

Summary
Conditional gene deletion in mice has contributed immensely to our understanding of many biological and biomedical processes. Despite an increasing awareness of non-protein coding functional elements within protein coding transcripts, current gene-targeting approaches typically involve simultaneous ablation of non-coding elements within targeted protein-coding genes. The potential for protein-coding genes to have additional non-coding functions necessitates the development of novel genetic tools capable of precisely interrogating individual functional elements. We present a strategy coupling Cre/loxP-mediated conditional gene disruption with faithful GFP-reporter expression in mice in which Cre-mediated stable inversion of a splice acceptor-GFP-splice donor cassette concurrently disrupts protein production and creates a GFP fusion product. Importantly, cassette inversion maintains physiologic transcript structure, thereby ensuring proper microRNA-mediated regulation of the GFP-reporter, as well as maintaining expression of non-protein coding elements. To test this potentially generalizable strategy, we generated and analyzed mice with this conditional knock-in reporter targeted to the Hmga2 locus.
Introduction
In recent years, methods allowing manipulation of genes of interest within living organisms have enabled a detailed understanding of numerous genes' function across diverse phyla. The ability to alter the mouse genome has been especially critical for the investigation of gene function in vivo and has contributed immensely to our understanding of many fundamental questions in developmental biology and biomedical sciences. In particular, the development of site-specific recombinase systems in mice, including the Cre/loxP and FLP/FRT systems, has allowed the inactivation of genes of interest with both spatial and temporal control. As a result, conditional gene disruption has become a critical tool for understanding gene function during development, homeostasis, as well as in disease states (Rajewsky, 2007; Schmidt-Supprian and Rajewsky, 2007). Fluorescent reporter alleles have proved to be another key resource in the dissection of gene function by allowing direct visualization and isolation of molecularly defined subsets of cells (Hadjantonakis et al., 2003).
The past decade has brought an increased awareness that protein-coding genes can also possess additional non-protein coding functions. Single transcriptional units with multiple functions suggest interesting mechanisms that influence the cellular state, but their very existence necessitates new genetic methods to specifically abrogate the protein-coding element without altering other functions of the transcript and to generate reporter alleles that remain subject to all endogenous forms of regulation. In developing a new allele system to couple specific disruption of protein-coding function with reporter expression we considered the potential non-coding functions of transcripts.
Approximately half of all microRNAs (miRNAs) are encoded within introns. Targeted deletion of several intronic miRNAs has resulted in severe phenotypes including embryonic lethality (Kuhnert et al., 2008; Miyaki et al., 2010; Nakamura et al., 2011; Wang et al., 2008; Zhao et al., 2007), while many other intronic miRNAs remain uncharacterized. Therefore, gene targeting which inadvertently disrupts the expression of intronic miRNAs could have unanticipated phenotypic consequences (Kuhnert et al., 2008; Osokine et al., 2008; Wang et al., 2008). Interestingly, rare exonic miRNAs have also been shown to have critical functions (Sundaram et al., 2013).
Further indicating the need for improved gene inactivation methods, recent work indicates that mRNAs may also directly influence miRNA function through competitive binding, suggesting that the relative abundance of one miRNA target may impact that miRNA's ability to repress other targets. Genetic deletion or truncation of transcripts that have this competitive endogenous RNA (ceRNA) activity could thus lead to inadvertent widespread alterations in the protein expression of conserved miRNA targets (Karreth et al., 2011; Kumar et al., 2014; Poliseno et al., 2010; Salmena et al., 2011; Tay et al., 2011). More generally, any titratable site-specific RNA-binding factor could be affected by the deletion of a highly expressed target transcript. Finally, recent reports of abundant circular RNA species comprised of exons of protein-coding genes with potential additional activity forewarn of further inadvertent changes in cellular state when transcripts are entirely ablated or truncated (Memczak et al., 2013; Salzman et al., 2012).
Additionally, the appropriate regulation of protein expression from reporter alleles can also be influenced by transcript structure. miRNAs, RNA binding proteins, and RNA secondary structure regulate transcript stability and protein expression. Thus, reporters that are embedded within the native transcript will more faithfully recapitulate the expression of the gene of interest. Indeed, reporter alleles with endogenous versus exogenous 3′UTRs can produce dramatically different expression patterns due to the presence or absence of appropriate miRNA–mediated regulation (Merritt et al., 2008; Yoo et al., 2009).
Collectively, the potential for transcripts to possess multiple functions indicates that more precise genome modification methods are required to avoid the potential phenotypic consequences of altering additional non-protein-coding functions of genes. Based on these considerations, we developed a novel murine allele system in which conditional disruption of protein-coding function is directly coupled to fluorescent marker expression without impacting other aspects of transcript function.
Results
A strategy to couple conditional gene disruption with GFP-reporter expression in mice
The alteration of gene function with temporal and spatial control as well as the use of fluorescent reporter alleles has allowed the detailed investigation of a plethora of developmental processes. Coupling conditional Cre/loxP-mediated gene disruption with fluorescent reporter expression would not only generate two useful tools from one gene targeting event, but would also allow single cell resolution of gene disruption, permit altered cells to be assessed relative to non-targeted cells, and enable the identification and isolation of cells that are attempting to express the disrupted gene.
