

Abstract
The origins of nonalcoholic fatty liver disease (NAFLD) may lie in early intrauterine exposures. Here we examined the maternal response to chronic maternal high-fat (HF) diet and the impact of postweaning healthy diet on mechanisms for NAFLD development in juvenile nonhuman primate (NHP) offspring at 1 year of age. Pregnant females on HF diet were segregated as insulin resistant (IR; HF+IR) or insulin sensitive (IS; HF+IS) compared with control (CON)-fed mothers. HF+IR mothers have increased body mass, higher triglycerides, and increased placental cytokines. At weaning, offspring were placed on a CON or HF diet. Only offspring from HF+IR mothers had increased liver triglycerides and upregulated pathways for hepatic de novo lipid synthesis and inflammation that was irreversible upon switching to a healthy diet. These juvenile livers also showed a combination of classical and alternatively activated hepatic macrophages and natural killer T cells, in the absence of obesity or insulin resistance. Our findings suggest that maternal insulin resistance, including elevated triglycerides, insulin, and weight gain, initiates dysregulation of the juvenile hepatic immune system and development of de novo lipogenic pathways that persist in vitro and may be an irreversible “first hit” in the pathogenesis of NAFLD in NHP.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease in children and adults (1,2). The clinical discovery of NAFLD in children has led to speculation that its origins may lie in early intrauterine exposures. Indeed, human infants born to obese and gestational diabetic mothers have increased liver fat compared with infants of normal mothers (3,4). While the epidemiologic association between increased maternal BMI and early childhood metabolic diseases has been well described (5–7), the mechanisms remain unclear. Studies in rodents have shown that maternal high-fat (HF) diet and insulin resistance during pregnancy leads to offspring obesity and hepatic lipid accumulation (8–11). However, rodent studies are limited in their ability to explain whether and how differences in maternal metabolism and intrauterine exposures may affect human development.
To study the effect of maternal HF diet and maternal phenotype on the offspring, we have used a nonhuman primate (NHP) model where female Japanese macaques are maintained on a chronic HF diet (12–15). We took advantage of the fact that some females on a HF diet become insulin resistant (IR; HF+IR) while other females remain insulin sensitive (IS; HF+IS) compared with control (CON) females. We previously showed that all maternal HF diet–exposed fetuses had increased liver triglyceride content (12,14). Further, when mothers on the HF diet were switched to a CON diet during the next pregnancy, fetal hepatic triglycerides were reduced (12). These findings demonstrate that intrauterine exposure to maternal HF diet, independent of maternal metabolic effects, increases fetal hepatic steatosis. Whether this is reversible postnatally is unknown but has important clinical implications for understanding the pathophysiology of NAFLD since liver damage in utero may prime the liver for the progression of metabolic disease.
In this study, we hypothesized that exposure to maternal HF diet during pregnancy through weaning would produce long-lasting effects on the offspring liver and may induce an inflammatory response in addition to steatosis noted earlier in third-trimester fetuses. Further, we tested if switching juvenile offspring exposed to maternal HF diet to a CON diet after weaning might alleviate the adverse consequences caused by exposure to maternal HF diet. Our results suggest that regardless of postweaning diet, juvenile NHP offspring from HF+IR, but not HF+IS, mothers develop increased hepatic de novo fatty acid synthesis and increased activated hepatic macrophages in the absence of obesity. Juvenile body weight, adiposity, and early signs of insulin resistance increased modestly on the postweaning HF diet, yet liver triglycerides and inflammation were not increased further. These data demonstrate that early exposure to a maternal HF+IR environment provokes the development of juvenile NAFLD that is irreversible in nondiabetic, preobese juvenile NHP.
Research Design and Methods
Maternal Diet Model
All animal procedures were in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Oregon National Primate Research Center. Adult female Japanese macaques were maintained on a CON (14.6% calories from fat) or HF (36.6% calories from fat) diet for 1–5 years prior to pregnancy (12–15). Body weight and baseline insulin, glucose, and triglyceride measurements were collected, and intravenous glucose tolerance tests (GTTs) were performed during the third trimester of each pregnancy (12). The area under the curve (AUC) for glucose and insulin was calculated from zero. Mothers were classified during pregnancy as HF+IR if they had an insulin AUC greater than two SDs above the mean of the CON mothers (Fig. 1A). Mothers with insulin AUC less than two SDs relative to the CON group were classified as HF+IS (12,13).
Figure 1.
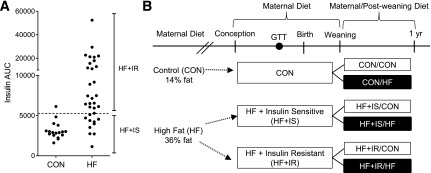
Study design and maternal HF diet classification. Females were placed on a CON or chronic HF diet beginning prior to pregnancy. Maternal metabolic phenotype was measured during the early third trimester. A: Based on GTTs during pregnancy, females were classified as HF and IR (HF+IR) if they had an insulin AUC greater than two SDs above CON females. The dashed line represents two SDs above the mean in the CON group. Females on the HF diet were IS (HF+IS) if they had an insulin AUC similar to CON females. B: Offspring were exposed to the maternal diet and maternal metabolic phenotype from conception to weaning. At weaning, around 7 months of age, offspring were placed on a postweaning CON (white bars) or HF (black bars) diet until the end of study at 1 year of age. Offspring diet treatment groups are indicated.
Offspring born to CON, HF+IS, and HF+IR dams were maintained with their mothers until weaning (7–8 months). At weaning, CON, HF+IS, and HF+IR offspring were placed on a CON or HF diet (13,15) (Fig. 1B). Body composition analysis (DEXA) was performed at 10 and 12 months of age (13). Daily growth and lean and fat mass (gain/loss) rates were calculated. At 1 year of age, juvenile animals were killed (13,15). Plasma glucose, insulin, nonesterified fatty acids, triglycerides, and cytokine concentrations were measured (12,13,16). HOMA-IR was calculated (13).
Liver Lipid Analysis
Liver triglyceride content was measured (Infinity Triglyceride Reagent) following lipid extraction (12). In a subset of offspring, lipid mass spectrometry fatty acid profiling was performed to measure triglyceride, diacylglyceride, and phospholipid concentrations and fatty acid composition (17).
