

Abstract
Initiating mechanisms that impair gluconeogenic enzymes and spare lipogenic enzymes in diet-induced obesity (DIO) are obscure. Here, we examined insulin signaling to Akt and atypical protein kinase C (aPKC) in liver and muscle and hepatic enzyme expression in mice consuming a moderate high-fat (HF) diet. In HF diet–fed mice, resting/basal and insulin-stimulated Akt and aPKC activities were diminished in muscle, but in liver, these activities were elevated basally and were increased by insulin to normal levels. Despite elevated hepatic Akt activity, FoxO1 phosphorylation, which diminishes gluconeogenesis, was impaired; in contrast, Akt-dependent phosphorylation of glycogenic GSK3β and lipogenic mTOR was elevated. Diminished Akt-dependent FoxO1 phosphorylation was associated with reduced Akt activity associated with scaffold protein WD40/Propeller/FYVE (WD40/ProF), which reportedly facilitates FoxO1 phosphorylation. In contrast, aPKC activity associated with WD40/ProF was increased. Moreover, inhibition of hepatic aPKC reduced its association with WD40/ProF, restored WD40/ProF-associated Akt activity, restored FoxO1 phosphorylation, and corrected excessive expression of hepatic gluconeogenic and lipogenic enzymes. Additionally, Akt and aPKC activities in muscle improved, as did glucose intolerance, weight gain, hepatosteatosis, and hyperlipidemia. We conclude that Akt-dependent FoxO1 phosphorylation occurs on the WD/Propeller/FYVE scaffold in liver and is selectively inhibited in early DIO by diet-induced increases in activity of cocompartmentalized aPKC.
Introduction
Insulin-resistant states of obesity, metabolic syndrome, and type 2 diabetes mellitus (T2DM) are pandemic in Western societies. Insulin resistance implies an impairment in glucose metabolism that initially increases insulin secretion. Insulin controls glucose metabolism: in liver, by activating Akt2, which diminishes glucose production at least partly by diminishing expression of gluconeogenic enzymes, and in muscle, by activating Akt2 and atypical protein kinase C (aPKC), which stimulate glucose uptake (1).
Paradoxically, in insulin-resistant states, some actions of insulin and/or other factors that have similar or overlapping actions are maintained, while other actions are impaired; this reflects that hyperinsulinemia owing to impaired glucose metabolism, or increases in factors that have insulin-like actions, can activate intact pathways. Thus, in liver, despite impaired regulation of gluconeogenesis, signaling pathways that regulate lipogenesis can remain open and contribute to clinical lipid abnormalities. Indeed, despite impaired Akt activation and increased expression of hepatic gluconeogenic enzymes, excessive aPKC activity and increased expression of lipogenic enzymes are seen in hepatocytes of T2DM humans (2) and livers of diabetic rodents (3–5) and high-fat-fed (HFF) mice (3,6). Moreover, in hepatocytes of type 2 diabetic humans, aPKC activity appeared to be at least partly elevated by hyperinsulinemia-dependent activation of insulin receptor substrate (IRS)-2–dependent phosphatidylinositol 3-kinase (PI3K) and generation of phosphatidylinositol-3,4,5-(PO4)3 (PIP3) (2), as observance of diabetes mellitus–induced increases in both aPKC activity and expression of lipogenic enzymes required that elevated insulin levels were maintained during prolonged incubations (2).
As another mechanism for provoking inordinate increases in hepatic aPKC activity in insulin-resistant states, certain lipids generated by dietary excesses, ceramides, and phosphatidic acid directly activate aPKC (1). Moreover, ceramide impairs hepatic Akt activation in mice fed 60% of calories from fat (7–9), and excessive hepatic aPKC activity contributes importantly to enhanced expression of lipogenic, proinflammatory, and gluconeogenic factors that promote obesity, hepatosteatosis, hyperlipidemia, and glucose intolerance in multiple models of insulin resistance (2–6). Activation of hepatic aPKC partly explains the paradox that hyperinsulinemic states characteristically have excessive hepatic production of insulin-dependent lipids, along with impaired ability of insulin to suppress hepatic glucose production. Further mechanistic insight into this paradox is herein provided by findings showing that, in initial stages of HFF, Akt-mediated activation of mTOR1C, which increases hepatic lipogenesis (10), is elevated, but in contrast, phosphorylation of FoxO1, which diminishes hepatic gluconeogenesis (11,12), is impaired.
In mice consuming a diet with 60% of calories from fat, impaired hepatic Akt activity/activation (7,8) can account for increased gluconeogenic enzyme expression and hepatic insulin resistance. To examine an earlier phase of diet-induced obesity (DIO), we used HFF mice consuming a “Western” diet with 40% of calories from milk fat and found that hepatic Akt2 activity/activation was increased but nevertheless accompanied by a defect in FoxO1 phosphorylation and impaired regulation of gluconeogenic enzyme expression. Moreover, the loss of Akt-dependent FoxO1 phosphorylation was apparently due to altered activities of Akt and aPKC bound to 40 kDa scaffold protein, WD40/Propeller-FYVE (WD40/ProF), which contains seven WD(trp-x-x-asp)-repeat proteins and one FYVE domain (domain in Fab1p, YOTB, Vac1p and EEA19 early endosome antigen-1) (13), and is required for Akt-mediated phosphorylation of FoxO1 in adipocytes (14). Thus, inhibition of hepatic aPKC in HFF mice diminished aPKC binding to WD40/ProF, restored WD40/ProF-associated Akt activity and FoxO1 phosphorylation, and diminished gluconeogenic enzyme expression. Consequently, hepatic lipogenic enzyme expression diminished, insulin activation of both Akt and aPKC in muscle improved, and problems of glucose intolerance, hyperlipidemia, hepatosteatosis, and weight gain were obviated.
