

Abstract
Centrosome amplification is a hallmark of cancer. However, despite significant progress in recent years, we are still far from understanding how centrosome amplification affects tumorigenesis. Boveri's hypothesis formulated more than 100 years ago was that aneuploidy induced by centrosome amplification promoted tumorigenesis. Although the hypothesis remains appealing 100 years later, it is also clear that the role of centrosome amplification in cancer is more complex than initially thought. Here, we review how centrosome abnormalities are generated in cancer and the mechanisms cells employ to adapt to centrosome amplification, in particular centrosome clustering. We discuss the different mechanisms by which centrosome amplification could contribute to tumour progression and the new advances in the development of therapies that target cells with extra centrosomes.
Keywords: centrosome amplification, centriole, clustering, cancer, aneuploidy, microtubules
1. Introduction
The centrosome is the main microtubule (MT)-organizing centre in animal cells, playing important roles in polarity, migration and cell division. The centrosome consists of a pair of orthogonally positioned centrioles, embedded in a complex proteinaceous structure, the pericentriolar material (PCM) [1]. In differentiated cells, the mother centriole, the older of the two centrioles, functions as the basal body that assembles the primary cilium, which, among other roles, can function as a centre for cellular signalling [2]. During the cell cycle, centrosomes duplicate only once during S phase to ensure that at mitotic onset a cell carries two centrosomes that will form the poles of the mitotic spindle [3]. Although Theodor Boveri originally described the centrosome as ‘the organ for cell division’, work in Drosophila and mammalian cells showed that cells without centrioles can assemble bipolar spindles [4–6], in part due to chromatin-mediated MT nucleation during mitosis. However, the idea that centrosomes are dispensable for mitosis is still controversial. Previous work showed that acentrosomal haploid cell cultures obtained from unfertilized Drosophila eggs are aneuploid, suggesting that centrosomes might be important to maintain genetic stability [7]. Supporting this idea, recent work demonstrated that permanent centriole loss in vertebrate DT40 cells leads to chromosome instability and aneuploidy [8].
A century ago, Boveri proposed that increased numbers of centrosomes cause cancer [9]. This was a bold move, given that he had never actually worked with cancer cells. Based on his observation that the sperm provided the functional centrosome early during embryogenesis, Boveri created dispermic eggs containing multiple centrosomes. These eggs, harbouring extra centrosomes, underwent multipolar mitoses and division of cells into three or more highly aneuploid progeny. These progeny all displayed different developmental characteristics, leading to the famous conclusion that chromosomes transmit these cellular traits [10,11]. This idea was the foundation for his later proposal for the driving role of aneuploidy in tumorigenesis [9]. The model that centrosome amplification caused improper chromosome segregation during mitosis, which triggered malignancy, had important contributions from his contemporaries Gino Galeotti and David von Hansemann. Both Galeotti and Hansemann, by observation of tumour histology, noted that abnormal mitotic figures are common features of cancer cells. Galeotti also recognized that abnormal mitoses were more frequently present in rapidly developing tumours [12]. Hansemann's work highlighted the presence of asymmetric cell divisions with abnormal distribution of chromosomes to daughter cells, which he termed asymmetric karyokinisis [13]. Although Hansemann reported that the presence of these abnormalities was common in carcinomas, he stated in his monograph of 1902 that a cancer diagnosis should not be made based solely on asymmetric nuclear divisions. In fact, Hansemann remarked that because these ‘faulty’ mitoses could be also observed in benign lesions or in tissue overgrowth, they were unlikely to be the cause of cancer [14]. Thus, from the earliest studies, opinions about whether chromosome segregation errors might cause cancer were divided: Boveri was in favour and Hansemann was opposed.
It was not only Hansemann who remained sceptical about the role of abnormal mitoses in cancer: indeed for many years the cancer field focused on the discovery of cancer-causing mutations in oncogenes and tumour suppressors as the drivers of tumorigenesis. It was not until the late 1990s, with the observation that loss of the tumour suppressor p53 was associated with centrosome amplification, that centrosome defects returned to the limelight [15]. Following this discovery, the work of many researchers established centrosome abnormalities as a common feature of all major classes of human cancer.