Gene Expression
RNA was extracted from liver and omental adipose tissue samples, and gene expression was measured using real-time PCR (LightCycler 480, Roche) as described (18) (see Supplementary Table 1 for primers). Results were normalized to 18s rRNA.
Primary Hepatocyte and Hepatic Macrophage Studies
Primary hepatocytes and hepatic macrophages were isolated from a subset of offspring (18). Hepatocytes were plated in complete DMEM (5.5 mmol/L glucose supplemented with 2 mmol/L glutamine, 2 mmol/L lactate, 1 mmol/L pyruvate, 1× nonessential amino acids, 100 units/mL penicillin–streptomycin, 1 nmol/L insulin, 100 nmol/L dexamethasone, and 10% FBS) on collagen-coated plates. After a 4-h attachment period, cells were washed and media was replaced with serum-free DMEM plus 0.2% BSA (SF) with or without 1 nmol/L insulin and 100 nmol/L dexamethasone, as described below.
For hepatic macrophage isolations, the supernatant from the hepatocyte isolation containing nonparenchymal cells was filtered and spun at 300× g to pellet nonparenchymal cells. Cells were resuspended in Histodenz (24%), and gradients were prepared and spun at 1,500× g for 20 min (19). Cells at the interface were collected, washed, and plated on tissue culture plates in complete DMEM with 55 μmol/L 2-mercaptoethanol. After 1–2 h attachment, nonadherent cells were aspirated and media was replaced on adherent cells, representing hepatic macrophages (20).
Inflammation Gene Expression Studies
Hepatocytes incubated in SF media and hepatic macrophages incubated in complete media with 2-mercaptoethanol were studied 24 h after isolation. Cells were incubated with SF media (basal), lipopolysaccharide (LPS; 100 ng/mL), or palmitate (PALM; 200 nmol/L) for 3 h of treatment and then harvested for RNA isolation and gene expression.
Lipogenic Gene Expression Studies
Lipogenic gene expression was measured 24 h after isolation following 8 h of treatment with SF media (basal, 5.5 mmol/L glucose) or high-nutrient media (SF media + 25 mmol/L glucose, 500 nmol/L dexamethasone, 100 nmol/L insulin). Cells were collected for RNA isolation and analyzed for gene expression.
Statistical Analysis
Maternal data were analyzed by mixed-model ANOVA with fixed effect of maternal diet (CON, HF+IS, HF+IR) and random effect of mother using SAS (PROC MIXED). Offspring data were analyzed by two-way ANOVA with fixed effects of maternal diet, postweaning diet, and interaction. Both models accounted for the inequality of variances between maternal groups. Individual posttest comparisons were made for comparisons of interest and when the interaction was significant (P < 0.15). Isolated cell data were analyzed similarly by one- or two-way ANOVA. Statistical significance was declared at P < 0.05 and P < 0.15 for interactions.
Results
GTTs Identify IR and IS Mothers
Using GTTs, we identified 25 pregnant females on the chronic HF diet demonstrating insulin resistance (HF+IR) (Fig. 1A). The remaining 10 females on the HF diet were IS (HF+IS). Maternal body weight was 20–30% higher in HF+IR mothers compared with HF+IS and CON mothers (Table 1). Fasting insulin concentrations were increased fourfold in HF+IR and 50% in HF+IS mothers compared with CON. Fasting glucose concentrations and glucose AUC during the GTT were similar between HF+IR and HF+IS mothers. Fasting triglyceride concentrations were increased in HF+IR mothers compared with HF+IS or CON mothers. Thus some mothers on the HF diet (HF+IR) have insulin resistance and higher triglycerides yet remain glucose tolerant.
Table 1.
Maternal characteristics during pregnancy in each maternal group
| CON | HF+IS | HF+IR | Maternal* | |
|---|---|---|---|---|
| n | 18 | 10 | 25 | |
| Years on HF diet | 2.4 ± 0.3 | 3.3 ± 0.3 | ||
| GTT glucose AUC | 7,534 ± 237a | 6,078 ± 333b | 6,911 ± 320a,b | <0.005 |
| GTT insulin AUC | 3,136 ± 261a | 3,711 ± 380a | 13,967 ± 2,267b | <0.0005 |
| Body weight (kg) | 9.4 ± 0.4a | 10.3 ± 0.5a | 12.3 ± 0.5b | <0.0005 |
| Glucose (mg/dL) | 42.7 ± 1.5 | 38.0 ± 2.6 | 45.4 ± 2.4 | 0.12 |
| Insulin (μU/mL) | 11.7 ± 1.6a | 18.5 ± 3.6a | 59.6 ± 10.3b | <0.001 |
| Leptin (ng/mL)† | 33.7 ± 8.2 | 54.1 ± 18.2 | 54.4 ± 10.1 | 0.24 |
| Glycerol (mg/mL) | 0.10 ± 0.02a | 0.18 ± 0.02a | 0.20 ± 0.03b | <0.01 |
| Triglycerides (mg/dL) | 61.66 ± 4.46a | 66.43 ± 6.61a | 84.96 ± 6.36b | <0.05 |
Differences between maternal groups are indicated by different superscript letters (P < 0.05).
Measurements in a subset of dams.
Postweaning Diet Modifies Offspring Growth and Plasma Profiles
Offspring from CON, HF+IS, and HF+IR females were maintained on their respective maternal diets or were switched to a CON or HF diet at weaning until 1 year of age (Fig. 1B). The postweaning HF diet had a modest effect on body weight and weight gain (Table 2). Offspring on the postweaning HF diet gained more weight per day compared with offspring on the CON postweaning diet. This was primarily due to increased lean rather than fat mass accretion. Retroperitoneal adipose mass tended to be increased in animals consuming the postweaning HF diet, independent of maternal group; however, body fat was low for all offspring at only 1–5% (Table 2).
Table 2.