Research Design and Methods
aPKC Inhibitors
PKC-ι inhibitor [1H-imidazole-4-carboxamide,5-amino]-2,3-dihydroxy-4-hydroxymethyl-cyclopentyl-[1R-(1a,2b,3b,4a)] (ICAP) was synthesized by Southern Research (Birmingham, AL) or United Chemical Resources (Birmingham, AL) (>95% purity). Note: ICAP is inactive, but, like AICAR (identical to ICAP except that AICAR has a ribose instead of a cyclopentyl ring), is converted intracellularly by adenosine kinase to the active compound, [1H-imidazole-4-carboxamide,5-amino]-[2,3-dihydroxy-4-[(phosphono-oxy)methyl]cyclopentane-[1R-(1a,2b,3b,4a)] (ICAPP) (15). Also note: 1) ICAP is only slightly less potent than ICAPP in in vivo studies, but ICAP synthesis is easier and less costly; 2) ICAPP/ICAP inhibits recombinant PKC-ι/λ but not PKC-ζ, PKC-α, PKC-β, PKC-δ, PKC-ε, or PKC-θ (2); 3) in isolated human hepatocytes, ICAPP/ICAP inhibits aPKC but not Akt2 or AMPK, and moreover, increases FoxO1 phosphorylation (2,15); and 4) in intact mice, ICAPP/ICAP inhibits aPKC in liver but not in muscle or adipose tissue (5).
Pan-aPKC inhibitor 2-acetyl-1,3-cyclopentanedione (ACPD) (purchased from Sigma-Aldrich, St. Louis, MO) inhibits recombinant PKC-ι/λ and PKC-ζ equally but not recombinant PKC-α, PKC-β, PKC-δ, or PKC-ε (Supplementary Figs. 1 and 2). Like ICAPP/ICAP, ACPD inhibits aPKC but not Akt2 or AMPK in human hepatocytes (15) (Supplementary Fig. 3) and inhibits aPKC in liver but not in muscle of intact mice (Supplementary Fig. 4). The reason for hepatic selectivity of ACPD and ICAP during in vivo treatment is uncertain.
Mouse Studies
C57Bl/6/SV129 male mice (age 3–5 months) were obtained from a colony maintained in the Tampa VA Vivarium. Mates from each litter were equally distributed to four groups that were studied over a 10-week period. Mice were fed (diets from Harlan Industries, Madison, WI) a low-fat diet (with 10% of calories from fat and containing 210 g/kg casein, 3 g/kg l-cystine, 500 g/kg corn starch, 100 g/kg maltodextrin, 39 g/kg sucrose, 20 g/kg anhydrous milk fat, 20 g/kg lard, 20 g/kg soybean oil, 35 g/kg cellulose, 35 g/kg mineral mix, 15 g/kg vitamin mix, and 0.01 g/kg antioxidant) or a high-fat diet (with 40% of calories from milk fat and containing 200 g/kg casein, 3 g/kg l-cystine, 150 g/kg corn starch, 120 g/kg maltodextrin, 216 g/kg sucrose, 200 g/kg anhydrous milk fat, 10 g/kg corn oil, 1.5 g/kg cholesterol, 50 g/kg cellulose, 35 g/kg mineral mix, 4 g/kg calcium carbonate, 10 g/kg vitamin mix, and 0.04 g/kg antioxidant (for fatty acid composition, see Sajan et al. [6]). Mice were also injected subcutaneously once daily with vehicle or aPKC inhibitor ICAP (1 mg/kg) or ACPD (10 mg/kg body wt). During the 9th week, glucose tolerance was measured after an overnight fast by injection at zero time of 2 mg glucose/kg body wt i.p. and measurement of blood glucose values at 0, 30, 60, 90, and 120 min as previously described (16). At the end of the 10th week, mice were treated acutely for 15 min with or without insulin (1 unit/kg body wt i.p.) and killed. Tissues were rapidly removed for subsequent analyses. Note that treatment of mice for 10 weeks with aPKC inhibitors ACPD and ICAP did not appear to have any detrimental effects on liver (as per serum aspartate aminotransferase and alanine aminotransferase) or renal (as per serum blood urea nitrogen and creatinine) function.
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committees of the University of South Florida College of Medicine and the James A. Haley Veterans Administration Medical Center Research and Development Committee (Tampa, FL).
Lysate Preparation
As previously described (2–6,15), liver and muscle samples were homogenized in ice-cold buffer containing 0.25 mol/L sucrose, 20 mmol/L Tris/HCl (pH 7.5), 2 mmol/L EGTA, 2 mmol/L EDTA, 1 mmol/L phenlysulfonlyfluoride, 20 μg/mL leupeptin, 10 μg/mL aprotinin, 2 mmol/L Na4P2O7, 2 mmol/L Na3VO4, 2 mmol/L NaF, and 1 μmol/L microcystin and then supplemented with 1% TritonX-100, 0.6% Nonidet, and 150 mmol/L NaCl and cleared by low-speed centrifugation.
aPKC, PKC, and Akt Assays
aPKCs were immunoprecipitated from lysates with rabbit polyclonal antiserum (Santa Cruz Biotechnologies, Santa Cruz, CA), which recognizes C-termini of PKC-ζ and PKC-λ/ι. Immunoprecipitates were collected on Sepharose-A/G beads (Santa Cruz Biotechnologies) and assayed as described (2–6,15). aPKC activation was also assessed by immunoblotting for phosphorylation of the auto(trans)phosphorylation site Thr555/560 in PKC-ι/ζ, required for, and reflective of, activation (1).
Akt2 enzyme activity was assayed in immunoprecipitates using a kit purchased from Millipore as previously described (2–6,15). Akt activity was also assessed by immunoblotting for phosphorylation of Ser473-Akt. AMP-activated protein kinase (AMPK) activity was assayed as previously described (2–6,15).
