarzer 2007). As shown by the solid line, C12 (10 μM) caused, after a delay of about 30 mins, redoxer to become reduced and redoxcyto to become oxidized; after about 60 mins, redoxer and redoxcyto reached similar values. Data show the time course of responses to C12 for redoxcyto and redoxer from five cells each. Average values (+/− SD) for redoxer (triangles) and redoxcyto (squares) are shown for control cells in the steady state and then after 1 hr treatment with C12 (10 or 50 μM). Avg +/− SD, n = 6 expts, 47 cellular measurements of redoxcyto and 67 cellular measurements of redoxer.
Effects of C12 on caspase, Δψmito and redoxer in JME and CFTR-corrected JME cells
Data to this point showed that C12 triggered apoptosis in JME (ΔF508CFTR) cells and that similar activation of caspases and release of cytoC occurred in Calu-3 (wtCFTR) cells. This indicated that CFTR was likely not an important regulator of C12-triggered apoptosis. We performed direct tests of the role of CFTR by comparing responses in JME cells that were treated with adenoviral vectors expressing lacZ or CFTR cDNA. As shown previously (Hybiske et al, 2007), these adenoviruses infect approximately 90% of JME cells. The adv-lacZ and adv-CFTR-infected cells were used for comparisons to quantitate caspase activation, depolarization of Δψmito, and permeabilization of the ER as measured by reduction of redoxer.
As shown in Fig. 7A, caspase 3/7 activities in untreated JME-lacZ and JME-CFTR cells were very similar in control, untreated cells and then during 20 mins, 60 mins and 120 mins treatment with 50 μM C12. The somewhat different rates of C12-triggered apoptosis in these experiments (Fig. 7A) compared to those reported in Fig. 1A likely reflect slightly different cell densities and, therefore, different access of C12 to the cells. Although there was a statistically larger activation of caspase 3/7 at time zero and after 60 mins of C12 in JME-CFTR compared to JME-lacZ cells, the difference was only about 10%, and there were no differences in caspase activation at either 20 or 120 mins. C12 also caused the same rates of depolarization of Δψmito (Fig. 7B). The larger and faster depolarization of Δψmito in these experiments (i.e., compared to responses in Fig. 2) likely resulted solely from the different cell densities in the two sets of experiments.
Fig. 7. Similar effects of C12 on caspase (A), Δψmito (B) and redoxer (C) in JME and CFTR-corrected JME cells.

5JME cells expressing lacZ or CFTR (adenovirus infections) were grown on coverglasses or well plates and exposed to C12 (50 μM) for times to 2 hrs. A. Activation of caspase 3/7 by 50 μM C12 was measured in JME-lacZ and JME-CFTR cells at t = 0, 20, 60 and 120 mins. Avg +/− SD (n = 3) of caspase activity measured in RLU at each time point. * p < 0.05. Other points were insignificantly different from each other. B. Time course of effect of C12 on Δψmito. JME-lacZ and JME-CFTR cells were loaded with JC-1, washed and then exposed to C12 (50 μM, arrow). Nuclear fluorescence (expressed as percentage of maximal JC-1 fluorescence observed during treatment with 10 μM FCCP at the end of the experiment) increased rapidly in both JME-lacZ and JME-CFTR cells. Inset shows average rates (+/− SD, n = 3) of C12-triggered decrease of JC-1 fluorescence in JME-lacZ and JME-CFTR cells. There was no difference in these rates (p > 0.40). C. Time course of effect of C12 on redoxer. JME-lacZ and JME-CFTR cells that had been transfected with ER-roGFP were exposed to C12 (50 μM, arrows). C12 caused the ER redox potential (resulting from reduction of oxidized environment experienced by ER-roGFP) to become reduced at the same rate in JME-lacZ and JME-CFTR cells. The strong reductant DTT (1 mM) was added as shown to induce maximal reduction of the ER-roGFP ratio. Inset shows average rates (+/− SD, n = 3) of C12-triggered decrease of ER-roGFP fluorescence ratio in JME-lacZ and JME-CFTR cells. There was no difference in these rates (p > 0.09).
Finally, C12 caused equivalent reduction of redoxer resulting from leakage of roGFP from the ER into the cytosol (Fig. 7C) in JME-lacZ and JME-CFTR cells.
Overall, these comparisons of caspase activation, Δψmito depolarization and reduction of redoxer showed that CFTR played no significant role in controlling C12-activated apoptosis events in CF (JME+LacZ) and CFTR-corrected CF cells (JME+CFTR).