Juvenile offspring characteristics at 1 year of age
| CON/CON | CON/HF | HF+IS/CON | HF+IS/HF | HF+IR/CON | HF+IR/HF | Diet effects |
|||
|---|---|---|---|---|---|---|---|---|---|
| Maternal | Postweaning | INT | |||||||
| Number of offspring | 21 | 9 | 5 | 5 | 9 | 16 | |||
| Growth | |||||||||
| Necropsy age (months)* | 12.8 ± 0.1 | 13.7 ± 0.3 | 13.3 ± 0.3 | 13.3 ± 0.4 | 13.5 ± 0.3 | 13.1 ± 0.3 | 0.96 | 0.51 | <0.05 |
| Body weight (g)* | 2,495 ± 65 | 2,788 ± 96 | 2,620 ± 100 | 2,580 ± 116 | 2,644 ± 78 | 2,905 ± 105 | 0.23 | <0.05 | 0.21 |
| Liver weight (g)* | 67 ± 2.3 | 71 ± 2.9 | 67 ± 3.7 | 63 ± 2.1 | 72 ± 3.3 | 72 ± 2.0 | 0.06 | 0.90 | 0.27 |
| Retroperitoneal adipose weight (g)* | 489 ± 50 | 644 ± 56 | 568 ± 176 | 546 ± 94 | 442 ± 37 | 724 ± 84 | 0.96 | 0.10 | 0.41 |
| DEXA weight (g)† | 2,535 ± 65 | 2,764 ± 104 | 2,628 ± 166 | 2,532 ± 196 | 2,731 ± 90 | 2,958 ± 115 | 0.10 | 0.28 | 0.51 |
| DEXA lean mass (g)† | 2,427 ± 66 | 2,611 ± 98 | 2,484 ± 146 | 2,418 ± 195 | 2,602 ± 82 | 2,815 ± 112 | 0.09 | 0.29 | 0.59 |
| DEXA fat mass (g)† | 49.3 ± 5.3 | 85.6 ± 11.0 | 71.9 ± 15.9 | 44.8 ± 8.5 | 55.9 ± 17.3 | 68.2 ± 12.6 | 0.70 | 0.49 | <0.05 |
| DEXA fat (% weight)† | 2.0 ± 0.2 | 3.1 ± 0.4 | 2.7 ± 0.4 | 1.8 ± 0.4 | 2.0 ± 0.6 | 2.3 ± 0.4 | 0.52 | 0.59 | <0.05 |
| Body weight gain (g/day)† | 1.9 ± 0.7 | 5.7 ± 0.6 | 1.1 ± 1.4 | 4.6 ± 0.5 | 3.0 ± 0.6 | 4.9 ± 0.5 | 0.46 | <0.001 | 0.36 |
| Lean mass gain (g/day)† | 2.6 ± 0.6 | 4.8 ± 0.7 | 1.6 ± 0.8 | 4.4 ± 0.4 | 3.5 ± 0.6 | 4.8 ± 0.4 | 0.19 | <0.001 | 0.47 |
| Fat mass gain (g/day)† | −0.9 ± 0.3 | 0.6 ± 0.3 | −0.7 ± 0.5 | 0.0 ± 0.1 | −0.7 ± 0.4 | −0.2 ± 0.1 | 0.66 | <0.01 | 0.30 |
| Plasma* | |||||||||
| Glucose (mg/dL) | 51.3 ± 3.2 | 60.5 ± 3.7 | 56.0 ± 4.4 | 53.0 ± 3.0 | 61.1 ± 4.4 | 58.9 ± 3.2 | 0.33 | 0.67 | 0.24 |
| Insulin (mU/mL) | 4.0 ± 0.6 | 8.4 ± 1.4 | 6.4 ± 2.1 | 6.7 ± 0.9 | 5.8 ± 1.8 | 10.5 ± 2.5 | 0.59 | <0.05 | 0.25 |
| HOMA-IR | 0.5 ± 0.1 | 1.3 ± 0.2 | 0.9 ± 0.4 | 0.9 ± 0.1 | 0.9 ± 0.3 | 1.7 ± 0.4 | 0.50 | 0.06 | 0.19 |
| Glucose:insulin ratio | 17.7 ± 2.4 | 9.8 ± 2.4 | 11.5 ± 3.6 | 8.4 ± 1.1 | 17.3 ± 3.6 | 9.8 ± 1.8 | 0.25 | <0.01 | 0.59 |
| Glycerol (mg/mL) | 0.08 ± 0.02 | 0.14 ± 0.07 | 0.07 ± 0.03 | 0.07 ± 0.02 | 0.16 ± 0.06 | 0.06 ± 0.01 | 0.27 | 0.55 | 0.06 |
| Triglycerides (mg/dL) | 41.8 ± 3.3 | 45.2 ± 10.9 | 54.8 ± 11.7 | 39.8 ± 7.5 | 33.9 ± 6.4 | 41.2 ± 4.0 | 0.34 | 0.80 | 0.35 |
| Nonesterified fatty acid (mEq/L) | 0.81 ± 0.08 | 0.57 ± 0.13 | 0.63 ± 0.06 | 0.70 ± 0.11 | 0.72 ± 0.12 | 0.66 ± 0.08 | 0.96 | 0.35 | 0.31 |
| Hepatic lipids‡ | |||||||||
| Triglycerides | 23.5 ± 4.6 | 55.6 ± 22.3 | 114.8 ± 23.0 | 103.9 ± 38.7 | 0.08 | 0.78 | 0.57 | ||
| Diacylglycerides | 0.87 ± 0.13 | 1.21 ± 0.20 | 1.62 ± 0.19 | 1.46 ± 0.19 | <0.05 | 0.66 | 0.28 | ||
| Phospholipids | 218.3 ± 17.8 | 238.0 ± 20.4 | 183.0 ± 12.5 | 223.5 ± 9.7 | 0.11 | <0.05 | 0.49 | ||
Measured at time of necropsy.
DEXA measurements at 12 months and growth rates calculated from DEXA analysis at 10 and 12 months.
Liver triglycerides, diacylglycerides, and phospholipids measured by mass spectrometry in a subset of offspring (n = 3–8 per group), expressed as μg fatty acid per mg liver weight. INT, interaction.
Fasting plasma glucose, glycerol, triglyceride, and nonesterified fatty acid concentrations in juvenile offspring were similar among all groups (Table 2). Offspring fasting insulin concentrations and HOMA-IR calculations were twofold higher, and glucose:insulin ratios were 50% lower on the postweaning HF diet, with HF+IR/HF offspring having the highest insulin concentrations. Notably, HOMA-IR was improved in HF+IR/CON offspring, suggesting that insulin resistance was reversed by switching to the CON diet at weaning. Thus the postweaning HF diet had modest effects on inducing insulin resistance and increasing body weight.