2. Landscape of centrosome abnormalities in human tumours
The prevalence and complexity of centrosomal abnormalities in human tumours is highlighted in a recent review that summarizes the existing clinical data concerning centrosome defects in cancer [16]. Centrosomal abnormalities have been described in a variety of solid tumours, including breast, prostate, colon, ovarian and pancreatic cancer [17–20], as well as haematological malignancies such as multiple myeloma, non-Hodgkin's and Hodgkin's lymphomas, acute and chronic myeloid leukaemia [21,22].
Centrosome abnormalities can be detected in early low-grade lesions in some tumours, such as breast cancer and several gastrointestinal cancers [23–25], suggesting the possibility of a role in tumour initiation, although the idea remains controversial. However, in most human cancers centrosome amplification has been associated with high-grade tumours and poor prognosis [16]. In some tumours, such as urothelial cancers, centrosome amplification is a strong predictor of tumour recurrence, highlighting its potential as a biomarker for advanced disease [26]. Also in breast, prostate and head and neck tumours, centrosome amplification is correlated with lymph node and distant metastasis, further reinforcing its association with disease progression [27–29]. Understanding the nature of this association, whether it is direct or indirect, could have a major impact in the developing therapies and new biomarkers.
3. Types of centrosomal defects
(a). Structural defects
Centrosomal defects in human cancers can be classified as structural or numerical aberrations [30] (figure 1). Structural defects can be divided into two groups: defects in centriole structure and defects in the amount of PCM. The most straightforward structural defects to identify are alterations in centriole size, usually scored as an increase in length but could also include increased variability in centriole length. The origins of centriole structural defects in cancer are still unclear, but changes in the expression of genes involved in controlling centriole structure are one appealing idea. For example, over- or under-expression of centrosomal components can lead to alterations in the centriole structure; for example, such as CPAP/SAS-4, whose overexpression increases centriole length, can affect centriole structure in several model systems [31–34].
Figure 1.
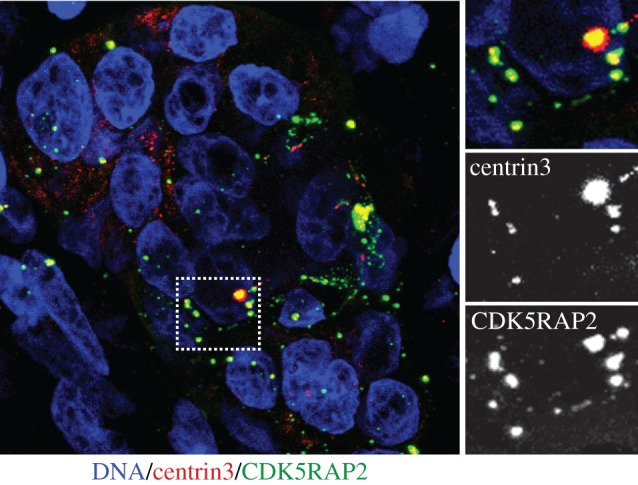
Centrosome abnormalities in high-grade serous ovarian cancer. Tissue sections of approximately 20 μm thickness were immunostained with antibodies against CDK5RAP2 (green) and centrin-3 (red). DNA is stained with Hoechst (blue). Area marked with rectangle is shown at higher magnification on right to illustrate numerical and structural aberrancies. In particular, note major variations in centrosome size and shape. Images shown are maximum intensity projections. Image is courtesy of Gayathri Chandrasekaran and Fanni Gergely.
Defining what constitutes a centriole structural defect in cancer is, however, not an easy task. First, because the size of centrioles (0.2–0.5 µm long) is close to the optical resolution of a standard light microscope, centriole length measurement requires either specialized fluorescence techniques or electron microscopy. Second, the common assays that are the basis for classifying tumours as having ‘structural’ defects can be difficult to interpret. For example, increased amount of PCM, deduced from measurement of the volume/diameter of the centrosome using a pericentriolar marker [27,35], has been considered a structural defect [30]. However, there can be two interpretations for a cell displaying an increased amount of PCM. One possibility is that PCM is indeed increased, which can be considered a structural alteration of the centrosome [27]. Alternatively, this could reflect supernumerary centrosomes that are typically clustered during interphase, and thus in fact be a ‘numerical defect’ [23,27,36]. These possibilities can only be distinguished with bona fide centriole labelling. The reverse misclassification can also occur since in vitro, increased centriole length was suggested to lead to centriole fragmentation [32]. Thus, it is not trivial to ascertain the origin of centrosome ‘structural’ alterations simply from fixed cell imaging, and this issue is further complicated by the fact that many primary tumour studies only examine PCM markers and not centriole markers. For these reasons, better methods to assess and classify centrosome abnormalities systematically are needed to understand better the landscape of centrosome abnormalities in human tumours and to gain insight into how these centrosomal defects arise.