Several genetic approaches in mice have been previously developed to inactivate a gene of interest and express a reporter gene including the combination of Cre-reporter alleles with floxed alleles (Alexander et al., 2009; Lao et al., 2012; Srinivas et al., 2001); mosaic analysis with double markers (MADM) (Tasic et al., 2012; Zong et al., 2005) ; systems based on the inversion of gene trap-like elements (Mandalos et al., 2012; Schnutgen et al., 2005; Schnutgen et al., 2003; Schnutgen and Ghyselinck, 2007; Xin et al., 2005) and floxing of the entire gene followed by a 3′ reporter (Moon et al., 2000; Potocnik et al., 2000; Theis et al., 2001; Wellershaus et al., 2008). Each of these methods has their unique strengths, but system-specific limitations have in all cases negated their widespread application (Table S1).
We devised a strategy based on the insertion of an inverted splice acceptor-eGFP-splice donor (SA-GFP-SD) cassette into an intron of the target gene. Pairs of heterologous loxP sites (2272 loxP and 5171 loxP) (Lee and Saito, 1998; Stern et al., 2008) flank the SA-GFP-SD cassette, thus allowing two sequential Cre-mediated recombination events to stably invert the SA-GFP-SD cassette. The inversion should disrupt gene function concomitantly generate a GFP reporter (Figure 1A and Figure S1). The first recombination event inverts the intervening sequence between either the 5171 loxP or the 2272 loxP sites and generates one of two intermediates in which alternate loxP variants are now in a head-to-tail orientation. The second recombination event then deletes the sequence between these sites leaving a final stably inverted cassette with one 2272 loxP site and one incompatible 5171 loxP site (Figure 1A and Figure S1). Most importantly, we engineered a splice donor site following the stop codon of GFP to allow the transcript to continue through the remaining exons and 3′UTR to the endogenous polyadenylation sites (Figure 1A, B). Because the reading frame at the end of the first exon of a gene of interest can be in any of the three reading frames, we generated base vectors with GFP in each reading frame to generate this style of conditional knock-in (CK) allele (Figure S1).
Figure 1. A strategy to integrate conditional gene disruption with GFP-reporter expression within an otherwise full-length transcript.
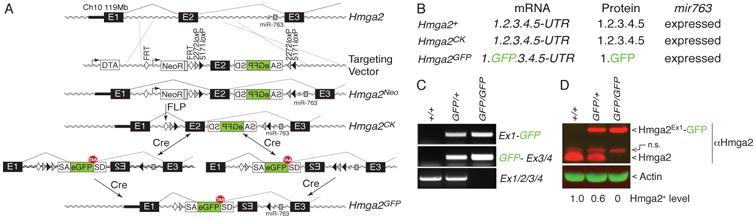
(A) Schematic of the Hmga2 targeting to create a Cre-regulated gene-trap allele that is both a conditional null allele and a conditional GFP-reporter. (B) Anticipated expression of the Hmga2 mRNA, protein, and intronic miRNA (miR-763) from the wild type (Hmga2+), targeted (Hmga2CK), and inverted (Hmga2GFP) alleles. (C) RT-PCR analysis of MEFs of the indicated Hmga2 genotypes. The splice-acceptor-GFP-splice donor in the Hmga2GFP allele generates the expected transcript containing exon 1 to GFP (Ex1-GFP) and GFP to exons 3 and 4 (GFP-Ex3/4) while completely disrupting the full length Hmga2 transcript (Ex1/2/3/4). (D) Western blot analysis of MEFs shows that the Hmga2GFP allele generates an Hmga2Ex1-GFP fusion that is detected by both the anti-Hmga2 antibody (which recognizes an epitope in the N-terminal portion of Hmga2) and the anti-GFP antibody. No endogenous Hmga2 is detected in Hmga2GFP/GFP MEFs. Actin shows equal loading. Wild-type Hmga2+ protein quantification is shown. A non-specific band (n.s.) is indicated. Representative of 4 independent experiments.
Generation of an Hmga2CK allele
To test our strategy to link Cre-mediated gene disruption with GFP reporter expression, we generated a CK allele of the chromatin-associated protein Hmga2 (Hmga2CK). We chose this gene because of our interest in Hmga2 in cancer metastasis as well as its dramatic role in body size regulation and potential role in stem cell biology (Nishino et al., 2008; Winslow et al., 2011; Xiang et al., 1990; Yu et al., 2007; Zhou et al., 1995). The Hmga2 genomic locus is quite large (∼116kb), has all exons in the same frame, has an intronic microRNA (miR-763), and produces a transcript that is highly regulated by endogenous miRNAs. Despite the difficulty of accounting for these common elements with existing genetic targeting approaches, these characteristics made Hmga2 an excellent candidate for our CK allele system. The Hmga2 locus was targeted with the SA-GFP-SD cassette in the reverse orientation (Figure 1A). Mice generated from correctly targeted ES cells were bred with mice expressing FLPe-recombinase to delete the neomycin-resistance cassette, thereby generating the Hmga2CK allele (Farley et al., 2000) (Figure 1A). To characterize the Hmga2 allele after cassette inversion, Hmga2CK mice were then bred with mice expressing Cre-recombinase to generate a germline Hmga2GFP allele (Wellershaus et al., 2008) (Figure 1A).
Cre-mediated stable inversion of the SA-GFP-SD cassette disrupts protein expression
The Hmga2GFP allele generated a transcript in which exon 2 was replaced by GFP, resulting in a protein product containing the N-terminus of Hmga2 fused to GFP (Figure 1B-D and Figure S1I). Although the splice acceptor within our cassette is well characterized and is capable of generating null-like alleles (Carette et al., 2009), it remained possible that some Hmga2 transcripts from the Hmga2GFP allele could splice from exon 1 to exon 3. In Hmga2GFP/GFP embryos and murine embryonic fibroblasts (MEFs), no evidence of an mRNA generated from exon 1 splicing to exon 3 was detected by RT-PCR (data not shown). Additionally, while splicing from exon 1 to exon 3 would create an in-frame protein product, even very long exposure times gave no indication of any truncated Hmga2 protein product in Hmga2GFP/GFP embryos or MEFs (Figure 1D, Figure S2A,B, Figure S3C, and data not shown).