Maternal Phenotype Dictates Hepatic Lipid Accumulation in Offspring
Liver triglyceride content was 50% higher in animals from HF+IR mothers compared with offspring from CON and HF+IS mothers, regardless of postweaning diet (Fig. 2A). Similarly, cytoplasmic lipid droplets were visible in livers from HF+IR offspring even on a CON postweaning diet compared with the offspring livers from HF+IS or CON mothers (Fig. 2B). Likewise, hepatic diacylglycerides were higher in juveniles born to HF+IR mothers compared with CON offspring regardless of postweaning diet (Table 2). These data strongly implicate maternal metabolic phenotype as the primary driver of persistent juvenile hepatic lipid content.
Figure 2.

Effect of maternal and postweaning HF diet on juvenile offspring hepatic lipid accumulation and lipogenic capacity. A: Liver triglyceride concentrations measured in offspring from CON, HF+IS, and HF+IR mothers (horizontal axis labels) on the postweaning CON (white bars) or HF (black bars) diet. In panels A and C, when the main effect of maternal group was significant, comparisons between groups are indicated: *P < 0.05 vs. maternal CON and **P < 0.05 vs. maternal HF+IS. B: Histological analysis of livers stained with hematoxylin and eosin. C: Hepatic mRNA expression of lipid metabolism genes (n = 21, 9, 5, 5, 9, and 16 per group). The main effect of postweaning CON versus HF diet is shown as #P < 0.05. D: Primary hepatocytes were treated for 8 h with high glucose and lipogenic hormones (insulin and dexamethasone). Gene expression results are expressed as fold increase relative to basal treatment (n = 2 CON/CON in white bars, 3 HF+IR/CON in black bars). *P < 0.05 vs. CON/CON; #P < 0.10 vs. CON/CON.
Fatty acid profiling was performed to determine whether maternal or postweaning diet affected lipid composition in the juvenile liver. The composition of fatty acids in hepatic triglycerides and diacylglycerides (Supplementary Table 2) reflected the composition of the postweaning diet (14). Relative saturated and monounsaturated fatty acids were increased, polyunsaturated fatty acids were decreased, and the ratio of n-6:n-3 fatty acids was increased in hepatic triglycerides and diacylglycerides on the postweaning HF diet (Supplementary Table 2). Increased diacylglyceride saturation has been implicated in insulin resistance (17,21–23); however, animals from HF+IR mothers had lower total diacylglyceride saturation compared with juvenile livers from HF+IS and CON mothers (Supplementary Table 2). Hepatic phospholipids were increased by 20% in all offspring on the postweaning HF diet compared with the CON diet (Table 2). Thus hepatic lipid composition was primarily reliant on diet and occurred independently of maternal phenotype.
Maternal Insulin Resistance Programs Increased Hepatic Lipogenic Gene Expression in Offspring
To investigate transcriptional mechanisms for hepatic steatosis in HF+IR offspring, we analyzed hepatic gene expression (Fig. 2C and D). Expression of the lipogenic transcription factor SREBP1 was twofold higher in the offspring of HF+IR mothers compared with HF+IS and CON offspring. The SREBP1 and insulin target genes for lipogenesis, FAS and ACC1, were also increased in HF+IR offspring compared with CON and HF+IS offspring (Fig. 2C). Other genes in the glycerol phosphate pathway for de novo diacylglyceride and triglyceride synthesis (24), including DGAT1, LPIN1, GPAT1, and AGPAT1, were increased in HF+IR offspring, regardless of postweaning diet (Fig. 2C). Expression of the fatty acid transporter CD36 was increased both by maternal and postweaning HF diet (Fig. 2C), suggesting increased lipid uptake. Expression of the lipid export gene APOB was also higher in HF+IR juvenile livers (Fig. 2C), which may reflect a compensatory mechanism in response to increased hepatic lipids.
We next determined whether the responsiveness to lipogenic stimuli was increased in isolated hepatocytes from juvenile animals born to HF+IR mothers. Hepatocytes were treated with high glucose, insulin, and dexamethasone. Since juveniles born to HF+IR mothers have increased hepatic lipids even on a CON postweaning diet, we studied hepatocytes from these offspring and CON/CON offspring. Compared with stimulated hepatocytes from CON offspring, stimulated HF+IR/CON hepatocytes had a greater increase in SREBP1, SREBP2, FAS, ACC1, DGAT1, and LPIN1 expression (Fig. 2D). Thus maternal insulin resistance has a long-lasting effect on juvenile liver lipogenic capacity that persisted in isolated hepatocytes and despite a healthy diet intervention after weaning.
Juvenile Livers from IR Mothers Display Inflammation
HF+IR mothers may confer an effect on the adaptive immune system in the offspring liver that plays a role in the evolution of NAFLD from simple steatosis to nonalcoholic steatohepatitis. To explore this, we characterized inflammatory gene expression in the juvenile livers. In whole liver tissue, juvenile offspring born to HF+IR mothers had increased expression for CD45, a marker for immune cells; CD11B and CD68, both canonical macrophage markers; and CD1D expression, a marker for natural killer T (NKT) cells, suggestive of increased immune cell numbers (Fig. 3). Expression of these immune cell markers persisted despite switching to a healthy CON diet postweaning. Although there was no change in expression of MCP1, we did find increased expression of the MCP1 chemokine receptor, CCR2, in HF+IR livers, suggesting potential for increased macrophage recruitment (25).
Figure 3.
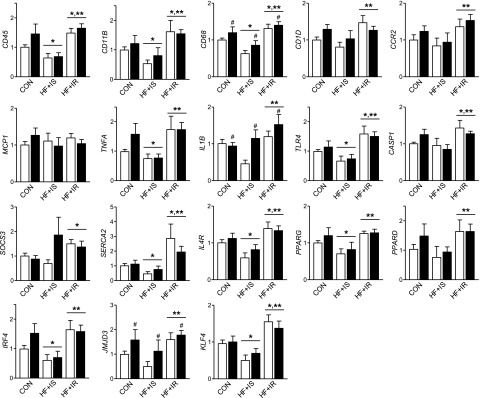
Effect of maternal and postweaning HF diet on offspring hepatic inflammation and stress pathways. Hepatic mRNA expression measured in offspring from CON, HF+IS, and HF+IR mothers (horizontal axis labels) on the postweaning CON (white bars) or HF (black bars) diet (n = 21, 9, 5, 5, 9, and 16 per group). When the main effect of maternal group was significant, comparisons between groups are indicated: *P < 0.05 vs. maternal CON and **P < 0.05 vs. maternal HF+IS. The main effect of postweaning CON versus HF diet is shown as #P < 0.05.