PAO1wt but not PAO1lasI biofilms trigger C12-like changes in Δψmito and Cacyto in JME cells
As shown in Fig. 8A, exposure of JME cells to established PAO1wt biofilms caused JC-1 fluorescence measured over the nucleus to increase up to a plateau; further increase in JC-1 fluorescence occurred following FCCP. These results were similar to the effects of adding 50 μM C12 to JME cells (Figs. 2 and 7). In contrast, PAO1ΔlasI biofilms had no detectable effect on JC-1 fluorescence, while further addition of PAO1wt caused maximal increase in JC-1 signal, i.e., there was no further effect of FCCP (Fig. 8B). These data were consistent with the idea that C12 in PAO1 biofilms triggered depolarization of Δψmito in JME cells. The data were also consistent with the idea that some other factor produced in biofilms may synergize with C12 to trigger maximal depolarization of Δψmito in JME cells.
Fig. 8. PAO1wt vs. PAO1lasI biofilms on Δψmito in JC-1-loaded JME cells.

Biofilms were prepared by vortexing one biofilm-covered mesh into 500 μl of Ringer’s, and then adding 10 μL of this suspension to the 100 μl of Ringer’s bathing the JC-1-loaded JME cells. Fluorescence (ex: 490 nm, em: 520–560 nm) was measured over nuclei. A. PAO1wt biofilm caused immediate increase of JC-1 fluorescence, and this was increased further by FCCP (10 μM). B. PAO1ΔlasI biofilm did not affect JC-1 fluorescence, but further addition of PAO1wt biofilm caused a large increase in JC-1 fluorescence that was not affected further by FCCP (10 μM). Traces show average JC-1 fluorescence for >10 cells. Results typical of three similar experiments for both PAO1wt and PAO1lasI.
Similar experiments were performed to test biofilms on Cacyto in fura-2-loaded JME cells. PAO1wt biofilms caused small, slow increases in fura-2 ratio (Fig. 9A), while PAO1ΔlasI biofilms caused a decrease or had no effect on fura-2 ratio (Fig. 9B). The small changes triggered by PAO1wt biofilms were roughly equal to the effects of 10 μM C12 (Fig. 4A).
Fig. 9. PAO1wt vs. PAO1lasI biofilms on Cacyto in fura-2-loaded JME cells.

Biofilms were prepared by vortexing one biofilm-covered mesh into 500 μl of Ringer’s, and then adding 10 μL of this suspension to the 100 μl of Ringer’s bathing the fura-2-loaded JME cells. Fluorescence ratios (ex: 350/380 nm, em: >510 nm) were measured and expressed relative to maximum ratios measured at the end of the experiment. A. PAO1wt biofilm caused immediate increase of fura-2 ratio. B. PAO1ΔlasI biofilm caused no increase of fura-2 ratio. Traces show average fura-2 ratio for >10 cells. Results typical of four similar experiments for both PAO1wt and PAO1lasI.
DISCUSSION
C12 triggers apoptosis; early events likely begin in the mitochondria and ER
A major conclusion from this study is that, in addition to causing disruption of structures of ER and mitos and loss of cell viability (Kravchenko et al, 2006; Shiner et al, 2006), C12 rapidly triggers events associated with the intrinsic pathway leading to apoptosis: depolarization of Δψmito and release of cytoC from mitos into the cytosol; activation of caspases 3/7, 8 and 9; blebbing of plasma membranes; cell shrinkage; and condensation of nuclei. Activation of the caspases occurred similarly to that stimulated by staurosporine, the better characterized trigger of apoptosis (Eckenrode et al, 2010). In addition, C12 also elicited changes in the ER: rapid release of Ca2+ followed by condensation/fragmentation and leakage of roGFP and oxidized contents into the cytosol. The oxidation of the cytosol and reduction of the ER that accompanied this leakage of ER contents into the cytosol appeared often to occur before roGFP began leaking into the cytosol, indicating that C12-triggered increases in ER permeability occurred gradually. The role of cytosolic oxidation and leakage of large ER proteins into the cytosol in subsequent apoptotic steps are unknown, but could, as proposed previously (Wang et al, 2011), provide a point of no return in apoptosis, similar to release of cytoC from mitos. Previous work (Schwarzer et al, 2010) showed that C12 did not cause cameleon D1 (60 kDa) to leak from the ER (Schwarzer et al, 2010), while roGFP (28 kDa) consistently leaked from the ER into the cytosol, consistent with data showing that thapsigargin-triggered permeabilization of the ER had a size cut off of 62 kDa (Wang et al, 2011).