Akt and aPKC activities associated with WD40/ProF were measured by incubating WD40/ProF immunoprecipitates with or without aPKC inhibitor ACPD or with or without Akti inhibitor (Calbiochem, La Jolla, CA) to define aPKC and Akt activities, respectively.
Western Analyses
Western analyses were conducted as previously described (2–6,15) using rabbit anti–phospho-Ser473-Akt antiserum, rabbit anti–glyceraldehyde-phosphate dehydrogenase (GAPDH) antiserum, rabbit anti-WD40/ProF antiserum, and rabbit anti-aPKC antiserum (Santa Cruz Biotechnologies, Santa Cruz, CA); rabbit anti–phospho-Thr560/555–PKC-ζ/λ/ι antiserum (Invitrogen, Carlsbad, CA); rabbit anti–p-Ser256-FoxO1 and anti-FoxO1 antiserum (Abnova, Walnut, CA); mouse monoclonal anti–PKC-λ/ι antibodies (Transduction Labs, Bedford, MA); and rabbit anti–phospho-Ser9-GSK3β antiserum, rabbit anti–phospho-Ser2448-mTOR antiserum, and mouse anti-Akt antibodies (Cell Signaling Technologies, Danvers, MA). Samples from experimental groups were compared on the same blots and corrected for recovery as needed by measurement of GAPDH immunoreactivity.
Measurements of Serum Triglycerides, Cholesterol, Free Fatty Acids, Insulin, and Glucose
Serum triglycerides, insulin, and glucose levels were measured as previously described (5,6,16).
mRNA Measurements
As previously described (2,4–6,15), tissues were added to Trizol reagent (Invitrogen) and RNA was extracted and purified with RNEasy Mini Kit and RNAase-Free DNase Set (Qiagen, Valencia, CA), quantified (A260/A280), checked for integrity by electrophoresis on 1.2% agarose gels, and quantified by real-time RT-PCR, using TaqMan reverse transcription reagent and SYBR Green kit (Applied Biosystems, Carlsbad, CA) with mouse nucleotide primers and horseradish-peroxidase transferase as an internal recovery standard.
Nuclear Preparations
Nuclei were prepared as previously described (4).
Ceramide Species Quantitation
Ceramide species were measured by liquid chromatography tandem mass spectrometry analysis of lipid extracts of liver lysates by Lipidomics Shared Resource, Medical University of South Carolina (Charleston, SC).
Statistical Evaluations
Data are expressed as mean ± SEM, and P values were determined by one-way ANOVA and least significant multiple-comparison methods.
Results
Effects of HFF on Activities of aPKC and Akt2 in Liver and Muscle in Low-Fat-Fed and HFF Mice
As seen in Figs. 1A and B and 2A and C, in control low-fat-fed (LFF) mice, insulin provoked rapid increases in activity of both aPKC and Akt2 in liver and muscle. In HFF mice, however, basal/resting and insulin-stimulated activities of aPKC and Akt2 were diminished in muscle, but in liver, basal/resting activities of aPKC and Akt2 were elevated, presumably reflecting hyperinsulinemia and possibly other activators, and increased after acute insulin treatment to levels comparable with those of LFF mice. Thus, because of elevated baselines, insulin-induced increments in hepatic Akt2 and aPKC activities were of lesser magnitude in HFF mice. Nevertheless, insulin signaling to hepatic aPKC and Akt2 was largely intact in HFF mice.
Figure 1.

Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on basal and insulin-stimulated activities of aPKC and Akt2 in liver (A) and muscle (B). Over 10 weeks, mice were fed a low-fat (10% of calories from fat) (L) or high-fat (40% of calories from milk fat) (H) diet and treated with or without aPKC inhibitor (daily injections of 1 mg/kg body wt s.c. ICAP or 10 mg/kg body wt s.c. ACPD). Before killing, fed mice were treated for 15 min with or without insulin (1 unit/kg body wt i.p.). Liver and hind limb muscles were harvested and examined for immunoprecipitable aPKC and Akt2 activity. Values are means ± SEM of six determinations. *P < 0.05; **P < 0.01; ***P < 0.001 for indicated comparisons. Letters above bars indicate the following: a, P < 0.05; b, P < 0.01; and c, P < 0.001 for insulin-stimulated versus resting/basal values in corresponding treatment groups. immunoppt, immunoprecipitate.
Figure 2.
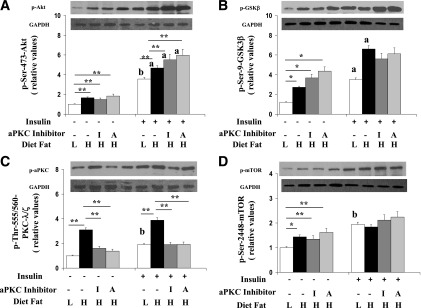
Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on hepatic resting/basal and insulin-stimulated phosphorylation of Ser473-Akt (A), Ser9-GSK3β (B), Thr555/560–PKC-λ/ζ (C), and Ser2448-mTOR (D). As in Fig. 1, over 10 weeks, mice were fed low-fat (L) or high-fat (H) diets and treated with or without aPKC inhibitor; 15 min before killing, fed mice were treated with or without insulin (1 unit/kg body wt). Liver was harvested and examined for immunoreactivity of indicated signaling factors. Bargram values are mean ± SEM of six determinations. *P < 0.05 and **P < 0.01 for indicated comparisons. Letters above bars indicate as follows: a, P < 0.05 and b, P < 0.01 for insulin-stimulated versus resting/basal values in corresponding treatment groups. Representative immunoblots are shown for indicated phosphoproteins and GAPDH loading controls.