Expression of the proinflammatory cytokines TNFA and IL1B were higher in the liver from HF+IR offspring compared with HF+IS or CON/CON offspring (Fig. 3). Intriguingly, TLR4 expression, an innate immune response receptor, was increased by 50% in the HF+IR juvenile liver compared with CON and HF+IS offspring and did not increase further with postweaning HF diet (Fig. 3). Expression of caspase 1 (CASP1) and SOCS3 was also increased in HF+IR liver, consistent with increased cytokine activation. Expression of SERCA2, a marker of ER stress, was increased in HF+IR livers (Fig. 3). Thus juvenile livers from HF+IR mothers showed increased activation of the innate immune system and classical macrophage activation as described in human NAFLD (26,27).
We also found evidence for alternatively activated macrophage pathways in the liver from animals born to HF+IR mothers (Fig. 3). Expression of the interleukin (IL)-4 receptor (IL4R) was increased in the HF+IR liver, suggesting increased responsiveness to IL-4, which induces alternatively activated macrophages (28). We also found increased expression of PPARG, PPARD, KLF4, IRF4, and JMJD3, factors promoting alternatively activated macrophages (28), in HF+IR offspring. We conclude that there was increased innate immune activation and alternative macrophage activation in the juveniles born to HF+IR mothers, independent of postweaning dietary manipulation.
Isolated Hepatic Macrophages From Offspring of IR Mothers Show a Combination of Classical and Alternative Activation
Given the gene expression pattern in whole liver tissue suggesting increased macrophage activation, we wanted to determine the specific activation phenotype of hepatic macrophages to inflammatory signals. Hepatic macrophages and hepatocytes were isolated from the livers of CON/CON and HF+IR/CON juvenile offspring. Hepatic macrophages had higher expression of CD45, CD11B, CD68, TLR4 (Fig. 4A, Supplementary Table 3), IL1B, TNFA, IL6, and MCP1 (compare Fig. 4B and C) compared with hepatocytes. Moreover, hepatic macrophages from HF+IR/CON offspring had higher basal expression of CD45, CD68, and TLR4 compared with CON/CON cells (Fig. 4A) and tended to have higher expression of JMJD3, KLF4, PPARD, PPARG, CCR2 (Fig. 4A), and IL4R (Fig. 4B). These results suggest that offspring born to HF+IR mothers and weaned to a healthy diet have increased recruitment (CCR2), increased innate activation (TLR4), and an alternative activation macrophage phenotype (PPARG, PPARD, IL4R) (25,28).
Figure 4.

Effect of maternal HF diet on hepatic macrophage and hepatocyte inflammatory activation. Hepatic macrophages and hepatocytes were isolated from juvenile livers and studied after 3 h of treatment with PALM and LPS compared with basal treatment (n = 2 CON/CON, 3 HF+IR/CON). A: Expression of canonical macrophage and activation markers in hepatic macrophages from CON/CON (white bars) and HF+IR/CON (dashed bars) offspring. Results shown are least square means and SEM for each offspring group. *P < 0.05 for main effect of offspring group; #P < 0.15. B: Expression of inflammatory genes in hepatic macrophages from CON/CON (white bars) and HF+IR (dashed bars) offspring following basal, LPS, and PALM treatment. C: Expression of inflammatory genes in hepatocytes from CON/CON (white bars) and HF+IR/CON (black bars) offspring following basal, LPS, and PALM treatment. In panels B and C, all results are expressed relative to hepatocyte basal CON/CON group. When the main effect of treatment was significant, comparisons between treatments are indicated: *P < 0.05 vs. BASAL. The main effect of maternal group (CON/CON versus HF+IR/CON) is shown as #P < 0.05.
Next we challenged hepatic macrophages in vitro with PALM or LPS to see whether maternal HF diet increased their responsiveness to stimuli. All cells had a robust increase in expression of the proinflammatory cytokines IL1B, TNFA, IL6, and MCP1 (Fig. 4B). Importantly, this increase in IL1B and TNFA was greater in hepatic macrophages from HF+IR compared with CON offspring, consistent with increased TLR4 expression in these cells (Fig. 4A). Expression of the cytokine signaling suppressor, SOCS3, tended to be higher in HF+IR/CON macrophages (Fig. 4B). Interestingly, in response to PALM and LPS, hepatic macrophages from HF+IR/CON juveniles had a fivefold increase in IL4R expression (Fig. 4B), suggesting increased alternative macrophage activation. These results suggest that maternal insulin resistance programs an inflammatory response that includes a combination of classic and alternative activation in hepatic macrophages.
In contrast to macrophages, PALM treatment in hepatocytes had a small effect on inducing IL1B and TNFA expression and lack of effect on IL6, MCP1, SOCS3, and IL4R expression, regardless of maternal diet group (Fig. 4C). In both CON/CON and HF+IR/CON hepatocytes, LPS, relative to PALM, elicited a greater inflammatory response as evidenced by increased expression of IL1B, TNFA, IL6, MCP1, and SOCS3; however, the fold induction was much less in hepatocytes compared with macrophages (compare Fig. 4B and C). These data support that exposure to the maternal HF+IR phenotype may prime hepatic macrophages, more so than hepatocytes, toward increased responsiveness to proinflammatory stimuli.
No Change in Adipose Tissue Inflammation or Circulating Cytokines in Juvenile Offspring
We measured adipose tissue inflammatory gene expression and circulating cytokines to assess systemic inflammation. In adipose tissue, expression of inflammation genes was not affected by maternal or postweaning diet (Table 3). Plasma cytokine concentrations were also not affected by maternal or postweaning diet in juvenile offspring (Table 3). These data suggest that maternal insulin resistance primes the liver for proinflammatory activation of resident hepatic macrophages, in the absence of adipocyte inflammation.
Table 3.