Although still speculative, the effects of C12 on ER and mitos combined with previous observations may provide a hint as to the molecular mechanism of C12 action. Previous work showed that Bax and Bak inhibit (Oakes et al, 2005) and Bcl-2 and Bcl-xl stimulate the IP3R (White et al, 2005; Oakes et al, 2005, Palmer et al, 2004). Bcl-2 and Bcl-xl also inhibit the proapoptotic actions of Bax and Bak (Chipuk and Greeen 2008), which, upon oligomerization, form large pores (Mikhailov et al, 2003; Perkins et al, 2009). Bax and Bak appear also to control Δψmito (Wang, et al, 2000; Degterev et al, 2001; Milanesi et al, 2006). Our current working hypothesis is that C12 disrupts interactions between Bax/Bak and Bcl-2/Bcl-xl in both ER and mitos, freeing up Bcl-2 and Bcl-xl to interact with and activate IP3R while also leading to oligomerization of Bax and Bak to permit loss of cytoC from mitos and roGFP from the ER into the cytosol. This model would explain C12-induced rapid increases in Cacyto and depolarization of Δψmito and slower increases in permeability of both the ER and mitos to proteins. Downstream activation of caspases would follow release of cytoC from mitos and of large proteins and oxidized contents from the ER into the cytosol. This model is, of course, still a working hypothesis that does not fit all the data, e.g. differences in Bax/Bak expression have been observed in CF vs CFTR-corrected cells (Jungas et al, 2002).
C12 and biofilms in CF
Our experiments showed that key events associated with C12-triggered apoptosis were unaffected by the expression of CFTR in JME cells: activation of caspase 3/7, depolarization of Δψmito and reduction of redoxer. We conclude that C12-triggered apoptosis occurs similarly in CF and nonCF airway epithelia. Because C12 triggers cAMP production and activation of CFTR (Schwarzer 2010), it is likely that CFTR will have been active in the JME-CFTR cells but not in the JME-lacZ cells during exposure to C12 and activation of apoptosis. In contrast, previous studies have found that wtCFTR expression decreases apoptosis in multiple cell types (Gottlieb and Dosanjh 1996; Jungas et al, 2002; Boncoeur et al, 2006; Keribiriou et al, 2009). A possible explanation for these different results is that protection by CFTR is irrelevant during the rapid activation of apoptosis triggered by C12, while activators like thapsigargin, oxidizers and cycloheximide that take longer to work (1–2 days) are inhibited by CFTR. Related possibilities are that C12 has unique proapoptotic effects (e.g., release of ER contents and oxidation of the cytosol or Bax/Bak/Bcl) not regulated by CFTR. Although CFTR appears not to play a role in regulating C12-triggered apoptosis, CFTR may play a protective role against P. aeruginosa, arising from its preventing P. aeruginosa infections and biofilm formation: both flagellin and C12 released from P. aeruginosa will activate CFTR-dependent Cl− and fluid secretion into the airways that may facilitate bacterial flushing (Illek et al, 2008). In CF individuals this P. aeruginosa-induced flushing will be missing so bacteria will accumulate in the airway surface liquid, leading eventually to biofilm formation.
Application of PAO1 biofilms to JME cells showed that the biofilms elicited lasI-dependent effects on Cacyto and Δψmito that were similar to effects of 10–50 μM C12. The Cacyto response triggered by PAO1wt biofilm was similar to that elicited by 10 μM C12, while the Δψmito response was similar to that activated by 50 μM C12. These data were consistent with the idea that C12 in PAO1wt biofilms was sufficient to trigger apoptosis-related events in airway epithelial cells, though it is also possible that other differences between PAO1wt and PAO1lasI biofilms contributed to the Cacyto and Δψmito responses. 10–50 μM is higher than previous chemical measurements of [C12] in CF sputum (Erickson et al, 2002; Chambers et al, 2005), indicating that [C12] may be higher in biofilms formed in CF airway mucus, as proposed by Bjarnsholt and Givskov (2007). These higher [C12] could trigger apoptosis in epithelial cells, macrophages and neutrophils in biofilm-infested airways. Such effects could contribute to CF pathogenesis by both increasing bacterial binding and retention in the airways (Lee et al, 1999) and also altering the normal balance between apoptosis of neutrophils and their phagocytic removal by macrophages.