(b). Numerical defects
Numerical aberrations, such as centrosome amplification, are the most frequently described centrosomal defects in cancer. A number of mechanisms can lead to centrosome amplification, including cytokinesis failure, mitotic slippage, cell–cell fusion, overduplication of centrioles and de novo centriole assembly [37]. The large number of underlying mechanisms may explain the diversity of proteins, including tumour suppressor and oncogenes, associated with centrosome amplification in cancer [38].
One major route for centrosome amplification is deregulation of the centrosome duplication cycle, which is in part controlled by the tight regulation of many of its components. Thus, many positive and negative regulators of centrosome duplication prevent centrosome amplification in normal cells [39]. This tightly controlled cycle relies on a surprisingly small number of core proteins that have been conserved during evolution [40]. One important core protein is the Polo-like kinase 4 (Plk4; SAK in Drosophila melanogaster, and ZYG-1 is the functional homologue in Caenorhabditis elegans), the master regulator of centrosome duplication (figure 2) [41]. Plk4 activity is a critical factor that regulates centriole number, as excess Plk4 activity leads to extra centrioles [42,43], whereas its depletion causes decrease in centriole numbers [4,42,44]. To ensure proper centriole duplication, the levels of Plk4 are regulated, mostly through SCFβTrCP/ubiquitin-dependent proteolysis [45–47], which is controlled, at least in part, by its own autophosphorylation [48–50]. Another important core protein is SAS-6, essential for the formation of the cartwheel structure that ensures the centriole's ninefold symmetry and whose levels are also controlled by proteolysis [51–57]. After initial formation, centrioles are stabilized and elongate until G2 phase, a process that involves CPAP/SAS-4 [31–34,58]. Centriole length is controlled by proteins such as CP110 and Cep97, which associate with the distal ends of the centrioles and likely function as capping proteins [32,33,59]. CP110 levels are also regulated by SCFcyclinF/ubiquitin-dependent proteolysis which is counteracted by the deubiquitinating enzyme USP33 [60], further highlighting the important role that proteolysis plays in this process. After centriole duplication, the centrosome undergoes maturation during G2/M phases leading to the increase accumulation of PCM, a process dependent upon Plk1 and Aurora A kinases [61,62]. At end of M phase, the protein link between the two centrioles is severed though the activities of Plk1 and separase, a process termed disengagement that primes the centrioles for duplication in the following S phase [63].
Figure 2.
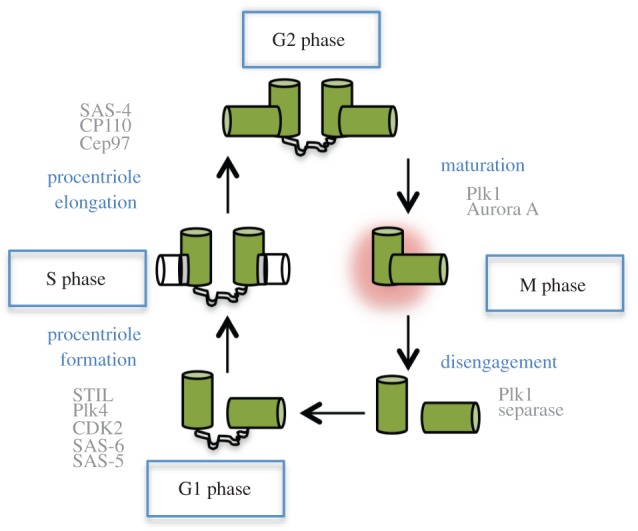
The centrosome duplication cycle. The master regulator of centriole duplication, the kinase Plk4, is recruited to the centrioles at the end of mitosis by the scaffolding protein CEP152/Asterless. Plk4 activity initiates the formation of the procentriole in S phase that forms adjacent to the pre-existing centriole. The centriole ninefold symmetry is established by the cartwheel, a structure formed through the oligomerization of SAS-6. During S phase CDK2/cyclin E activity is also important for centriole assembly together with the SCF complex which plays important roles in regulating the ubiquitination and proteasome-dependent degradation of many centriolar components. Procentriole elongation is promoted by recruitment of CPAP/SAS-4 which assembles and stabilizes the MTs that form the ninefold outer structure of the procentrioles. Elongation of the procentriole continues through G2 phase and is controlled by the capping proteins CP110 and Cep97 which regulate centriole size. Centrosome maturation is induced later in G2 phase and is promoted by Aurora A and Plk1, leading to increased PCM accumulation. Later, in M phase, centrioles disengage by a mechanism involving the activation of separase by the APC complex and the kinase Plk1. Centriole disengagement is an important licensing factor for centriole duplication, but exactly how this mechanism is regulated is still unclear.