Circulating cytokines and adipose tissue gene expression in juvenile offspring
| CON/CON | CON/HF | HF+IR/CON | HF+IR/HF | Diet effects |
|||
|---|---|---|---|---|---|---|---|
| Maternal | Postweaning | INT | |||||
| Adipose mRNA gene expression | |||||||
| n | 12 | 9 | 5 | 10 | |||
| CD68 | 1.0 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.1 | 0.8 ± 0.1 | 0.50 | <0.05 | 0.16 |
| IL6 | 1.0 ± 0.5 | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.4 ± 0.1 | 0.51 | 0.31 | 0.47 |
| TLR4 | 1.0 ± 0.1 | 0.8 ± 0.1 | 1.1 ± 0.1 | 0.9 ± 0.1 | 0.54 | <0.05 | 0.65 |
| MCP1 | 1.0 ± 0.2 | 0.7 ± 0.3 | 0.8 ± 0.1 | 0.5 ± 0.1 | 0.41 | 0.19 | 0.89 |
| IL1B | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.4 | 1.1 ± 0.2 | 0.44 | 0.66 | 0.87 |
| TNFA | 1.0 ± 0.3 | 0.9 ± 0.2 | 0.8 ± 0.1 | 0.7 ± 0.1 | 0.39 | 0.65 | 0.98 |
| NLRP3 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 2.0 ± 0.1 | 0.49 | 0.89 | 0.79 |
| SERCA2 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.2 | 0.06 | 0.89 | 0.96 |
| Plasma cytokine concentrations* | |||||||
| n | 4 | 5 | 3 | 4 | |||
| IL-2 | 226 ± 158 | 65 ± 17 | 57 ± 5 | 84 ± 29 | 0.38 | 0.43 | 0.27 |
| IL-8 | 7,133 ± 1,366 | 8,895 ± 336 | 8,868 ± 475 | 7,438 ± 943 | 0.87 | 0.85 | 0.09 |
| IL-12 | 1,142 ± 114 | 930 ± 73 | 991 ± 114 | 941 ± 103 | 0.49 | 0.22 | 0.43 |
| MCP1 | 1,541 ± 180 | 1,596 ± 85 | 1,570 ± 135 | 1,484 ± 101 | 0.75 | 0.90 | 0.59 |
| sCD40L | 5,305 ± 1,082 | 4,728 ± 516 | 4,790 ± 752 | 5,357 ± 155 | 0.95 | 0.99 | 0.54 |
| Transforming growth factor-α | 208 ± 64 | 216 ± 53 | 176 ± 20 | 137 ± 21 | 0.28 | 0.76 | 0.63 |
Values are arbitrary units. Other cytokines, granulocyte macrophage colony-stimulating factor, interferon-γ, IL-1β, IL-4, IL-5, IL-6, IL-10, tumor necrosis factor-α, and vascular endothelial growth factor, were below detectable levels. INT, interaction.
Maternal Metabolism Predicts Liver Outcomes in Offspring
Maternal insulin AUC during GTT, body weight, and plasma triglycerides were all positively related to offspring liver triglycerides (Fig. 5A). Interestingly, in the HF+IS and HF+IR offspring, maternal insulin AUC correlated more closely with offspring liver inflammatory markers, including IL1B, TLR4, and CD11B (R = 0.32–0.48) (Fig. 5B), than with hepatic triglyceride content (Fig. 5A) or SREBP1, SREBP2, or FAS expression (R = 0.11–0.16) (Fig. 5C). These relationships suggest the effects of maternal metabolism have more predictive power on the offspring’s hepatic immune system than on the de novo lipogenic pathway in development of juvenile NAFLD.
Figure 5.
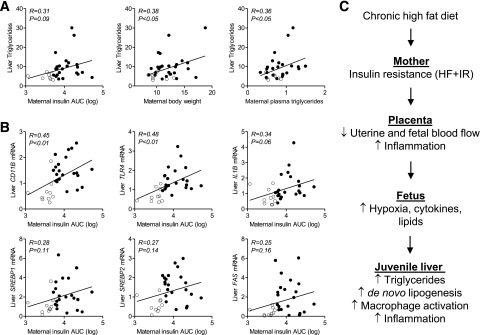
Relationship between maternal outcomes on the HF diet with juvenile offspring hepatic triglycerides and gene expression. Correlations between (A) juvenile offspring liver triglycerides and maternal insulin AUC (log transformed) during GTT, pregnancy body weight, and plasma triglycerides and (B) offspring inflammation and lipogenic gene expression with maternal insulin AUC. Open circles represent HF+IS (n = 9) and closed circles represent HF+IR (n = 25) maternal–offspring pairs. C: Mechanisms for NAFLD in HF+IR offspring. HF+IR placentas have increased transplacental lipids, decreased uterine and fetal blood flow, and increased placental inflammation, which increases fetal cytokine exposure. We speculate that this combination of lipid, cytokine, and hypoxia exposure in utero produced by the HF+IR mother programs the offspring liver for increased NAFLD at 1 year of age.
Discussion
We took advantage of the fact that not all NHP mothers consuming a chronic HF diet are obese, and like humans, some are significantly overweight and have exaggerated insulin resistance (e.g., HF+IR mothers) while others consuming the HF diet remain relatively metabolically normal (e.g., HF+IS mothers). Unlike juvenile offspring from HF+IS mothers, which largely resembled CON offspring, offspring from HF+IR mothers showed a pattern of hepatic gene expression, cytokines, and lipogenic responses consistent with the early stages of NAFLD. Importantly, the NAFLD phenotype persisted despite the absence of postnatal obesity, insulin resistance, or systemic or local adipose tissue inflammation. These findings indicate that maternal metabolic dysfunction programs the NAFLD phenotype and suggests that postnatal diet may be incapable of reversing or correcting this disorder in juvenile offspring.
The livers of HF+IR NHP juvenile offspring showed evidence for increased de novo lipogenic capacity. The genes SREBP1, SREBP2, FAS, ACC1, LPIN1, and DGAT1 were higher in the HF+IR offspring, despite consuming a healthy diet after weaning. Because the turnover rate of the liver triglyceride pool is ∼40 days or less in humans (29), the 5-month period where offspring are on the healthy CON postweaning diet should be sufficient for any maternally derived lipid to dissipate. Further, in isolated hepatocytes, the same lipogenic gene pattern was increased upon hormone stimulation, supporting that livers from HF+IR offspring have a gene profile favoring lipid synthesis and storage that persists postnatally. Increased hepatic steatosis has been reported in postnatal offspring in other rodent models of maternal obesity (9–11,30,31); however, most of these offspring also have increased adiposity, making the early origins of steatosis difficult to discern. We observed that in the NHP HF+IR offspring, steatosis is present when the animals are lean with no detectable systemic inflammation. This demonstrates that the mechanism(s) driving excess hepatic fat storage may be different than those underlying adipose tissue expansion and suggests that the early NAFLD phenotype in HF+IR offspring may be an early risk factor for other metabolic diseases, including cardiovascular disease, cancer, and obesity (32,33).