MATERIALS AND METHODS
Reagents
Unless otherwise specified, all reagents and chemicals were obtained from Sigma. C12 and staurosporine were dissolved in DMSO and frozen in separate vials and then thawed for single experiments. The cAMP-elevating agonist forskolin (Calbiochem) was prepared as a 20 mM stock solution in DMSO, and an aliquot was added at final concentrations of 2–50 μM. CFTR blocker CFTRinh172 was provided by Dr. Alan Verkman (University of California, San Francisco), prepared as a 20 mM stock solution in DMSO, and added to solutions at concentrations noted in the text. The Ca2+-ATPase blocker thapsigargin was prepared as a 1 mM stock in DMSO and used at 1–5 μM.
Epithelial cell culture
Cystic fibrosis airway cells JME/CF15, a continuous SV40 large T antigen-transformed nasal epithelial cell line homozygous for ΔF508 CFTR (Jefferson et al, 1990; Hybiske et al, 2007), were cultured in DMEM:F12 media containing 10% FBS, 2mM L-glutamine, 1% Pen/Strep, 10ng/ml EGF, μM hydrocortisone, 5μg/ml insulin, 5μg/ml transferrin, 30nM triiodothyronine, 180μM adenine and 5.5μM epinephrine. For some experiments, cells were passaged at 1:2 dilution, and the remaining cell suspension was seeded directly onto a 24-well or 12-well tissue culture plate (BD Falcon, Bedford, MA), onto a 96-well plate for caspase assays or onto coverglasses for imaging. Calu-3 cells, a human airway gland epithelial cell line of adenocarcinoma origin and characterized by high levels of endogenous CFTR expression (Haws et al, 1994; Shen et al, 1994), were cultured in ATCC Eagle’s minimum essential medium supplemented with 10% FBS, and 1% penicillin-streptomycin. Cells were grown on culture plates for caspase assays or seeded onto Snapwell inserts (0.4-um pore size) and then grown until cells formed confluent monolayers (transepithelial resistance > 500 ohm.cm2). During experiments, cells were incubated in Ringer’s solution containing (in mM): 145 mM NaCl, 2 mM KCl, 1.5 mM K2HPO4, 1 mM MgSO4, 10 mM HEPES, 2 mM CaCl2 and 10 mM glucose.
Adenovirus expression of lacZ or CFTR in JME cells
JME cells were split and plated on coverglasses or on culture plates for caspase assays, then treated with adenovirus (adv, 100 MOI) to express either lacZ or wtCFTR, as described previously (Hybiske et al, 2007). Cells were incubated with adv for a day, then cells were washed to remove adv and allowed to grow for two more days before being used for experiments. As shown previously, this procedure elicited expression in approximately 90% of the cells (Hybiske et al, 2007).
Immunofluorescence of cytochrome C
JME or Calu-3 cells cells grown on coverglasses or filters as described in the text were left untreated or incubated with C12 (50 μM) for times mentioned in the text; cells were then rinsed three times with PBS, fixed for 5 min with 3.7% formaldehyde in PBS, rinsed with PBS and permeabilized with 0.5% Triton X-100 (in PBS) for 15 min. After blocking with 1% BSA-PBS for 20 min, cells were incubated for 1 hr at room temperature with an anti-cytochrome C antibody (Santa Cruz Biotech), followed with rinses with 1% BSA-PBS. Finally, cells were incubated for 1 hr with an Alexa546-anti-rabbit secondary antibody (Molecular Probes, Eugene, OR). Images were obtained using standard widefield immunofluorescence imaging microcope (Nikon, Diaphot) or an inverted microscope (Nikon T2000) with a spinning disk confocal attachment (Solamere, Salt Lake City, UT).
Measuring Cacyto and Δψmito using fluorescent probes and imaging microscopy
For imaging experiments to measure cytosolic [Ca] (Cacyto) or mito membrane potential (Δψmito), cells were incubated with growth media containing either the Ca-sensitive probe fura-2/AM (2–5 μM) for 40–60 mins or Δψmito probe JC-1 (10 μM) for 10 mins at room temperature, and then washed three times with Ringer’s solution to remove the extra dye. Dye-loaded cells were mounted onto a chamber on the stage of either a wide field imaging microscope or a spinning disk confocal imaging microscope. Cells were maintained at room temperature during the experiment. Treatments with agonists were made by diluting stock solutions into Ringer’s solution at the concentrations stated in the text.