In some tumours, centrosome amplification may result from centriole overduplication, for example through the overexpression of centriolar proteins. Deregulation of the ubiquitin regulators could play an important role in this process by altering the stability of centriolar components. For example, downregulation of βTrCP leads to centrosome amplification through the stabilization of Plk4 [45,47,64,65], whereas overexpression of USP33 leads to increased CP110 levels and centrosome amplification [60]. Regulation of centriolar proteins can also be altered at the level of transcription. High-risk human papillomavirus (HPV)-associated tumours are perhaps the best example of the occurrence of centrosome overduplication in cancer. Overexpression of the HPV-16 viral E6 and E7 oncoproteins has been shown to disrupt host cell cycle checkpoints important for oncogenic transformation [66]. In addition, the HPV-16 E7 disrupts normal centriole duplication, inducing centrosome amplification through a process that involves increased Plk4 mRNA steady-state levels [67]. Plk4 mRNA levels are negatively regulated by p53 through the recruitment of HDAC (histone deacetylases) repressors to the promoter of PLK4 [68]. Thus, loss of p53 could potentially contribute to centrosome amplification through increased levels of Plk4. This idea would fit with the observation that p53 loss is associated with increased centrosome numbers in mouse fibroblasts [15]. However, recent analysis of brains of p53–/– mice revealed that these animals have normal centrosome number [69], arguing that p53 loss is not sufficient to generate centrosome amplification. Thus, p53 may play either a direct contributory role or only an indirect role in allowing cells to proliferate, enabling other mechanisms that more directly produce centrosome amplification [70,71] (discussed below). Clarifying the effects of p53 loss on the propensity for centrosome amplification in different cell types and genetic backgrounds will be an important future direction for the field.
Centrosome overduplication can also be induced by overexpression of PCM components, such as pericentrin [72]. Similarly, loss of the tumour suppressor BRAC1 leads to centrosome amplification via increasing the levels of the PCM component γ-tubulin [73]. Re-duplication of centrioles can also be observed in cells during prolonged G2 arrest by a mechanism involving the maturation and premature disengagement of the procentrioles promoted by Plk1 [74]. Thus pathological conditions, such as persistent DNA damage, that may increase the time cells spend in G2, could contribute to centrosome amplification in cancer cells.
Centrosome amplification can also originate from tetraploid cells, which themselves can be derived from cytokinesis failure, mitotic slippage, endoreduplication or cell–cell fusion [75]. Tetraploid cells, generated by cytokinesis failure, endore-duplication or cell–cell fusion, induce tumorigenesis [76–78]. Although the contribution of centrosome amplification was not assessed in these studies, cells derived from p53–/– tetraploid tumours exhibit high levels of centrosome amplification [76]. These observations contrast with recent in vitro experiments where the transient induction of cytokinesis failure did not lead to the long-term amplification of centrosomes in culture [79]. These observations illustrate the fact that there is no simple correspondence between the generation of extra centrosomes and the continued maintenance of supernumerary centrosomes. It is clear that on its own, centrosome amplification is deleterious, which is underscored by earlier work showing that newly generated tetraploid cells spontaneously lose extra centrosomes with continuous passage in culture [80]. For cytokinesis failure or other events that lead to tetraploidy to generate long-term centrosome amplification, other permissive conditions may need to coexist—either cell type or genetic changes.
Consistent with the generally deleterious consequences of centrosome amplification, overexpression of either Plk4 or SAS-6 decreased cell proliferation via the stabilization of p53 and the induction of p21 [81]. The growth disadvantage caused by centrosome amplification probably prevents the accumulation of cells with extra centrosomes in culture independently of the method used to generate them. Consistently, inhibition of p53 rescues the proliferation defects of cells with extra centrosomes, allowing cells to maintain high levels of centrosome amplification [81]. Stabilization of p53 provides a similar mechanism to limit the proliferation of aneuploid cells [82]. Although centrosome amplification generates aneuploidy [80], p53-dependent loss of proliferation observed in cells with extra centrosomes seems to be independent of aneuploidy. Indeed, extra centrosomes themselves can induce p53 stabilization through the activation of Hippo tumour suppressor pathway prior to cell division (N. Ganem, H. Cornils & D. Pellman 2013, unpublished data). Therefore, cells can respond to both aneuploidy and centrosome amplification by inducing a p53-dependent cell cycle arrest.