Cre-mediated stable inversion of the SA-GFP-SD cassette generates a GFP fusion protein
In addition to functionally inactivating a targeted gene, a major power of our system is its ability to simultaneously generate a conditional GFP reporter allele. To assess GFP expression, we performed a series of experiments on embryos and MEFs from mice carrying the Hmga2CK or Hmga2GFP alleles. First, we examined embryos from several developmental stages and were able to easily distinguish Hmga2+/+ from Hmga2GFP/+ and Hmga2GFP/GFP mice by virtue of their GFP fluorescence (Figure 2A and Figure S3A). Quantitative detection of the GFP signal confirmed that Hmga2GFP/GFP embryos are approximately twice as bright as Hmga2GFP/+ embryos (Figure S3A). Analysis of MEFs by flow cytometry also confirmed robust GFP expression from the Hmga2GFP allele (Figure 2B). Immunohistochemical staining of E14.5 embryos for Hmga2 indicated that while a majority of cells within the embryos express Hmga2, only a fraction of the cells within the fetal liver express high levels of Hmga2 (Figure 2C). Therefore, we used flow cytometry to determine whether the Hmga2GFP allele could identify the Hmga2-expressing cell population within the fetal liver. We found that a subpopulation of fetal liver that were negative for hematopoietic lineage markers produced the highest level of Hmga2-GFP, with most cells producing low levels of GFP and Hmga2GFP/GFP cells producing higher levels of GFP than Hmga2GFP/+ cells (Figure 2D, E). Neither Hmga2CK/+ nor Hmga2CK/CK embryos produced GFP as assessed by direct visualization under the fluorescence dissecting scope, flow cytometry on fetal liver cells and MEFs, or western blot analysis of embryos and MEFs (Figure 2F, S3A, S2G-S2H, and S3C, and data not shown). To verify the conditional nature of the allele, we infected Hmga2CK/CK MEFs with an adenoviral vector expressing Cre. Cre expression converted the CK alleles to their GFP conformation, with infected cells expressing high levels of GFP and lacking full-length Hmga2 protein (Figure 2F and Figure S3C).
Figure 2. Gene-trapped Hmga2 functions as a conditional reporter of protein expression.
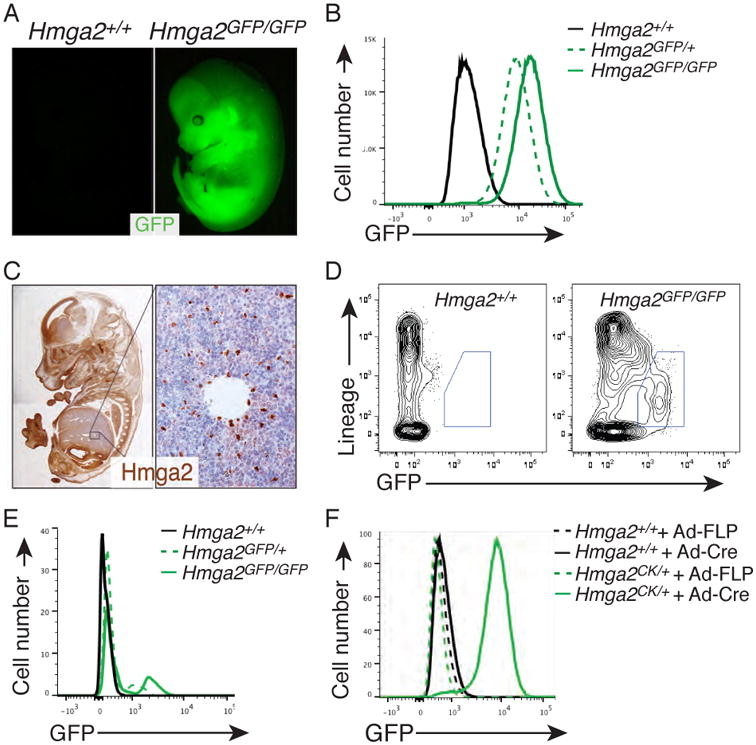
(A) Fluorescent images of Hmga2+/+ and Hmga2GFP/GFP E14.5 embryos. (B) Flow cytometric analysis of MEFs documents robust GFP expression and correspondingly higher expression in cells from Hmga2GFP/GFP mice. (C) Hmga2 is widely expressed in embryos, but only a subset of fetal liver cells express high levels of Hmga2. Immunohistochemical staining of an E14.5 embryo is shown. Enlarged area is indicated. Hematoxylin counterstained. (D) Flow cytometry analysis of fetal liver cells identifies a population of Hmga2hi Lineageneg cells. FSC/SSC gated cells are shown. (E) Overlay of the GFP intensity of fetal liver cells of the indicated genotypes. This histogram illustrates that most cells express a low level of Hmga2 and that GFP intensity increases in Hmga2GFP/GFP fetal liver cells. (F) Conditional expression of the GFP reporter in MEFs after infection of Hmga2CK/+ cells with Adenoviral-Cre (Ad-Cre). Hmga2+/+ MEFs and Adenoviral-FLP (Ad-FLP) infection are negative controls.