Fluorescence ratio imaging measurements of Cacyto or Δψmito were performed using equipment and methods reported previously (Fu et al., 2007; Hybiske et al., 2007; Schwarzer et al., 2007). Briefly, a Nikon Diaphot inverted microscope was used with a 40× Neofluar objective (1.4 NA) and a computer-controlled filter wheel (Lambda-10, Sutter Instruments, Novato, CA) that provided selective excitation and emission. A CCD camera collected emission images. For fura-2, excitation wavelengths were 350 ± 5 nm and 380 ± 5 nm, and emission was >510 nm. For JC-1, excitation was either 488 nm or 490 ± 10 nm and emission was 520–560 nm. JC-1 emission was quantitated from measurements of the nuclear region (observed as region with little or no JC-1 fluorescence). Axon Imaging Workbench 4.0 (Axon Instruments, Foster City, CA) controlled filters and collection of data. All images were corrected for background (region without cells). Data have been reported as relative ratios. For fura-2, maximum and minimum ratios were generated at the end of each experiment by treating cells with ionomycin (10 μM) + thapsigargin (5 μM) in solutions containing zero Ca2+ (+ 1 mM EGTA) or 10 mM Ca2+. JC-1 ratios associated with maximal depolarization of Δψmito were generated at the end of the experiment by treating cells with 10 μM FCCP.
ER morphology and redox potentials of the ER and cytosol
JME cells were transfected with a plasmid encoding redox-sensitive GFP (roGFP) targeted to the ER lumen or to the cytosol to measure redox potentials in the ER or cytosol (redoxer or redoxcyto). In some experiments, one batch of cells was transfected with cytosol-roGFP and another batch with ER-roGFP, and these cells were then grown on one coverglass to permit imaging both organelles at once. Confocal imaging to determine cellular and organelle morphology was performed using the spinning disk attachment and 488 nm laser (520–560 emission) and recorded using a CCD camera (Hamamatsu Orca) and in-house software. Ratiometric imaging of roGFP was performed using the widefield imagimg microscope described above. Cells were alternately excited at 385±5 nm and 474±5 nm, and emission (>510 nm) images wee collected and analyzed. Images were background subtracted, and normalized data were calibrated. At the end of experiments, 385/474 ratios were recorded during maximal oxidation (with 10 mM H2O2) and maximal reduction (10 mM DTT), and these were used with a previously determined in situ calibration curve to calculate redox potentials in the cytosol and ER (redoxcyto, redoxer) (Schwarzer et al, 2007).
Assays of caspases 3/7, 8 and 9
Caspase activities were measured by cell-based homogeneous luminescent assays for caspases 3/7, 8 and 9 (Caspase-Glo, Promega, Madison, WI), in which specific substrates that contain the tetrapeptide (DEVD for caspase 3/7, LETD for caspase 8 and LEHD for caspase 9) were cleaved by the activated caspases from the cells to release aminoluciferin reacting with the luciferase and resulting in the production of light. Calu-3 or JME cells were plated on a clear-bottom, white 96-well plate in 100 μl media per well for 4–5 days until they were confluent. During the experiment, cells were treated with different drugs in 37°C incubator for 20–240 mins, depending on the experiment, or were left untreated as controls. On the same plate, some wells without cells but 100 μl the same media served as blanks. After treatment, 100 μl reagent was added to each well with cells (treated or controls) and their media, or blank (media only). The plate was incubated at room temperature for 1 hour on a shaker, and the end-point luminescence was measured in a plate-reading luminometer (LmaxII 384, Molecular Devices, Sunnyvale, CA). Data were background (blank) subtracted and averaged.
Pseudomonas aerguinosa biofilms
P. aeruginosa biofilms are cultivated on sterile Hybond nylon membranes (1 cm2) placed onto LB plates and inoculated with an overnight culture of PAO1 wt or PAO1ΔlasI diluted to an OD600 of 0.1. Agar plates were then incubated at 37°C for 48 hrs (Lynch et al, 2007) to establish biofilms. As measured from colony counts, the biofilms grew to equivalent densities, PAO1: 4.87±0.23×1010 CFU/biofilm and PAO1ΔlasI: 4.27±0.38×1010 CFU/biofilm (p > 0.2). P. aeruginosa biofilms (PAO1 and PAO1ΔlasI) were applied to JME cells grown separately by either inverting the nylon membrane onto the cells or by vortexing the biofilms into Ringer’s solution (500 μl) and then adding 10 μl aliquots of this vortexed biofilm to the 100 μl bathing the epithelial cells. Experiments measured Cacyto and ψmito of confluent JME cells during exposure to PAO1 and PAO1ΔlasI biofilms. Results with the two methods were similar.
Acknowledgments
We thank Stanley Yen and Stacey Shuai for performing electrophysiology and caspase assays. This work was supported by grants from the Cystic Fibrosis Research, Inc (New Horizons Grant to TM and Postdoctoral fellowship to WK), Cystic Fibrosis Foundation (MACHENG09, ILLEKG08), Beverley M. Folger Foundation, Elizabeth Nash Foundation, and the NIH (AI075410 and U01HL098964 to SVL).