Given the potentially detrimental effects of centrosome amplification, it is somewhat surprising that it is a widespread characteristic of many human tumours. It is possible that centrosome amplification might confer some unknown advantage to the cells that is only revealed in vivo. Alternatively, but not exclusively, other events that allow cells to proliferate and maintain extra centrosomes must occur during cancer progression. This last possibility suggests that centrosome amplification per se may not have a role in tumour initiation and could explain why centrosome abnormalities are mostly associated with advanced tumour stages. However, we cannot exclude a role for centrosome amplification in tumour initiation that could be dependent on the cellular context, specific microenvironment and genetic background of the patient.
4. Coping with centrosome amplification: sticking together
Boveri's initial hypothesis that multipolar cell divisions induced by centrosome amplification cause aneuploidy and tumorigenesis has been recently questioned. Whether cells can survive the gross aneuploidy that would result from a multipolar division has always been a nagging question. Furthermore, an alternative outcome for cells with centrosome amplification is known: cells can cluster supernumerary centrosomes to assemble pseudo-bipolar spindles [83]. This observation supports early work that suggests that in human tumours multipolar anaphases are less frequent than multipolar metaphases, raising the possibility that these multipolar metaphases are transient [84]. Indeed, following cancer cells over time by live cell imaging clearly demonstrated that the progeny of cells undergoing a multipolar division are typically unviable, further suggesting that such chaotic cell divisions are dead ends [80]. It is therefore not surprising that cells have various mechanisms to limit the detrimental consequences of a multipolar mitosis, such as inactivation of centrosomes, centrosome loss and centrosome clustering [37].
Centrosome inactivation is characterized by a reduction of PCM levels and was observed in flies with amplified centrosomes [85]. In this model system, centrosomes that were scattered along the spindle had significantly lower levels of PCM and thus decreased amounts of associated MT asters, [85]. This is similar to what has been described in polyspermic newt eggs where the formation of a gradient of cyclin B and γ-tubulin distribution (higher in the animal hemisphere) ensures that only the centrosome associated with the principal sperm nucleus will contribute to the formation of the bipolar spindle by preferential accumulation of cyclin B and γ-tubulin [86].
Centrosome loss is also a mechanism used by cells to prevent centrosome amplification during fertilization in most metazoans, including C. elegans and Homo sapiens. Centrosome elimination during oogenesis ensures that fertilized zygotes contain the correct number of centrioles. Centrosome loss is also associated with a decrease in PCM and a loss of MT nucleating activity [87], probably leading to centriole disintegration. Although the mechanisms leading to centrosome loss are unclear, a recent report found that the loss of the helicase CGH-1 in C. elegans delays this process, probably by preventing the degradation of a specific maternal mRNA [87]. Nevertheless, it is still unknown whether mechanisms such as centrosome inactivation and centrosome loss exist in cancer cells.
Centrosome clustering is the best-characterized ‘coping’ mechanism for cells with extra centrosomes and is perhaps the most prevalent in cancer. Initially described in mouse neuroblastoma N1E-115 cells more than 30 years ago, centrosome clustering can be found in both interphase and mitotic cells [88,89]. Centrosome clustering allows cells to undergo a pseudo-bipolar mitosis despite high levels of centrosome amplification [89]. More recently, work from several groups has elucidated mechanisms that contribute to centrosome clustering [37,90–93].
(a). Mechanisms that contribute to centrosome clustering
Several microtubule-associated proteins (MAPs) have been described to be important for centrosome clustering. Indeed, the first molecule described to have a role in centrosome clustering was the minus-end-directed motor dynein [93]. Work that followed showed that another minus-end-directed motor HSET/KIFC1, and the Drosophila homologue Ncd, played essential roles in this process, possibly by promoting the crosslinking of antiparallel MTs between adjacent centrosomes [85,92]. Other MAPs such as TACC3 and ch-TOG also play a role in this process downstream of integrin-linked kinase (ILK) protein [94].