Cassette inversion maintains the physiologic expression of intronic miRNAs
Approximately half of known miRNAs are encoded within the introns of protein coding genes. While some gene inactivation strategies disrupt transcript expression, thereby disrupting both the protein-coding mRNA and the intronic miRNA, our system maintains the production of the full-length transcript, suggesting that intronic miRNAs should not be affected (Figure 1A, B). miR-763 resides within intron 3 of the Hmga2 locus, therefore we quantified miR-763 expression in MEFs and embryos carrying the Hmga2CK and Hmga2GFP alleles. Importantly, neither the Hmga2CK nor the Hmga2GFP allele affected miR-763 expression, confirming that our gene targeting strategy can specifically inactivate the protein-coding element without impacting other functional components within downstream introns (Figure S2I-S2L).
Cassette inversion maintains the full-length transcript allowing physiologic microRNA-mediated regulation of the GFP reporter
One theoretical advantage of generating a conditional allele using our general strategy is the ability to embed the GFP reporter within an otherwise full-length transcript, thereby ensuring that the GFP reporter remains under all the transcriptional and post-transcriptional regulatory control elements of the endogenous locus. In particular, the inclusion of a splice donor (SD) site after the stop codon of GFP allows transcription to continue all the way through the 3′ untranslated region, suggesting that the conditional GFP reporter should remain under physiologic miRNA regulation (Figure 1A, 1B, S1H, S3B, and S3D). To directly assess whether the Hmga2GFP allele maintains a physiologic and full-length transcript structure, we performed northern blot analyses using Hmga2- and GFP-specific probes. The composite Hmga2GFP transcript was detected as a single predominant band that was similar in size to the Hmga2wt counterpart indicating that cassette inversion embeds GFP within the full-length transcript (Figure S3E and S3F).
As Hmga2 is a well characterized let-7 miRNA target (Lee and Dutta, 2007; Mayr et al., 2007) and is regulated by let-7 in MEFs ((Viswanathan et al., 2008) and Figure 3C and S4), we performed a series of experiments to test whether the Hmga2GFP allele remains under let-7 control. Transfection of Hmga2GFP/+or Hmga2GFP/GFP MEFs with a synthetic let-7 mimic reduced GFP expression in a dose-dependent manner (Figure 3A-C, Figure S4A-S4B, and data not shown). Furthermore, the reduction of Hmga2Ex1-GFP protein mirrored that of endogenous Hmga2 after transfection of Hmga2GFP/+ MEFs with the let-7 mimic (Figure 3C). The Hmga2-GFP reporter was also increased by stable expression of the let-7 inhibitor Lin28 (Figure 3D and Figure S4C). Collectively, these results indicate that the Hmga2Ex1-GFP reporter generated after Cre-mediated cassette inversion remains under appropriate post-transcriptional 3′UTR regulation.
Figure 3. Hmga2Ex1-GFP remains under miRNA control.
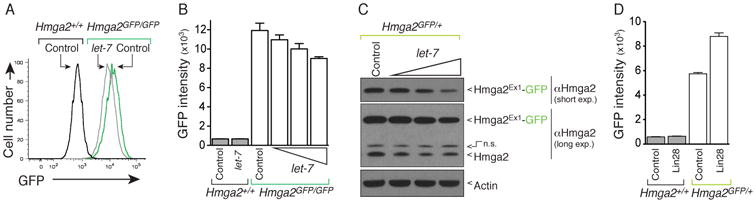
(A) Flow cytometric analysis of HmgaGFP/GFP MEFs after transfection with a let-7 mimic documents let-7-mediated reduction in GFP fluorescence. (B) Quantification of the GFP mean fluorescence intensity (MFI) after transfection with a control miRNA mimic or increasing amounts of let-7 mimic (2, 10, and 36 pmol - only 36pmol let-7 mimic is shown for Hmga2+/+ cells). Mean +/- SD of triplicate wells is shown. Data is representative of 4 experiments. (C) Let-7-mediated Hmga2Ex1-GFP protein reduction parallels that of full length Hmga2 in Hmga2GFP/+ MEFs. Cells were transfected with a control miRNA mimic (36 pmol) or increasing amounts of let-7 mimic (2, 10, and 36 pmol). Both Hmga2 and Hmga2Ex1-GFP expression decreases in a dose-dependent manner. A non-specific band (n.s.) is marked. Actin shows equal loading. (D) Quantification of GFP mean fluorescence intensity after stable retroviral Lin28 expression. Mean +/- SD of triplicate wells is shown.
Cassette inversion introduced a premature stop codon after the GFP coding sequence followed by several additional exons; therefore it is possible that the composite transcript could be subject to nonsense-mediated decay (NMD) (Chang et al., 2007). To directly compare the expression of the Hmga2wt and Hmga2GFP transcripts in isogenic cells, we treated Hmga2CK/CK cells with a Cre-expressing adenoviral vector to generate Hmga2GFP/GFP cells. qPCR and northern blotting for total Hmga2 indicate that the expression of Hmga2wt in Hmga2CK/CK cells is comparable to the expression of Hmga2GFP in Hmga2GFP/GFP cells. Additionally, inhibition of NMD induced comparable up-regulation of Hmga2GFP and Hmga2wt transcripts, had no effect on let-7-mediated repression of either Hmga2wt or Hmga2GFP mRNA or protein, and did not alter Lin28-induced up-regulation of Hmga2GFP protein (Figure S4).