Similar to Akt activation, resting hepatic IRS-1–dependent PI3K activity, the major activator of Akt during insulin action (17), trended upward, presumably reflecting hyperinsulinemia, and increased normally with acute insulin treatment (Supplementary Fig. 5). In contrast, insulin did not increase activity of IRS-1–dependent PI3K in muscles of HFF mice. Different from IRS-1–dependent PI3K, insulin-stimulated IRS-2–dependent PI3K activity was intact in muscle as well as liver in HFF mice. (Note: In liver, IRS-2–dependent PI3K participates along with IRS-1–dependent PI3K in Akt activation but is the sole activator of aPKC during insulin action [17]).
Effects of aPKC Inhibitors on Activities of aPKC and Akt2 in Liver and Muscle of HFF Mice
We treated HFF mice for 10 weeks with two small-molecule aPKC inhibitors, ICAP and ACPD, in doses that reduced basal/resting and exogenous insulin-stimulated hepatic aPKC activities to levels seen in control/LFF mice (Figs. 1A and 2C). In contrast, aPKC inhibitor treatment did not alter resting Akt2 activity in livers of HFF mice but enhanced insulin-stimulated Akt2 activity therein (Figs. 1A and 2A). In muscle, with aPKC inhibitor treatment, resting aPKC and Akt2 activities increased to, and insulin-stimulated activities approached, activities seen in control/LFF mice (Fig. 1B).
Effects of HFF on Expression of Hepatic Lipogenic and Gluconeogenic Enzymes
HFF provoked increases in hepatic mRNA and protein levels of lipogenic enzymes sterol receptor element–binding protein-1c (SREBP-1c) (note: protein as per active nuclear fragment) and fatty acid synthase (FAS) and gluconeogenic enzymes PEPCK and glucose-6-phosphatase (G6Pase), as measured in the fed state (Fig. 3A and B). However, with aPKC inhibitor treatment and reduction of hepatic aPKC activity, expression of mRNA and protein levels of SREBP-1c, FAS, PEPCK, and G6Pase in HFF mice were not significantly different from expression in control/LFF mice (Fig. 3A). Consonant with the idea that diminished hepatic aPKC activity was responsible for improvements in expression of lipogenic and gluconeogenic enzymes in HFF mice treated with aPKC inhibitors, aPKC is required for 1) feeding- and insulin-dependent increases in activity and expression of SREBP-1c, which increases expression of multiple lipogenic enzymes (2,4–6,15,18,19); and 2) fasting-dependent increases in expression of PEPCK and G6Pase (2,4–6,15).
Figure 3.

Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on mRNA (A) and immunoreactive protein (B) levels of lipogenic (SREBP-1c, FAS) and gluconeogenic (PEPCK, G6Pase) enzymes in livers of ad libitum–fed mice. As in Fig. 1, over 10 weeks, mice were consuming low-fat (L) and high-fat (H) diets and treated with or without aPKC inhibitor. After killing, liver tissue was examined for mRNA and protein levels of indicated enzymes. Values are mean ± SEM of 12 determinations. (Note: Acute 15-min insulin treatment as described in Fig. 1 did not alter mRNA and protein levels.) *P < 0.05; **P < 0.01; ***P < 0.001 for indicated comparisons. Representative immunoblots are shown for indicated proteins. Note that levels of the active SREBP-1c fragment were measured in nuclear preparations.
Effects of HFF on Phosphorylation of FoxO1 and Other Akt Substrates in Liver
In livers of T2DM humans (2,15) and rodents (3–6), Akt activity/activation is diminished and expression of PEPCK/G6Pase is understandably increased. In presently used HFF mice, however, the presence of increased PEPCK/G6Pase expression in the face of elevated hepatic Akt2 activity seemed at odds. This conundrum was resolved by finding that Akt2-dependent phosphorylation of Ser256-FoxO1, which, by its phosphorylation and nuclear exclusion mediates insulin-dependent decreases in gluconeogenic enzyme expression (11,12), was markedly diminished basally and virtually unresponsive to exogenous insulin treatment in livers of HFF mice (Fig. 4A).
Figure 4.
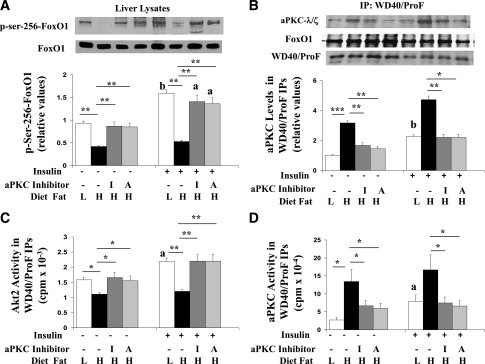
Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on phosphorylation of Ser256-FoxO1 in liver lysates (A); recovery of immunoreactivity of aPKC, FoxO1, and WD40/ProF in WD40/ProF immunoprecipitates (B); recovery of Akt enzyme activity in WD40/ProF immunoprecipitates (C); and recovery of aPKC enzyme activity in WD40/ProF immunoprecipitates (D). As in Fig. 1, over 10 weeks, mice were fed a low-fat (L) or high-fat (H) diet and treated with or without aPKC inhibitor; 15 min before killing, mice were treated with or without insulin (1 unit/kg body wt). Liver was harvested and examined for indicated signaling factors in liver lysates or WD40/ProF immunoprecipitates prepared from liver lysates. Bargram values are mean ± SEM of six determinations. *P < 0.05; **P < 0.01; ***P < 0.001 for indicated comparisons. Letters above bars indicate the following: a, P < 0.05 and b, P < 0.01 for insulin-stimulated versus basal/resting values in corresponding treatment groups. Note that FoxO1 and WD40/ProF levels were not altered by treatments. Also note that large amounts of immunoreactive immuno–γ globulins precluded accurate measurement of nearby Akt in WD40/ProF immunoprecipitates.