A functional spindle assembly checkpoint (SAC) was also shown to be required for clustering of supernumerary centrosomes prior to anaphase. Cells with extra centrosomes spend increased time in mitosis due to the formation of a transient multipolar spindle that delays the normal biorientation of chromosomes and thus the satisfaction of the SAC prior to centrosome clustering. Hence, decreased time in mitosis by inhibition of the SAC causes defects in centrosome clustering, promoting multipolar mitosis [85,92,95].
In addition, several proteins that promote the tension between kinetochores and spindle MTs seem to be important to maintain centrosome clustering in cancer cells. These include components of the chromosome passenger complex, sister chromatid cohesion, augmin complex and proteins involved in the kinetochore–MT attachment [96]. Similarly, the MAP and Ran GTPase effector HURP that has important roles in the stability of kinetochore fibres is also important for centrosome clustering [97]. Thus, a theme is emerging whereby the balance between forces that control the tension of the mitotic spindle apparatus play important roles in centrosome clustering. This is perhaps not surprising because of the known role of the forces acting on spindle poles in resisting the traction forces mediated by the motors Kid and CENP-E during chromosome alignment to maintain bipolarity [98,99]. Inhibition of NuMA in the human cancer cell line CFPAC1 leads to disrupted/multipolar spindles that are rescued if polar ejection forces mediated by the chromokinesin Kid are also inhibited [99]. Therefore, NuMA plays important roles in generating forces at the centrosome that are essential to antagonize polar ejection forces during chromosome alignment. Furthermore, bipolar spindles where both NuMA and Kid were simultaneously inhibited were sensitive to HSET/KIFC1, which is indispensable for bipolar spindle formation in these cells [99]. Thus, changing the balance of forces within the spindle alters the cellular requirements to establish and/or maintain bipolarity. Likewise, both ninein and CLASPs are required for spindle bipolarity by generating forces at the spindle poles that counteract the forces generated at the chromosomes by Kid and CENP-E during chromosome alignment [98]. Because of the MT nucleation capacity of extra centrosomes, centrosome amplification might change this fine balance, thus rendering cells with extra centrosomes more sensitive to proteins involved in maintaining spindle and kinetochore–MT tension.
The distribution of the adhesive contacts with the microenvironment appears to play also an important role in centrosome clustering [92]. This is achieved through the formation of retraction fibres during mitotic round-up that provide cortical cues at the cell cortex to promote spindle orientation [100]. Thus, the number and positioning of these cortical cues will dictate whether cells are able to cluster extra centrosomes [92]. This finding has interesting implications because it suggests that the tumour microenvironment can affect centrosome clustering and therefore the survival of cells containing extra centrosomes. It will be interesting to determine how different three-dimensional environments, for example with differing stiffness, might affect centrosome clustering and the survival of cells with extra centrosomes.
Altogether, it appears that cancer cells containing extra centrosomes hijack several existing properties of normal cells to promote centrosome clustering, perhaps explaining the prevalence of this clustering in cancer. Indeed, the normal pole focusing components are also required for centrosome clustering, and to date no unique components to promote centrosome clustering have been identified. However, in some cases, centrosome amplification generates an increased requirement for these normal components, raising the possibility of a new cancer-specific therapeutic strategy (see §6). For example, HSET/KIFC1 and HURP are not essential in normal somatic cells [101,102] but become indispensable for bipolar spindle assembly in cancer cells containing supernumerary centrosomes [92,97]. In addition, both HSET/KIFC1 and HURP are essential for bipolar spindle formation in acentrosomal spindles [97,101], suggesting that the forces involved in pole focusing in the absence of centrosomes are similar to the ones required to bundle extra centrosomes together. Furthermore, it is also possible that cancer cells with defective pole focusing mechanisms, independently of centrosome amplification, might also have increased requirement for some of these components (see §6).
5. Consequences of centrosome abnormalities
To date, our knowledge on how centrosome abnormalities contribute to tumorigenesis is still modest. This is particularly true in the case of structural centrosomal defects, partly because these are not always easy to classify. Intriguingly, loss of centrioles can lead to aneuploidy in DT40 cells [8]. In addition, the loss of centrioles proved to be tumorigenic in a fly model where neuroblasts are transplanted into the abdomen, possibly through the impairment of asymmetric cell division of neural stem cells. Thus, we should perhaps consider the possibility that not only extra centrosomes but also centrosome loss or decrease in PCM recruitment could lead to alterations that could contribute to tumorigenesis. Thus, although we focus this section on the consequences of centrosome amplification in tumours (summarized in figure 3), other centrosome abnormalities may be important.