Hmga2 protein is responsible for the phenotypes associated with Hmga2 deficiency
Previously, Hmga2 null alleles have been generated through spontaneous deletion, fortuitous insertional mutagenesis, and conventional gene targeting. In all cases, Hmga2 null mice displayed a dramatic dwarfism phenotype (Xiang et al., 1990; Zhou et al., 1995). To determine whether Hmga2GFP mice phenocopy Hmga2 null mice, we analyzed the growth of Hmga2+/+, Hmga2GFP/+, and Hmga2GFP/GFP mice. Consistent with the Hmga2-null phenotype, P0 Hmga2GFP/GFP mice were slightly smaller than Hmga2+/+ and Hmga2GFP/+ littermates (Figure 4A). Both male and female Hmga2GFP/GFP mice recapitulated the expected dwarfism phenotype with 12-week old mice being shorter and ∼50% the weight of control mice (Figure 4B-F). Additional aspects of the Hmga2 null phenotype were also observed in the Hmga2GFP/GFP mice, including a disproportionate reduction in fat tissue and pinnae size, and a relatively mild reduction in brain size (Nishino et al., 2008) (data not shown). Hmga2CK/CK mice were phenotypically indistinguishable from Hmga2+/+ control mice, indicating that the cassette itself does not dramatically affect the expression of the targeted allele prior to its inversion by Cre (Figure S2E and S2F).
Figure 4. Hmga2GFP mice recapitulate the null phenotype.
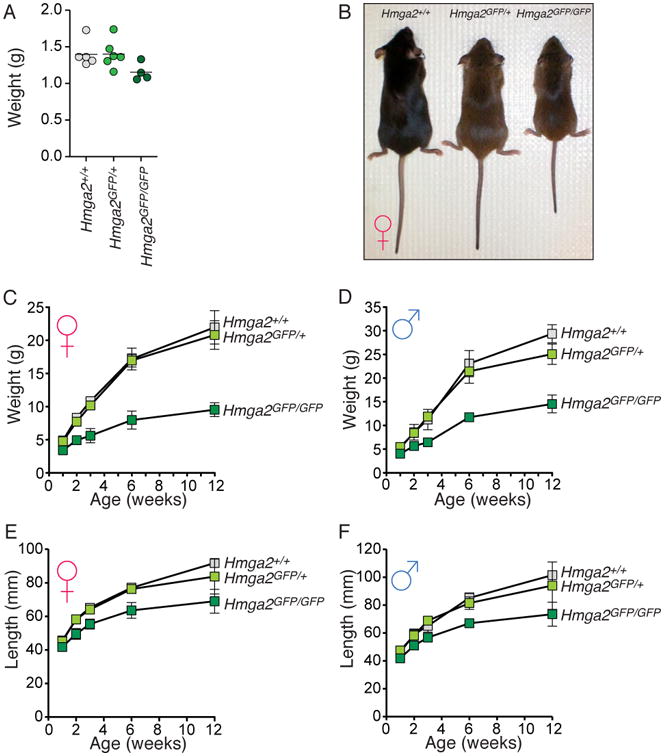
(A) P0 Hmga2GFP/GFP pups are ∼20% smaller than control littermates. Each dot represents a P0 mouse and the bar represents the mean. (B) Photo of 12-week old Hmga2+/+, Hmga2GFP/+, and Hmga2GFP/GFP female littermates. (C, D) Growth curves of female (C) and male (D) Hmga2+/+, Hmga2GFP/+, and Hmga2GFP/GFP mice. Mean +/- SD is shown for each time point. (E, F) Length curves of female (E) and male (F) Hmga2+/+, Hmga2GFP/+, and Hmga2GFP/GFP mice. Mean +/- SD is shown for each time point.
Discussion
The unequivocal coupling of gene disruption and reporter expression, unaltered expression of intronic microRNAs in the presence of disrupted protein-coding function, and the induction of a fluorescent reporter under the control of all endogenous genomic elements and 3′UTR control regions all support the broad use of CK alleles to better understand gene function in vivo. As miRNA-mediated control of protein expression and the role of ceRNA networks are almost completely unstudied in this context, combined approaches that employ both CK alleles and alleles that entirely abrogate transcript expression could represent an important future method to study the importance of ceRNA and other non-coding transcript functions in complex tissues and at the organismal level. This system also circumvents the major difficulties encountered with mosaic deletion resulting from ineffective Cre-ER or weak cell-type specific Cre alleles, since it allows cells that delete a targeted gene to be definitively distinguished from those that do not, and in fact allows for neighboring control and null cells to be analyzed within the same tissue. While other strategies to link gene disruption with reporter expression have been developed, none allow the generalizable and streamlined generation of alleles that specifically disrupt protein coding function while leaving all non-protein coding functions intact (Supplementary Table 1). The CK allele system will be widely applicable for creating Cre-regulated alleles to better understand the protein-coding element of genes during many stages of development, normal homeostasis, and diseases states.
The CK allele system should be amenable for targeting most genes as it employs a well-characterized splice acceptor that has been used for multiple genome-wide loss-of-function screens in haploid cells (Burckstummer et al., 2013; Carette et al., 2011). However, a few situations do exist that preclude the use of the CK technology. Introns flanked by non-canonical splice acceptor-donor pairs are rare, constituting less than one percent of introns, but would be insensitive to an inverted CK allele. Additionally, our system is also limited to genes that contain at least one intron and in which an exon1-GFP fusion would lack function.
mRNA stability is influenced by RNA sequence and structure, and both are altered in the CK allele after cassette inversion and incorporation of the GFP sequence. Additionally, interrupting the protein coding function by introducing a premature stop codon after GFP could induce nonsense mediated decay (Brogna and Wen, 2009). While we found that switching of the Hmga2wt transcript to Hmga2GFP did not reduce steady-state mRNA abundance, the effect of cassette inversion on transcript stability on additional targets will have to be determined empirically.