In contrast to FoxO1 phosphorylation but in keeping with increases in hepatic Akt2 activity in HFF mice, Akt-dependent phosphorylation of both Ser9-glycogen synthase kinase (GSK)-3β, which, by inhibiting GSK3β, mediates stimulatory effects on glycogen synthesis, and Ser2448-mTOR, which, by activating S6 kinase, mediates stimulatory effects on lipogenesis (10), was increased by HFF, as well as by exogenous insulin treatment and, moreover, was not altered by aPKC inhibitor treatment (Fig. 2B and D). Accordingly, the defect in FoxO1 phosphorylation in HFF mice was relatively specific and did not reflect a generalized defect in hepatic Akt-dependent phosphorylation.
Effects of aPKC Inhibitors on FoxO1 Phosphorylation in Livers of HFF Mice
Treatment of HFF mice with aPKC inhibitors fully or largely restored basal/resting and insulin-stimulated hepatic FoxO1 phosphorylation (Fig. 4A). This restoration suggested that activation of hepatic aPKC contributed to the impairment of FoxO1 phosphorylation in HFF liver. Moreover, the stimulatory effect of aPKC inhibitors on FoxO1 phosphorylation provided a reasonable explanation for the improvement/suppression of gluconeogenic enzyme expression in HFF mice (Fig. 3A).
Role of ProF in aPKC-Dependent Inhibition of FoxO1 Phosphorylation in Livers in HFF Mice
Findings that FoxO1 phosphorylation was diminished despite heightened Akt2 activity, and was increased in basal/resting conditions by aPKC inhibitors without change in overall cellular Akt2 activity, suggested that Akt-dependent FoxO1 phosphorylation and inhibition thereof by HFF-activated aPKC might be compartmentalized. It was therefore interesting that, in adipocytes, the scaffold protein WD40/ProF binds FoxO1 and activated forms of both Akt and aPKC and, moreover, is required for Akt-mediated phosphorylation of FoxO1 but not other Akt substrates, such as GSK3β and mTORC1 (14), i.e., a pattern of selective inhibition of Akt substrates identical to that observed above in HFF liver. Indeed, we found in liver that levels and activity of aPKC recovered in WD40/ProF immunoprecipitates were increased, particularly by HFF and to a lesser degree by exogenous insulin treatment (Fig. 4B and D); activity of Akt recovered in WD40/ProF immunoprecipitates was increased by insulin in control/LFF mice (Fig. 4C); basal/resting and insulin-stimulated activities of Akt associated with WD40/ProF were diminished by HFF (Fig. 4C); and treatment of HFF mice with aPKC inhibitors diminished aPKC binding to WD40/Prof (Fig. 4B and D) but simultaneously increased WD40/ProF-associated Akt activity (Fig. 4C) and total cellular FoxO1 phosphorylation (Fig. 4A), thereby decreasing gluconeogenic enzyme expression (Fig. 3A).
Effects of aPKC Inhibitors on Glucose and Lipid Metabolism in HFF Mice
With aPKC inhibitor–induced improvements in gluconeogenic enzyme suppression in liver and insulin signaling in muscle, glucose tolerance (Fig. 5A), fasting blood glucose levels (Fig. 5A), serum glucose levels in resting/fed conditions and after acute insulin treatment (Fig. 5B), and fed serum insulin levels (Fig. 5C) in HFF mice were reduced to levels comparable with those of control/LFF mice. On the other hand, fasting serum insulin levels were reduced but remained mildly elevated (Fig. 5C).
Figure 5.

Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on glucose tolerance in fasted mice (A), resting and insulin-stimulated serum glucose levels in fed mice (B), serum insulin levels in fasted and fed mice (C), and serum levels of triglycerides and cholesterol in fed mice (D). Over 10 weeks, mice were consuming low-fat (L) and high-fat (H) diets and treated with or without aPKC inhibitor, as in Fig. 1. At 9 weeks, after an overnight fast, mice were subjected to glucose tolerance testing (2 mg glucose/kg body wt i.p. and measurement of blood glucose at 0, 30, 60, 90, and 120 min). At 10 weeks, 15 min before killing, fed mice were treated with or without insulin (1 unit/kg body wt). Blood and sera were examined for indicated parameters. Values are means ± SEM of 12 determinations in panels A, C, and D (15-min insulin treatment did not alter serum lipid values) and mean ± SEM of 6 determinations in B. *P < 0.05 and ***P < 0.001 for indicated comparisons. HF, high fat; LF, low fat.
As with glucose homeostasis, treatment with aPKC inhibitors largely reversed/was prevented the following: 1) increases in serum triglyceride and cholesterol levels (Fig. 5D), 2) weight gain (Fig. 6A) (note: Food intake was unaltered [Fig. 6B], raising the possibility of an increase in caloric expenditure), 3) increases in epididymal and retroperitoneal fat depots (Fig. 6C), and 4) increases in hepatic triglycerides and fat content (Oil Red O staining) (Fig. 6D).
Figure 6.
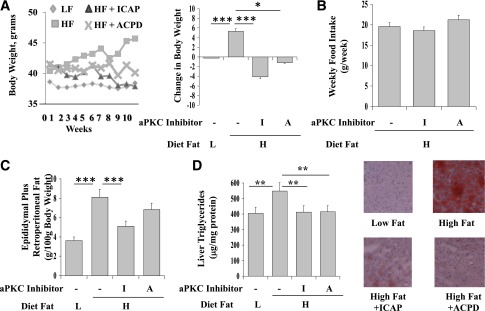
Effects of HFF and aPKC inhibitors ICAP (I) and ACPD (A) on body weight (A), food intake (B), fat pad weights (C), and hepatic triglyceride levels and fat contents as per Oil Red O staining (D). As in Fig. 1, over 10 weeks, mice were consuming low-fat (L) and high-fat (H) diets and treated with or without aPKC inhibitor. Values are mean ± SEM of 12 values. *P < 0.05; **P < 0.01; ***P < 0.001. HF, high fat; LF, low fat.