NAFLD is often associated with insulin resistance, and whether insulin resistance is a cause or consequence of hepatic steatosis remains debatable (22,34,35). In our juvenile offspring, continued HF diet exposure after weaning increased fasting insulin concentrations and insulin AUC during the GTT (13); however, this hyperinsulinemia did not accelerate hepatic steatosis further in the HF+IR/HF juveniles. Likewise, HF+IR/CON offspring, when switched to a healthy diet at weaning, showed a persistent increase in steatosis and de novo lipogenic gene expression, despite lower insulin levels and normal body weight compared with HF+IR/HF offspring on the postweaning HF diet. Thus neither increased insulin concentrations nor insulin resistance in the offspring explain increased de novo lipogenesis in the HF+IR juvenile offspring. Overall, this suggests that hepatic steatosis found in fetuses from HF-diet mothers is resistant to changes in diet after weaning and, therefore, in utero exposures from an HF+IR mother may lay the foundation for an “unhealthy” liver.
Inflammation is an important link in the progression of NAFLD, and steatosis is associated with increased macrophage activation (36–42). Offspring born to HF+IR mothers, regardless of postweaning diet, had increased hepatic macrophage numbers (increased CD11B and CD68 mRNA), which may reflect increased recruitment (increased CCR2 mRNA) (25). Here, hepatic macrophages in HF+IR offspring present with increased classical and alternative activation. Increased classical activation of hepatic macrophages in HF+IR offspring is supported based on increased expression of TLR4 in HF+IR macrophages and a greater proinflammatory cytokine response (IL1B, TNFA, IL6) upon treatment with PALM and LPS. Our whole liver gene expression results support this based on increased IL1B, TNFA, CASP1, and TLR4 expression in HF+IR offspring. Increased alternative activation is evidenced by increased expression of IL4R, PPARG, PPARD, IRF4, JMJD3, and KLF4. Alternative activation of macrophages involves PPARG and PPARD, which fulfill a protective function to increase fatty acid oxidation (28). We speculate that alternative activation of hepatic macrophages in HF+IR offspring serves an adaptive or reparative role in response to steatosis. However, alternative activation of macrophages has also been associated with pathological tissue remodeling and fibrosis (42). Moreover, alternative activation is found in models of insulin resistance and obesity in liver and adipose macrophages (28,37,42–44). We also found increased expression of CD1D, suggesting increased NKT cell activation, which is seen in human NAFLD (45). Thus our results support that juveniles born to HF+IR mothers develop increased classical and alternatively activated hepatic macrophages and NKT cell activation that in combination with increased de novo fatty acid synthesis, supports a NAFLD phenotype.
The mechanisms whereby excessive maternal insulin resistance programs immune cells and NAFLD in the juvenile liver are unclear. Liver Kupffer cell populations are established prior to birth and self-maintained postnatally (46). Thus fetal exposures associated with maternal insulin resistance may prime these resident immune cells, rendering them more responsive to stimulation as proinflammatory effector cells and/or differentiation into alternatively activated macrophages. Although we previously found increased fetal liver triglycerides in offspring from HF diet–fed mothers (12), we found no evidence for increased hepatic cytokines, suggesting that immune cells may not have matured fully or that the stimulus for inflammation may occur in the final month of gestation, when fetal growth accelerates. We previously demonstrated that only HF+IR mothers and their fetuses have reduced uterine and fetal blood flow. Further, 13 of 36 cytokines were significantly upregulated in the umbilical, but not maternal, circulation of HF+IR maternal–fetal pairs (16). This suggests that high maternal insulin resistance may trigger fetal/placental hypoxia (16,47) and inflammatory cytokine production. Indeed, there is increased inflammation in cord blood of infants born to obese mothers (48) and in sheep fetuses exposed to overfeeding (49). Furthermore, we recently demonstrated that lowering placental inflammation in an obese pregnant transgenic mouse model prevented NAFLD (50). Together our results suggest a model whereby maternal insulin resistance on a HF diet may produce exposure to increased transplacental lipids and hypoxia/inflammatory stimuli in the fetus that drives inflammation and subsequent juvenile NAFLD (Fig. 5C).
Importantly, since not all obese women have babies born with increased birth weight or with fatty livers (3,4,7), focusing on the metabolic conditions that provoke excessive insulin resistance during pregnancy, rather than obesity per se, could be an important new therapeutic target for lessening the transmission of metabolic dysfunction from mother to child and preventing future NAFLD in the next generation.
Supplementary Material
Article Information
Acknowledgments. The authors thank Diana Takahashi, Ashley Kostrba, Sarah Comstock, and the staff of the Oregon National Primate Research Center for assistance with animal studies. The authors also thank Rachel C. Janssen (University of Colorado Denver) for assistance with editing the manuscript.
Funding. Research support was provided by the National Institutes of Health (K01-DK-090199 to S.R.T., P51-OD-011092 as partial salary support for K.L.G., and R24-DK-090964 to K.L.G. and J.E.F.), the National Institutes of Health/National Center for Advancing Translational Sciences Colorado Clinical and Translational Science Institute grant UL1-TR-000154 from the Colorado Nutrition and Obesity Research Center (NORC) (P30-DK-048520), and the Academic Enrichment Fund of the University of Colorado School of Medicine (to K.C.E.K.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. S.R.T. designed the experiments, performed liver and cell experiments, analyzed data, wrote the manuscript, and reviewed the manuscript. K.C.B. performed liver and cell experiments, analyzed data, performed lipid profiling, and reviewed the manuscript. S.A.N. performed liver and cell experiments, analyzed data, and reviewed the manuscript. K.C.E.K. reviewed the manuscript. B.C.B. and G.I.S. performed lipid profiling and reviewed the manuscript. K.L.G. designed the experiments and reviewed the manuscript. J.E.F. designed the experiments, wrote the manuscript, and reviewed the manuscript. J.E.F. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0276/-/DC1.