Hmga2 is an important regulator of body size across a wide range of species including humans (Makvandi-Nejad et al., 2012; Weedon et al., 2007). Previous murine Hmga2 null alleles have all resulted from the abrogation of the entire Hmga2 transcript (Xiang et al., 1990; Zhou et al., 1995). That our Hmga2GFP allele recapitulates the null phenotypes confirms that most, if not all, of the developmental phenotypic consequences observed in these earlier models are driven by the loss of Hmga2 protein function rather than the function of miR-763 or disruption of a let-7-Hmga2 ceRNA network. Our results provide a loss-of-function complement to the increased body size in transgenic mice expressing increased levels of Hmga2 protein (Arlotta et al., 2000; Battista et al., 1999). Given that the molecular functions of Hmga2 during development and disease remains poorly characterized, our allele should aid in dissecting the role of this important chromatin-regulating protein.
Experimental Procedures
Creation of base targeting vectors
The allele design (termed the CK system) is based on the creation of an in-frame GFP fusion protein, therefore CK plasmids were generated for each of the three reading frames (Figure S1; pCK(+0), pCK (+1), and pCK (+2)). To generate these plasmids eGFP-splice acceptor (SA)-2272loxP-5171loxP fragments in each of the three reading frames were amplified with the addition of a 5′ multiple cloning site, a consensus splice donor (SD) site (in reverse: actcacctt) and a 3′ PacI site. These fragments were ligated into a plasmid backbone that contained a DTA expression cassette, a multiple cloning site, an FRT-flanked Neomycin-resistance (NeoR) gene, 2272loxP-5171loxP sites, and a final multiple cloning site (Figure 1A, Supplemental Methods and Figure S1).
Generation of CK alleles
Construction of a CK allele for any gene of interest requires the amplification and ligation of the targeting arms into the appropriate pCK plasmid. Having an exon within the region of the genome to be inverted is not required for the system to work, but an exon can be included if desired (as in the Hmga2CK allele). Reasons to include an exon may include: the ATG start site lies within the second exon, a potential alternate ATG start lies within the second exon, or if one simply desires to further guarantee gene disruption. If no exon is included, it would likely be preferable to insert the cassette in the first intron of the target gene to limit the length of the endogenous gene produced prior to splicing into the splice acceptor (SA)-GFP-splice donor (SD) cassette after Cre-mediated inversion. In cases where an exon is included within the region that will be inverted the overall conformation would likely be very similar to the Hmga2CK allele that we have generated. If an exon is to be included it will be necessary to clone three regions into the pCK vector in any order; otherwise only two regions will be required (Figure S1A and Supplemental Methods).
Hmga2 targeting and generation of Hmga2CK and Hmga2GFP mice
The Hmga2-targeting construct was linearized and electroporated into v6.5 ES cells (kind gift from Rudolph Jaenisch) using standard conditions. Neomycin (300μg/mL G418)-resistant colonies were picked, expanded, and screened by long-range PCR for correct targeting of the Hmga2 locus. 5/294 clones (1.7%) were correctly targeted. C57BL/6J blastocysts were injected with targeted ES cells resulting in several high-percentage chimeras. Targeted Hmga2Neo mice were bred to Rosa26FLPe mice (JAX Stock number 003946) to delete the Neomycin resistance (NeoR) cassette and generate the Hmga2CK allele. A subset of Hmga2CK mice was bred to β-actin-Cre mice to create mice with a germline Hmga2GFP allele (Figure 1A). The MIT and Stanford Institutional Animal Care and Use Committees approved all animal studies and procedures.
Generation of MEFs, adenoviral infection, and flow cytometry
Murine embryonic fibroblasts (MEFs) were generated from embryos between E12.5 and E16.5. To express Cre-recombinase in MEFs, cells were infected with either Adeno-Cre or Adeno-FLPo (control) purchased from the Gene Transfer Vector Core at the University of Iowa. Analysis of the cells was performed 3 days after infection. Data were collected on a BD LSR II analyzer (BD Biosciences) at the Stanford Shared FACS Facility. For the quantification of GFP in MEFs on a LSR II flow cytometer, cells were thoroughly trypsinized and stained with DAPI for the exclusion of dead cells.
Western blot analysis
MEFs were trypsinized and pelleted before being lysed. Denatured samples were separated by SDS-PAGE, followed by transfer to PVDF membranes. Membranes were stained with the indicated primary antibodies rabbit anti-Hmga2-P1 (BioCheck; 59170AP), rabbit anti-GFP (Cell Signaling Technology; 2956), and anti-β-actin antibody (Sigma-Aldrich; A5441). For LI-COR imaging, membranes were first stained with indicated primary antibodies followed by the staining with either the goat anti-mouse-IRDye 800CW (926-32210, LI-COR, Inc.) or the goat anti-rabbit-Alexa Fluor 680 (A-21109, Life Technologies, Inc.) for 1 hour at room temperature in the dark. After 3 washes (1X PBS, 0.05% Tween20), the membranes were imaged with the LI-COR imager LI-COR Odyssey.