Effects of Ceramide on Hepatic aPKC Activity in WT and HFF Mice
As ceramide provokes hepatic abnormalities in HFF mice (7–9) and directly activates aPKC (1,20,21), we questioned whether ceramide contributed to increases in aPKC activity in our HFF mice. Ceramide strongly activated aPKC when added to immunoprecipitates prepared from liver lysates of LFF/control mice but had only a weak effect on aPKC immunoprecipitated from lysates of HFF mice and diminished activity of aPKC immunoprecipitated from lysates of HFF mice treated acutely with insulin (Fig. 7). (Note: Ceramide has biphasic effects on aPKC activity, with inhibition after stimulation in dose-response studies [20,21]; similarly, PIP3 inhibits aPKC activity when added in excess [22]). On the other hand, acute insulin treatment, acting via PIP3, elicited further increases in aPKC activity in HFF mice (Fig. 7 [compare with Fig. 1A]). That ceramide levels were increased in HFF mice is shown in Fig. 7.
Figure 7.

A: Effects of ceramide on aPKC immunoprecipitated from liver lysates obtained from mice consuming low-fat (LF) and high-fat (HF) diets and treated with or without insulin (Ins) for 15 min prior to killing, as described in Figs. 1–6 (except that these mice were not treated with aPKC inhibitors). Assays of immunoprecipitated aPKC were conducted, with indicated concentrations of ceramide (Sigma-Aldrich). Values are mean ± SEM of three to five determinations. B: Effects of feeding HF and LF diets on hepatic levels of ceramide and sphingomyelin species. As in Fig. 1, over 10 weeks, mice were fed a low-fat or high-fat diet but were not treated with aPKC inhibitors. Bargram values are mean ± SEM of eight determinations. *P < 0.05. DH, dihydro; SM, sphingomyelin.
Discussion
Finding heightened hepatic Akt2 activity in the resting state and attainment of normal levels of Akt2 activity AFTER maximal insulin treatment, along with diminished FoxO1 phosphorylation and increased PEPCK/G6Pase expression, allowed us to localize an important defect in glucose metabolism specifically at the level of FoxO1 phosphorylation. This post-Akt2 signaling defect in mice consuming 40% of calories from fat contrasts with impaired insulin-stimulated hepatic Akt activation in mice consuming 60% of calories from fat (7,8) and presumably reflects an earlier stage of development of hepatic insulin resistance in HFF/DIO. On the other hand, variable increases in resting/basal Akt activity have been noted in rodents consuming diets containing 60% of calories from fat over 4 weeks (23,24). Thus, alterations in Akt activity may vary, depending on the intensity and length of HFF.
The finding that two inhibitors of hepatic aPKC restored FoxO1 phosphorylation in our HFF/DIO mice suggested that the excessive activation of hepatic aPKC observed in these mice was responsible for the selective impairment in FoxO1 phosphorylation. Further support comes from findings in studies of WD40/ProF, which, in adipocytes, serves as a scaffold protein required for both insulin/aPKC-dependent vesicle-associated membrane protein-2 phosphorylation and glucose transport (25) and insulin/Akt-dependent phosphorylation of FoxO1 but not GSK3β and mTOR (14). Accordingly, in liver, we found the following: aPKC activity in WD40/ProF immunoprecipitates was increased by HFF and insulin, Akt2 activity in WD40/ProF immunoprecipitates was increased by insulin but diminished by HFF, and aPKC inhibitors diminished aPKC and increased Akt2 activity to levels comparable with those of LFF mice.
Interestingly, HFF-induced impairment of hepatic FoxO1 phosphorylation was relatively specific and did not involve Akt substrates GSK3β and mTOR, in accordance with Akt substrate specificity observed in studies of WD40/ProF knockdown in adipocytes (14). It therefore appears that WD40/ProF provides a functional compartment or platform that specifically enables FoxO1 phosphorylation in multiple insulin-sensitive tissues. That excessive HFF-induced activation of aPKC in this hepatic compartment diminished cocompartmentalized Akt activity and its action on total cellular FoxO1 and gluconeogenic enzyme expression underscores the importance of this compartment/platform in regulating hepatic glucose metabolism and its vulnerability to excessive aPKC activation. Further studies are needed to see whether Akt substrates other than FoxO1 are phosphorylated on the WD40/ProF platform and whether other functions of insulin that require Akt and FoxO1 phosphorylation are abrogated by excessive aPKC activation.
Also note that aPKC directly binds, phosphorylates, and inhibits Akt (26–28), and the abundance and proximity of aPKC to Akt on the seven-bladed propellers of the WD40/ProF scaffold may have further amplified the ability of aPKC to specifically influence WD40/ProF-associated Akt activity and Akt-dependent FoxO1 phosphorylation in livers of HFF mice. In this regard, relief of inhibitory effects of aPKC on Akt activation presumably contributed importantly to the enhancement of insulin-stimulated activity/phosphorylation of total hepatic Akt2 in aPKC inhibitor–treated HFF mice, but interestingly, this enhancement did not alter GSK3β/mTOR phosphorylation.
That WD40/ProF is involved in Akt2-mediated phosphorylation of hepatic FoxO1 is noteworthy, as FoxO1 mediates insulin effects on hepatic gluconeogenesis (11,12), a key element in glucose homeostasis. Also note that, as insulin regulation of PEPCK and G6Pase expression appears to require only relatively low levels of Akt activity (10), this efficiency may reflect the ability of the WD40/ProF scaffold to facilitate Akt-dependent FoxO1 phosphorylation. Nevertheless, as seen here, this coupling efficiency is abrogated by HFF, apparently through inordinate aPKC activation and subsequent binding of active aPKC to WD40/ProF.