References
- Anderson GG, Moreau-Marquis S, Stanton BA, O’Toole GA. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect Immun. 2008;76:1423–33. doi: 10.1128/IAI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnsholt T, Givskov M. Quorum-sensing blockade as a strategy for enhancing host defences against bacterial pathogens. Philos Trans R Soc Lond B Biol Sci. 2007;362:1213–22. doi: 10.1098/rstb.2007.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnsholt T, van Gennip M, Jakobsen TH, Christensen LD, Jensen PØ, Givskov M. In vitro screens for quorum sensing inhibitors and in vivo confirmation of their effect. Nat Protoc. 2010;5:282–93. doi: 10.1038/nprot.2009.205. [DOI] [PubMed] [Google Scholar]
- Boncoeur E, Tabary O, Bonvin E, Muselet C, Fritah A, Lefait E, Redeuilh G, Clement A, Jacquot J, Henrion-Caude A. Oxidative stress response results in increased p21WAF1/CIP1 degradation in cystic fibrosis lung epithelial cells. Free Radic Biol Med. 2006;40:75–86. doi: 10.1016/j.freeradbiomed.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Chambers CE, Visser MB, Schwab U, Sokol PA. Identification of N-acylhomoserine lactones in mucopurulent respiratory secretions from cystic fibrosis patients. FEMS Microbiol Lett. 2005;244:297–304. doi: 10.1016/j.femsle.2005.01.055. [DOI] [PubMed] [Google Scholar]
- Charlton TS, de Nys R, Netting A, Kumar N, Hentzer M, Givskov M, Kjelleberg S. A novel and sensitive method for the quantification of N-3-oxoacyl homoserine lactones using gas chromatography-mass spectrometry: application to a model bacterial biofilm. Environ Microbiol. 2000;2:530–541. doi: 10.1046/j.1462-2920.2000.00136.x. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen LD, van Gennip M, Jakobsen TH, Givskov M, Bjarnsholt T. Imaging N-acyl homoserine lactone quorum sensing in vivo. Methods Mol Biol. 2011;692:147–57. doi: 10.1007/978-1-60761-971-0_11. [DOI] [PubMed] [Google Scholar]
- Davis BM, Jensen R, Williams P, O’Shea P. The interaction of N-acylhomoserine lactone quorum sensing signaling molecules with biological membranes: implications for inter-kingdom signaling. PLoS One. 2010;5:e13522. doi: 10.1371/journal.pone.0013522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest. 1995;96:2204–10. doi: 10.1172/JCI118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285:13678–84. doi: 10.1074/jbc.M109.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson DL, Endersby R, Kirkham A, Stuber K, Vollman D, Rabin HR, Mitchell I, Storey DG. Pseudomonas aeruginosa quorum-sensing systems may control virulence factor expression in the lungs of patients with cystic fibrosis. Infect Immun. 2002;70:1783–1790. doi: 10.1128/IAI.70.4.1783-1790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Bettega K, Carroll S, Buchholz KR, Machen TE. Role of Ca2+ in responses of airway epithelia to Pseudomonas aeruginosa, flagellin, ATP, and thapsigargin. Am J Physiol Lung Cell Mol Physiol. 2007;292:L353–64. doi: 10.1152/ajplung.00042.2006. [DOI] [PubMed] [Google Scholar]
- Gerasimenko J, Ferdek P, Fischer L, Gukovskaya AS, Pandol SJ. Inhibitors of Bcl-2 protein family deplete ER Ca2+ stores in pancreatic acinar cells. Pflugers Arch. 2010;460:891–900. doi: 10.1007/s00424-010-0859-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb R, Dosanjh A. Mutant cystic fibrosis transmembrane conductance regulator inhibits acidification and apoptosis in C127 cells: Possible relevance to cystic fibrosis. Proc Natl Acad Sci USA. 1996;93:3587–91. doi: 10.1073/pnas.93.8.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009;21:871–7. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Haws C, Finkbeiner WE, Widdicombe JH, Wine JJ. CFTR in Calu-3 human airway cells: channel properties and role in cAMP-activated Cl-conductance. Am J Physiol. 1994;266:L502–12. doi: 10.1152/ajplung.1994.266.5.L502. [DOI] [PubMed] [Google Scholar]
- Hybiske K, Fu Z, Schwarzer C, Tseng J, Do J, Huang N, Machen TE. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1250–60. doi: 10.1152/ajplung.00231.2007. [DOI] [PubMed] [Google Scholar]