Upf1 knock down
The small interfering RNA (siRNA) specific for murine Upf1 transcripts was purchased from Life Technologies (siRNA ID: s72879). The procedure to deliver the control and Upf1 siRNA into MEFs were described as above. Transfectants were harvested 3 days post-transfection and lysed for quantification of mRNA. For functional validation of Upf1 knockdown, Gas5 transcript was used as described by Keeling et al. (Keeling et al., 2013)
Northern blot analysis
Northern blots were performed using standard methods. Briefly, radiolabeled probes were generated by random hexamer-primed synthesis using a PCR-amplified cDNA sequences using DNA polymerase I Klenow fragment and alpha-32P-dCTP. Probes were purified using a spin column kit (Nucleotide Removal Kit, QIAGEN). Membranes were pre-washed, hybridized with radiolabeled probe for 16-20 hours at 42°C, washed twice with 2× SSC and 0.1% SDS at 42°C for 20 minutes, and twice with 0.1× SSC and 0.1% SDS at 55°C for 20 minutes. Membranes were imaged on a Molecular Dynamics Typhoon 9400 Imager (Amersham/GE Healthcare).
Let-7 transfection and retroviral Lin28 expression
To deliver control (mirVana negative control mimic) and/or let-7 (mirVana mmulet-7a-5p miRNA mimic) miRNA mimics into MEFs, Lipofectamine RNAiMAX was used according to manufacturer's description (Life Technologies). To normalize total input mimics, compensatory levels of control mimic were added with titrated let-7 mimic. Overexpression of the let-7 inhibitor Lin28 (Viswanathan et al., 2008) was achieved by transducing MEFs with a retroviral vector (MSCV) harboring the Lin28 cDNA (Addgene, pMSCV-mLin28A/NeoR, Plasmid No. 26357)(Viswanathan et al., 2009). An MSCV-NeoR vector without Lin28 was used as a negative control. Transduced MEFs were cultured in the presence of G418 (0.5 mg/ml) for 3 days to allow complete enrichment of transductants. Selected MEFs were assessed for GFP and Hmga2 levels using flow cytometry and western blot analysis.
Supplementary Material
Highlights.
Genetic system to coupling gene disruption with GFP expression
Preserves function of all non-protein-coding elements after recombination
Allows tracking of cells harboring targeted protein deficiency
Acknowledgments
We thank Pauline Chu for histology, the Stanford Shared FACS Facility for expert assistance, Patrick Stern and Xun Zeng for reagents, and Tracy Staton, Deborah Caswell, and Matthew Scott for critical reading of the manuscript. We thank the Koch Institute Swanson Biotechnology Center (SBC) for technical support, specifically Denise Crowley at the Hope Babette Tang (1983) Histology Facility, Noranne Enzer, John Mkandawire and Peimin Qi at the Rippel Mouse ES Cell and Transgenics Core Facility, and Scott Malstrom at the Applied Therapeutics and Whole Animal Imaging facility. S-H.C and CH.C. are Stanford University School of Medicine Dean's Postdoctoral Fellows. C-H.C. is additionally funded by an American Lung Association Fellowship. V.I.R is supported by a Walter V. and Idun Berry Postdoctoral Fellowship. D.M.F is supported by the National Institutes of Health grant R00-CA158581. T.J. is a Howard Hughes Medical Institute Investigator and a Daniel K. Ludwig Scholar. This work was supported by the National Institutes of Health grant R00-CA151968 and a Pancreas Cancer Action Network-AACR Career Development Award, in memory of Skip Viragh (13-20-25-WINS) (to M.M.W.), and in part by the MIT Cancer Center support grant (P30-CA14051) and the Stanford Cancer Institute support grant (P30-CA124435) from the National Cancer Institute.
Footnotes
The authors declare no completing financial interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander CM, Puchalski J, Klos KS, Badders N, Ailles L, Kim CF, Dirks P, Smalley MJ. Separating stem cells by flow cytometry: reducing variability for solid tissues. Cell Stem Cell. 2009;5:579–583. doi: 10.1016/j.stem.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Tai AK, Manfioletti G, Clifford C, Jay G, Ono SJ. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas. J Biol Chem. 2000;275:14394–14400. doi: 10.1074/jbc.m000564200. [DOI] [PubMed] [Google Scholar]
- Battista S, Fidanza V, Fedele M, Klein-Szanto AJ, Outwater E, Brunner H, Santoro M, Croce CM, Fusco A. The expression of a truncated HMGI-C gene induces gigantism associated with lipomatosis. Cancer Res. 1999;59:4793–4797. [PubMed] [Google Scholar]
- Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nature structural & molecular biology. 2009;16:107–113. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- Burckstummer T, Banning C, Hainzl P, Schobesberger R, Kerzendorfer C, Pauler FM, Chen D, Them N, Schischlik F, Rebsamen M, et al. A reversible gene trap collection empowers haploid genetics in human cells. Nature methods. 2013;10:965–971. doi: 10.1038/nmeth.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Wuethrich I, Blomen VA, Varadarajan M, Sun C, Bell G, Yuan B, Muellner MK, Nijman SM, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nature biotechnology. 2011;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annual review of biochemistry. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Hadjantonakis AK, Dickinson ME, Fraser SE, Papaioannou VE. Technicolour transgenics: imaging tools for functional genomics in the mouse. Nature reviews Genetics. 2003;4:613–625. doi: 10.1038/nrg1126. [DOI] [PubMed] [Google Scholar]
- Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, DeNicola G, Webster KA, Weiss D, Perez-Mancera PA, et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147:382–395. doi: 10.1016/j.cell.2011.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Wang D, Dai Y, Murugesan S, Chenna B, Clark J, Belakhov V, Kandasamy J, Velu SE, Baasov T, et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PloS one. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Hampton J, Stankunas K, Asano T, Chen CZ, Kuo CJ. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989–3993. doi: 10.1242/dev.029736. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM, Downward J. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–217. doi: 10.1038/nature12785. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lao Z, Raju GP, Bai CB, Joyner AL. MASTR: a technique for mosaic mutant analysis with spatial and temporal control of recombination using conditional floxed alleles in mice. Cell Rep. 2012;2:386–396. doi: 10.1016/j.celrep.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Saito I. Role of nucleotide sequences of loxP spacer region in Cre-mediated recombination. Gene. 1998;216:55–65. doi: 10.1016/s0378-1119(98)00325-4. [DOI] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, Loredo AI, Bellone RR, Mezey JG, Brooks SA, et al. Four loci explain 83% of size variation in the horse. PLoS One. 2012;7:e39929. doi: 10.1371/journal.pone.0039929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandalos N, Saridaki M, Harper JL, Kotsoni A, Yang P, Economides AN, Remboutsika E. Application of a novel strategy of engineering conditional alleles to a single exon gene, Sox2. PLoS One. 2012;7:e45768. doi: 10.1371/journal.pone.0045768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol. 2008;18:1476–1482. doi: 10.1016/j.cub.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaki S, Sato T, Inoue A, Otsuki S, Ito Y, Yokoyama S, Kato Y, Takemoto F, Nakasa T, Yamashita S, et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010;24:1173–1185. doi: 10.1101/gad.1915510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon AM, Boulet AM, Capecchi MR. Normal limb development in conditional mutants of Fgf4. Development. 2000;127:989–996. doi: 10.1242/dev.127.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol. 2011;31:3019–3028. doi: 10.1128/MCB.05178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osokine I, Hsu R, Loeb GB, McManus MT. Unintentional miRNA ablation is a risk factor in gene knockout studies: a short report. PLoS Genet. 2008;4:e34. doi: 10.1371/journal.pgen.0040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocnik AJ, Brakebusch C, Fassler R. Fetal and adult hematopoietic stem cells require beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity. 2000;12:653–663. doi: 10.1016/s1074-7613(00)80216-2. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. From a dream to reality. European journal of immunology. 2007;37(Suppl 1):S134–137. doi: 10.1002/eji.200737819. [DOI] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- Schnutgen F, De-Zolt S, Van Sloun P, Hollatz M, Floss T, Hansen J, Altschmied J, Seisenberger C, Ghyselinck NB, Ruiz P, et al. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc Natl Acad Sci U S A. 2005;102:7221–7226. doi: 10.1073/pnas.0502273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnutgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, Ghyselinck NB. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol. 2003;21:562–565. doi: 10.1038/nbt811. [DOI] [PubMed] [Google Scholar]
- Schnutgen F, Ghyselinck NB. Adopting the good reFLEXes when generating conditional alterations in the mouse genome. Transgenic Res. 2007;16:405–413. doi: 10.1007/s11248-007-9089-8. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern P, Astrof S, Erkeland SJ, Schustak J, Sharp PA, Hynes RO. A system for Cre-regulated RNA interference in vivo. Proc Natl Acad Sci U S A. 2008;105:13895–13900. doi: 10.1073/pnas.0806907105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram GM, Common JE, Gopal FE, Srikanta S, Lakshman K, Lunny DP, Lim TC, Tanavde V, Lane EB, Sampath P. ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing. Nature. 2013;495:103–106. doi: 10.1038/nature11890. [DOI] [PubMed] [Google Scholar]
- Tasic B, Miyamichi K, Hippenmeyer S, Dani VS, Zeng H, Joo W, Zong H, Chen-Tsai Y, Luo L. Extensions of MADM (Mosaic Analysis with Double Markers) in Mice. PLoS One. 2012;7:e33332. doi: 10.1371/journal.pone.0033332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, de Wit C, Schlaeger TM, Eckardt D, Kruger O, Doring B, Risau W, Deutsch U, Pohl U, Willecke K. Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis. 2001;29:1–13. doi: 10.1002/1526-968x(200101)29:1<1::aid-gene1000>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O'Sullivan M, Lu J, Phillips LA, Lockhart VL, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Lettre G, Freathy RM, Lindgren CM, Voight BF, Perry JR, Elliott KS, Hackett R, Guiducci C, Shields B, et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet. 2007;39:1245–1250. doi: 10.1038/ng2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellershaus K, Degen J, Deuchars J, Theis M, Charollais A, Caille D, Gauthier B, Janssen-Bienhold U, Sonntag S, Herrera P, et al. A new conditional mouse mutant reveals specific expression and functions of connexin36 in neurons and pancreatic beta-cells. Exp Cell Res. 2008;314:997–1012. doi: 10.1016/j.yexcr.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Winslow MM, Dayton TL, Verhaak RG, Kim-Kiselak C, Snyder EL, Feldser DM, Hubbard DD, DuPage MJ, Whittaker CA, Hoersch S, et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature. 2011;473:101–104. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Benson KF, Chada K. Mini-mouse: disruption of the pygmy locus in a transgenic insertional mutant. Science. 1990;247:967–969. doi: 10.1126/science.2305264. [DOI] [PubMed] [Google Scholar]
- Xin HB, Deng KY, Shui B, Qu S, Sun Q, Lee J, Greene KS, Wilson J, Yu Y, Feldman M, et al. Gene trap and gene inversion methods for conditional gene inactivation in the mouse. Nucleic Acids Res. 2005;33:e14. doi: 10.1093/nar/gni016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Staahl BT, Chen L, Crabtree GR. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature. 2009;460:642–646. doi: 10.1038/nature08139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Zhou X, Benson KF, Ashar HR, Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature. 1995;376:771–774. doi: 10.1038/376771a0. [DOI] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.