The idea that aPKC limits Akt action on FoxO1 may seem counterintuitive, as it implies that insulin itself restrains FoxO1 phosphorylation by coactivating hepatic aPKC with Akt. However, in normal conditions, restraining effects of insulin/aPKC on Akt-dependent FoxO1 phosphorylation are only partly effective but perhaps necessary to prevent hypoglycemia that might otherwise occur if Akt operated on FoxO1 without restraint. In support of the possibility that aPKC tonically restrains Akt effects on FoxO1, inhibition of aPKC increases FoxO1 phosphorylation and diminishes gluconeogenic enzyme expression in fasting conditions and in the absence of either HFF or exogenous insulin treatment (2–6,10). As another possibility, increases in aPKC activity induced by diet-related lipids may be more marked and better targeted to WD40/ProF and therefore more effective in uncoupling Akt and FoxO1 than those induced by normal feeding-related increases in insulin secretion.
We speculate (Fig. 8) that hepatic aPKC activity was increased initially in our HFF mice by diet-derived lipids, ceramides, and/or phosphatidic acid and, secondarily, from hyperinsulinemia after impaired FoxO1 phosphorylation and increased gluconeogenesis. Supportive of this hypothesis, ceramide, which provokes hepatic abnormalities in HFF mice (7–9), activated aPKC in immunoprecipitates from LFF liver, but poorly in immunoprecipitates from HFF liver, suggesting that the ceramide was already exerting a near-maximal effect. However, acute insulin treatment provoked further increases in hepatic aPKC activity, indicating that aPKC was not maximally activated by diet-dependent factors. Also note that ceramide and PIP3 activate aPKC by binding to separate sites (1,20,21), and additive effects are not surprising.
Figure 8.

Development of hepatic and secondary systemic insulin resistance in DIO. In response to dietary excess, availability of lipids that directly activate aPKC, e.g., ceramide and phosphatidic acid, increases. Subsequent activation of hepatic aPKC increases binding of aPKC to ProF, a scaffolding protein that couples Akt and FoxO1, and this leads to impaired ability of Akt2 to phosphorylate FoxO1 on Ser256; as a result, expression of PEPCK and G6Pase and hepatic glucose output increase. Ensuing increases in blood glucose levels stimulate insulin secretion, and both glucose and insulin, as well as fatty acids, increase phosphatidic acid production via the de novo pathway. Increased insulin secretion activates hepatic Akt2, as well as aPKC, which together increase hepatic lipid production, thereby providing more substrates for phosphatidic acid and ceramide synthesis. In short, a vicious cycle is set up for lipid production and aPKC activation. This cycle is abetted in human (but not rodent) liver by virtue of the fact that increased aPKC activity provokes increases in levels of PKC-ι mRNA and protein (2). As a by-product of increases in circulating levels of liver-derived lipids and cytokines, insulin signaling in muscle and certain other tissues (e.g., adipose tissue [data not shown]) is impaired, adding further to diminished glucose disposal and systemic insulin resistance.
Importantly, insulin signaling to both Akt2 and aPKC in muscle improved with inhibition of hepatic aPKC. Thus, in our HFF/DIO model, abnormalities in insulin signaling in muscle appeared to be largely dependent on aPKC-dependent hepatic abnormalities, e.g., increased secretion of hepatic lipids, proinflammatory cytokines, and/or other factors. These improvements in muscle accompanying aPKC inhibitor treatment probably contributed importantly to improved glucose tolerance in HFF mice. Moreover, improvements in body weight and/or energy balance (not measured), may have contributed to improvements in muscle and/or liver in HFF mice after treatment with aPKC inhibitors.
Despite the fact that aPKC inhibitors kept clinical parameters of glucose and lipid metabolism at or close to normal in our HFF mice, serum insulin levels were improved but not fully normalized. The reason for this is uncertain, but the residual hyperinsulinemia may have been needed to activate spare insulin receptors partially downregulated in amount or activity by diet-induced increases in conventional/novel PKCs or other factors. In any case, the marked improvements in glucose and lipid metabolism suggested that the residual hyperinsulinemia was functionally sufficient, and we did not use higher doses of aPKC inhibitors to drive down serum insulin levels.
Finally, as to the paradox in insulin-resistant states that stimulatory effects of insulin and/or other factors on hepatic lipogenesis may be excessive when inhibitory effects of insulin or other factors on FoxO1 and hepatic gluconeogenesis are deficient, the present findings show that, in “initial” or “early” stages of HFF/DIO, i.e., when hepatic Akt2 activation is still intact, at least part of this paradox may reflect impaired ability of Akt to phosphorylate FoxO1, coupled with normal or excessive ability of Akt and aPKC, as activated by insulin and/or other factors, to phosphorylate mTORC1/S6kinase or other lipogenic factors. Later, as hepatic Akt activation by insulin is impaired, continued increases in hepatic aPKC, along with a modicum of basal Akt, or continued increases in resting Akt activity (see 23,24) or other factors that activate mTOR/S6 kinase may be sufficient to maintain increases in hepatic lipogenesis. In both situations, reduction of hepatic aPKC activity by dietary or other means appears to be an important therapeutic goal.
Supplementary Material
Article Information
Funding. This study was supported by funds from the Department of Veterans Affairs Merit Review Program and a National Institutes of Health grant (7RO1DK 065969-09) to R.V.F.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.P.S., M.L.S., R.A.I., and M.L. conducted studies and assays, assembled data, and assisted with interpretation of data. M.E.A.-D. screened and ranked potential inhibitory compounds binding to PKC-ι, and assisted with interpretation of data. R.V.F. conceived, designed, and directed the studies; analyzed data; and wrote the manuscript. R.V.F. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1863/-/DC1.