- Illek B, Fu Z, Schwarzer C, Banzon T, Jalickee S, Miller SS, Machen TE. Flagellin-stimulated Cl-secretion and innate immune responses in airway epithelia: role for p38. Am J Physiol Lung Cell Mol Physiol. 2008;295:L531–42. doi: 10.1152/ajplung.90292.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahoor A, Patel R, Bryan A, Do C, Krier J, Watters C, Wahli W, Li G, Williams SC, Rumbaugh KP. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J Bacteriol. 2008;190:4408–15. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson DM, Valentich JD, Marini FC, Grubman SA, Iannuzzi MC, Dorkin HL, Li M, Klinger KW, Welsh MJ. Expression of normal and cystic fibrosis phenotypes by continuous airway epithelial cell lines. Am J Physiol. 1990;259:L496–505. doi: 10.1152/ajplung.1990.259.6.L496. [DOI] [PubMed] [Google Scholar]
- Jungas T, Motta I, Duffieux F, Fanen P, Stoven V, Ojcius DM. Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277:27912–8. doi: 10.1074/jbc.M110288200. [DOI] [PubMed] [Google Scholar]
- Kerbiriou M, Teng L, Benz N, Trouvé P, Férec C. The calpain, caspase 12, caspase 3 cascade leading to apoptosis is altered in F508del-CFTR expressing cells. PLoS One. 2009;4:e8436. doi: 10.1371/journal.pone.0008436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisits MJ, Parsek MR. Does Pseudomonas aeruginosa use intercellular signalling to build biofilm communities? Cell Microbiol. 2006;8:1841–9. doi: 10.1111/j.1462-5822.2006.00817.x. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, et al. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321:259–63. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, et al. N-(3-oxo-acyl)homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. J Biol Chem. 2006;281:28822–30. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- Lee A, Chow D, Haus B, Tseng W, Evans D, Fleiszig S, et al. Airway epithelial tight junctions and binding and cytotoxicity of Pseudomonas aeruginosa. Am J Physiol. 1999;277:L204–17. doi: 10.1152/ajplung.1999.277.1.L204. [DOI] [PubMed] [Google Scholar]
- Li L, Hooi D, Chhabra SR, Pritchard D, Shaw PE. Bacterial N-acylhomoserine lactone-induced apoptosis in breast carcinoma cells correlated with down-modulation of STAT3. Oncogene. 2004;23:4894–902. doi: 10.1038/sj.onc.1207612. [DOI] [PubMed] [Google Scholar]
- Li H, Wang L, Ye L, Mao Y, Xie X, Xia C, et al. Influence of Pseudomonas aeruginosa quorum sensing signal molecule N-(3-oxododecanoyl) homoserine lactone on mast cells. Med Microbiol Immunol. 2009;198:113–21. doi: 10.1007/s00430-009-0111-z. [DOI] [PubMed] [Google Scholar]
- Lynch SV, Dixon L, Benoit MR, Brodie EL, Keyhan M, Hu P, et al. Role of the rapA gene in controlling antibiotic resistance of Escherichia coli biofilms. Antimicrob Agents Chemother. 2007;51:3650–8. doi: 10.1128/AAC.00601-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace PD, Riedl SJ. Molecular cell death platforms and assemblies. Curr Opin Cell Biol. 2010;22:828–36. doi: 10.1016/j.ceb.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Sheridan JA, Blohmke CJ, Turvey SE, Hancock RE. The Pseudomonas aeruginosa autoinducer 3O-C12 homoserine lactone provokes hyperinflammatory responses from cystic fibrosis airway epithelial cells. PLoS One. 2011;6:e16246. doi: 10.1371/journal.pone.0016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. Association of Bax and Bak homo-oligomers in mitochondria. Bax requirement for Bak reorganization and cytochrome c release. J Biol Chem. 2003;278:5367–76. doi: 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, Semenzato G, Cesura AM, Pinard E, Bernardi P. The mitochondrial effects of small organic ligands of BCL-2: sensitization of BCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6-trione derivative. J Biol Chem. 2006;281:10066–72. doi: 10.1074/jbc.M513708200. [DOI] [PubMed] [Google Scholar]
- Moreau-Marquis S, Bomberger JM, Anderson GG, Swiatecka-Urban A, Ye S, O’Toole GA, Stanton BA. The DeltaF508-CFTR mutation results in increased biofilm formation by Pseudomonas aeruginosa by increasing iron availability. Am J Physiol Lung Cell Mol Physiol. 2008;295:L25–37. doi: 10.1152/ajplung.00391.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau-Marquis S, O’Toole GA, Stanton BA. Tobramycin and FDA-approved iron chelators eliminate Pseudomonas aeruginosa biofilms on cystic fibrosis cells. Am J Respir Cell Mol Biol. 2009;41:305–13. doi: 10.1165/rcmb.2008-0299OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez R, Sancho-Martínez SM, Novoa JM, López-Hernández FJ. Apoptotic volume decrease as a geometric determinant for cell dismantling into apoptotic bodies. Cell Death Differ. 2010;17:1665–71. doi: 10.1038/cdd.2010.96. [DOI] [PubMed] [Google Scholar]
- Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–10. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–42. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- Pacheco AR, Sperandio V. Inter-kingdom signaling: chemical language between bacteria and host. Curr Opin Microbiol. 2009;2:192–198. doi: 10.1016/j.mib.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A. 2004;101:17404–9. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JP, Passador L, Iglewski BH, Greenberg EP. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1995;92:1490–4. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro CM, McKay RR, Hosoki E, Bird GS, Putney JW., Jr Effects of elevated cytoplasmic calcium and protein kinase C on endoplasmic reticulum structure and function in HEK293 cells. Cell Calcium. 2000;27:175–85. doi: 10.1054/ceca.2000.0108. [DOI] [PubMed] [Google Scholar]
- Schwarzer C, Illek B, Suh JH, Remington SJ, Fischer H, Machen TE. Organelle redox of CF and CFTR-corrected airway epithelia. Free Radic Biol Med. 2007;43:300–16. doi: 10.1016/j.freeradbiomed.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer C, Wong S, Shi J, Matthes E, Illek B, Ianowski JP, et al. Pseudomonas aeruginosa Homoserine lactone activates store-operated cAMP and cystic fibrosis transmembrane regulator-dependent Cl-secretion by human airway epithelia. J Biol Chem. 2010;285:34850–63. doi: 10.1074/jbc.M110.167668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BQ, Finkbeiner WE, Wine JJ, Mrsny RJ, Widdicombe JH. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl-secretion. Am J Physiol. 1994;266:L493–501. doi: 10.1152/ajplung.1994.266.5.L493. [DOI] [PubMed] [Google Scholar]
- Shiner EK, Terentyev D, Bryan A, Sennoune S, Martinez-Zaguilan R, Li G, et al. Pseudomonas aeruginosa autoinducer modulates host cell responses through calcium signalling. Cell Microbiol. 2006;8:1601–10. doi: 10.1111/j.1462-5822.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- Smith RS, Fedyk ER, Springer TA, Mukaida N, Iglewski BH, Phipps RP. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-kappa B and activator protein-2. J Immunol. 2001;167:366–74. doi: 10.4049/jimmunol.167.1.366. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Meyer T. Calcium-induced restructuring of nuclear envelope and endoplasmic reticulum calcium stores. Cell. 1997;89:963–71. doi: 10.1016/s0092-8674(00)80281-0. [DOI] [PubMed] [Google Scholar]
- Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, et al. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun. 2003;71:5785–93. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gennip M, Christensen LD, Alhede M, Phipps R, Jensen PØ, Christophersen L, et al. Inactivation of the rhlA gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. APMIS. 2009;117:537–46. doi: 10.1111/j.1600-0463.2009.02466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikström E, Bui L, Konradsson P, Magnusson KE. Role of calcium signalling and phosphorylations in disruption of the epithelial junctions by Pseudomonas aeruginosa quorum sensing molecule. Eur J Cell Biol. 2010;89:584–97. doi: 10.1016/j.ejcb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Vikström E, Tafazoli F, Magnusson KE. Pseudomonas aeruginosa quorum sensing molecule N-(3 oxododecanoyl)-l-homoserine lactone disrupts epithelial barrier integrity of Caco-2 cells. FEBS Lett. 2006;580:6921–6928. doi: 10.1016/j.febslet.2006.11.057. [DOI] [PubMed] [Google Scholar]
- Wagner VE, Iglewski BH. P. aeruginosa biofilms in CF infection. Clin Rev Allergy Immunol. 2008;35:124–134. doi: 10.1007/s12016-008-8079-9. [DOI] [PubMed] [Google Scholar]
- Wang J-L, Liu D, Zhang Z-J, Shan S, Han X, Srinivasula SM, et al. Proc Natl Acad Sci U S A. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ. 2011;18:38–47. doi: 10.1038/cdd.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–8. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P, Cámara M. Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr Opin Microbiol. 2009;12:182–191. doi: 10.1016/j.mib.2009.01.005. [DOI] [PubMed] [Google Scholar]