References
- 1.Welsh JA, Karpen S, Vos MB. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988-1994 to 2007-2010. J Pediatr 2013;162:496–500 [DOI] [PMC free article] [PubMed]
- 2.Agopian VG, Kaldas FM, Hong JC, et al. Liver transplantation for nonalcoholic steatohepatitis: the new epidemic. Ann Surg 2012;256:624–633 [DOI] [PubMed] [Google Scholar]
- 3.Brumbaugh DE, Tearse P, Cree-Green M, et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr 2013;162:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Modi N, Murgasova D, Ruager-Martin R, et al. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res 2011;70:287–291 [DOI] [PubMed] [Google Scholar]
- 5.Lawlor DA, Relton C, Sattar N, Nelson SM. Maternal adiposity—a determinant of perinatal and offspring outcomes? Nat Rev Endocrinol 2012;8:679–688 [DOI] [PubMed] [Google Scholar]
- 6.Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol 2010;299:R711–R722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamnes Køpp UM, Dahl-Jørgensen K, Stigum H, Frost Andersen L, Næss O, Nystad W. The associations between maternal pre-pregnancy body mass index or gestational weight change during pregnancy and body mass index of the child at 3 years of age. Int J Obes (Lond) 2012;36:1325–1331 [DOI] [PubMed] [Google Scholar]
- 8.Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 2008;294:R528–R538 [DOI] [PubMed] [Google Scholar]
- 9.Oben JA, Mouralidarane A, Samuelsson AM, et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol 2010;52:913–920 [DOI] [PubMed] [Google Scholar]
- 10.Bruce KD, Cagampang FR, Argenton M, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009;50:1796–1808 [DOI] [PubMed] [Google Scholar]
- 11.Isganaitis E, Woo M, Ma H, et al. Developmental programming by maternal insulin resistance: hyperinsulinemia, glucose intolerance, and dysregulated lipid metabolism in male offspring of insulin-resistant mice. Diabetes 2014;63:688–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCurdy CE, Bishop JM, Williams SM, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009;119:323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comstock SM, Pound LD, Bishop JM, et al. High-fat diet consumption during pregnancy and the early post-natal period leads to decreased α cell plasticity in the nonhuman primate. Mol Metab 2012;2:10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant WF, Gillingham MB, Batra AK, et al. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS ONE 2011;6:e17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grant WF, Nicol LE, Thorn SR, Grove KL, Friedman JE, Marks DL. Perinatal exposure to a high-fat diet is associated with reduced hepatic sympathetic innervation in one-year old male Japanese macaques. PLoS ONE 2012;7:e48119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frias AE, Morgan TK, Evans AE, et al. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 2011;152:2456–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergman BC, Perreault L, Hunerdosse DM, Koehler MC, Samek AM, Eckel RH. Increased intramuscular lipid synthesis and low saturation relate to insulin sensitivity in endurance-trained athletes. J Appl Physiol (1985) 2010;108:1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thorn SR, Brown LD, Rozance PJ, Hay WW, Jr, Friedman JE. Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 2013;62:65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Kasmi KC, Anderson AL, Devereaux MW, et al. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 2012;55:1518–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nnalue NA, Shnyra A, Hultenby K, Lindberg AA. Salmonella choleraesuis and Salmonella typhimurium associated with liver cells after intravenous inoculation of rats are localized mainly in Kupffer cells and multiply intracellularly. Infect Immun 1992;60:2758–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farese RV, Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 2012;15:570–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bergman BC, Perreault L, Hunerdosse DM, Koehler MC, Samek AM, Eckel RH. Intramuscular lipid metabolism in the insulin resistance of smoking. Diabetes 2009;58:2220–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res 2004;43:134–176 [DOI] [PubMed] [Google Scholar]
- 25.Obstfeld AE, Sugaru E, Thearle M, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010;59:916–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mouralidarane A, Soeda J, Visconti-Pugmire C, et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013;58:128–138 [DOI] [PubMed] [Google Scholar]
- 27.Syn WK, Oo YH, Pereira TA, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol 2011;6:275–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayol SA, Simbi BH, Fowkes RC, Stickland NC. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 2010;151:1451–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr 2009;102:514–519 [DOI] [PubMed] [Google Scholar]
- 32.Paradis V, Zalinski S, Chelbi E, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 2009;49:851–859 [DOI] [PubMed] [Google Scholar]
- 33.Santoro N, Caprio S. Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis in obese adolescents: A looming marker of cardiac dysfunction. Hepatology 2014;59:372–374 [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab 2013;24:4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clementi AH, Gaudy AM, van Rooijen N, Pierce RH, Mooney RA. Loss of Kupffer cells in diet-induced obesity is associated with increased hepatic steatosis, STAT3 signaling, and further decreases in insulin signaling. Biochim Biophys Acta 2009;1792:1062–1072 [DOI] [PMC free article] [PubMed]
- 37.Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol 2010;298:G107–G116 [DOI] [PubMed] [Google Scholar]
- 38.Tateya S, Rizzo NO, Handa P, et al. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes 2011;60:2792–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westerbacka J, Kolak M, Kiviluoto T, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007;56:2759–2765 [DOI] [PubMed] [Google Scholar]
- 40.Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab 2012;15:635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005;11:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol 2009;51:212–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014;59:130–142 [DOI] [PubMed] [Google Scholar]
- 44.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246 [DOI] [PubMed] [Google Scholar]
- 45.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 2013;10:627–636 [DOI] [PubMed] [Google Scholar]
- 46.Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013;38:79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilkening RB, Meschia G. Current topic: comparative physiology of placental oxygen transport. Placenta 1992;13:1–15 [DOI] [PubMed] [Google Scholar]
- 48.Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008;29:274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu MJ, Du M, Ford SP. Impacts of maternal obesity on placental and gut inflammation and health. J Anim Sci. 15 Nov 2013 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 50.Heerwagen MJ, Stewart MS, de la Houssaye BA, Janssen RC, Friedman JE. Transgenic increase in N-3/n-6 Fatty Acid ratio reduces maternal obesity-associated inflammation and limits adverse developmental programming in mice. PLoS ONE 2013;8:e67791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.