This research does not represent the views of the Department of Veterans Affairs or the U.S. Government.
References
- 1.Farese RV, Sajan MP. Metabolic functions of atypical protein kinase C: “good” and “bad” as defined by nutritional status. Am J Physiol Endocrinol Metab 2010;298:E385–E394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sajan MP, Farese RV. Insulin signalling in hepatocytes of humans with type 2 diabetes: excessive production and activity of protein kinase C-ι (PKC-ι) and dependent processes and reversal by PKC-ι inhibitors. Diabetologia 2012;55:1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Standaert ML, Sajan MP, Miura A, et al. Insulin-induced activation of atypical protein kinase C, but not protein kinase B, is maintained in diabetic (ob/ob and Goto-Kakazaki) liver. Contrasting insulin signaling patterns in liver versus muscle define phenotypes of type 2 diabetic and high fat-induced insulin-resistant states. J Biol Chem 2004;279:24929–24934 [DOI] [PubMed] [Google Scholar]
- 4.Sajan MP, Standaert ML, Rivas J, et al. Role of atypical protein kinase C in activation of sterol regulatory element binding protein-1c and nuclear factor kappa B (NFkappaB) in liver of rodents used as a model of diabetes, and relationships to hyperlipidaemia and insulin resistance. Diabetologia 2009;52:1197–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sajan MP, Nimal S, Mastorides S, et al. Correction of metabolic abnormalities in a rodent model of obesity, metabolic syndrome, and type 2 diabetes mellitus by inhibitors of hepatic protein kinase C-ι. Metabolism 2012;61:459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sajan MP, Standaert ML, Nimal S, et al. The critical role of atypical protein kinase C in activating hepatic SREBP-1c and NFkappaB in obesity. J Lipid Res 2009;50:1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab 2009;297:E211–E224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ussher JR, Koves TR, Cadete VJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes 2010;59:2453–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest 2011;121:4222–4230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A 2010;107:3441–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitamura Y, Accili D. New insights into the integrated physiology of insulin action. Rev Endocr Metab Disord 2004;5:143–149 [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 2007;6:208–216 [DOI] [PubMed] [Google Scholar]
- 13.Fritzius T, Burkard G, Haas E, et al. A WD-FYVE protein binds to the kinases Akt and PKCzeta/λ. Biochem J 2006;399:9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritzius T, Moelling K. Akt- and Foxo1-interacting WD-repeat-FYVE protein promotes adipogenesis. EMBO J 2008;27:1399–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sajan MP, Ivey RA, 3rd, Farese RV. Metformin action in human hepatocytes: coactivation of atypical protein kinase C alters 5′-AMP-activated protein kinase effects on lipogenic and gluconeogenic enzyme expression. Diabetologia 2013;56:2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farese RV, Sajan MP, Yang H, et al. Muscle-specific knockout of PKC-λ impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest 2007;117:2289–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo S, Copps KD, Dong X, et al. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol Cell Biol 2009;29:5070–5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumoto M, Ogawa W, Akimoto K, et al. PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest 2003;112:935–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi CM, Kondo T, Sajan MP, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKClambda/ζ. Cell Metab 2006;3:343–353 [DOI] [PubMed] [Google Scholar]
- 20.Wang G, Silva J, Krishnamurthy K, Tran E, Condie BG, Bieberich E. Direct binding to ceramide activates protein kinase Czeta before the formation of a pro-apoptotic complex with PAR-4 in differentiating stem cells. J Biol Chem 2005;280:26415–26424 [DOI] [PubMed] [Google Scholar]
- 21.Müller G, Ayoub M, Storz P, Rennecke J, Fabbro D, Pfizenmaier K. PKC ζ is a molecular switch in signal transduction of TNF-α, bifunctionally regulated by ceramide and arachidonic acid. EMBO J 1995;14:1961–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Standaert ML, Bandyopadhyay G, Perez L, et al. Insulin activates protein kinases C-ζ and C-λ by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem 1999;274:25308–25316 [DOI] [PubMed] [Google Scholar]
- 23.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 2005;146:1473–1481 [DOI] [PubMed] [Google Scholar]
- 24.Liu H-Y, Hung T, Wen,G-B, et al. Increased basal level of Akt-dependent insulin signaling may be responsible for the development of insulin resistance. Am J Physiol Metab 2009;297:E898–E906 [DOI] [PMC free article] [PubMed]
- 25.Fritzius T, Frey AD, Schweneker M, Mayer D, Moelling K. WD-repeat-propeller-FYVE protein, ProF, binds VAMP2 and protein kinase Czeta. FEBS J 2007;274:1552–1566 [DOI] [PubMed] [Google Scholar]
- 26.Konishi H, Kuroda S, Kikkawa U. The pleckstrin homology domain of RAC protein kinase associates with the regulatory domain of protein kinase C ζ. Biochem Biophys Res Commun 1994;205:1770–1775 [DOI] [PubMed] [Google Scholar]
- 27.Doornbos RP, Theelen M, van der Hoeven PC, van Blitterswijk WJ, Verkleij AJ, van Bergen en Henegouwen PM. Protein kinase Czeta is a negative regulator of protein kinase B activity. J Biol Chem 1999;274:8589–8596 [DOI] [PubMed] [Google Scholar]
- 28.Weyrich P, Neuscheler D, Melzer M, Hennige AM, Häring HU, Lammers R. The Par6alpha/aPKC complex regulates Akt1 activity by phosphorylating Thr34 in the PH-domain. Mol Cell Endocrinol 2007;268:30–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.