Figure 3.

Consequences of centrosome amplification in cancer. A model of how centrosome amplification could contribute to tumorigenesis is illustrated in this figure. Most of our understanding of the consequences of centrosome amplification came from studying mitosis. It is known that extra centrosomes can affect cells by promoting chromosome missegregation and also by impairing asymmetric cell division in Drosophila neuroblasts. It is also becoming clear that the role of extra centrosomes is not limited to mitosis. Centrosome amplification can affect cilia signalling in interphase cells. In addition, increased MT nucleation in cells with extra centrosomes, which are clustered in interphase, can also alter the regulation of Rho GTPases and thus affect the migration and invasive properties of cells. It is also possible that supernumerary centrosomes could affect polarity and signalling.
(a). Consequences of centrosome amplification during mitosis
(i). Chromosome segregation
The correlation between centrosome amplification and aneuploidy has long been known in human tumours [16,103]. However, because multipolar cell divisions, driven by centrosome amplification, are detrimental for cell proliferation and cannot explain the observed rates of chromosome instability (CIN), another mechanism was required to explain this correlation. An alternative mechanism has now been proposed [80,104]. During the process of clustering supernumerary centrosomes, multipolar spindles are transiently formed. This multipolar intermediate favours the formation of defective attachments of chromosomes to the spindle MTs, known as merotelic kinetochore–MT attachments [80,104]. Because merotelic attachments can escape the robust control of the SAC, they are an important source of aneuploidy because cells undergoing the missegregation event do not arrest and are thus not culled from the population [105].
Aneuploidy seems to play a multifaceted role in cancer; whereas in some mouse models aneuploidy per se can promote tumorigenesis, in some conditions higher levels of aneuploidy can function as a tumour suppressor [106]. How aneuploidy contributes to tumorigenesis is still unclear, but the idea that on-going aneuploidy, often termed CIN, could provide a platform that allows tumours to evolve is very appealing [106]. In addition, recent work suggests that lagging chromosomes during mitosis, e.g. as a consequence of centrosome amplification, could be a source of DNA damage, for example when trapped at the cytokinetic furrow [107]. Moreover, lagging chromosomes can be encapsulated into micronuclei causing abnormal DNA replication and extensive DNA damage [108]. The generation of DNA damage in micronuclei is related, at least in part, to their fragile nuclear envelopes which can undergo abrupt loss of integrity [109]. This generation of extensive DNA damage provides a mechanism by which mitotic errors can lead to mutations. Because the damage is restricted to the missegregated chromosome [108], its localization might explain ‘chromothripsis’ a process where one or a few chromosomes or chromosome arms appear to have shattered, with some of the fragments having been stitched together in random order and orientation [110].
(ii). Asymmetric cell division
Although centrosome amplification could contribute to tumorigenesis via the generation of aneuploidy, it is also clear that extra centrosomes may impact cell physiology in ways that are independent of chromosome segregation. In fact, elegant work in Drosophila has shown that centrosome amplification can induce tumorigenesis by a mechanism that is likely independent of aneuploidy [85]. Transplantation of neuroblasts carrying extra centrosomes into the abdomen of adult flies results in the generation of tumours that grow rapidly and kill the recipient host. Interestingly, these tumours show only minimal aneuploidy [85] and induction of aneuploidy alone does not induce tumours in this model system [111], indicating that other mechanisms are probably responsible for this phenotype. Indeed, neuroblasts containing extra centrosomes show defects in asymmetric cell division, despite efficient clustering of extra centrosomes [85]. These defects were previously shown to induce the expansion of the neuronal stem cell compartments, leading to tumours [112]. This type of mechanism could, in principle, be applicable to human tumours thought to originate from a small population of stem-like cells, if these cells similarly undergo asymmetric cell division.
The ability to manipulate centrosome number by controlled expression of Plk4 [42] has opened new ways to address how centrosome amplification affects development and tumorigenesis in vivo. One recent study described the first such mouse model [69]. In contrast to the experiments in the fly, induction of centrosome amplification in the developing mouse brain is not sufficient to induce tumours. Instead, owing to the accumulation of highly aneuploid cells, mouse neuronal stem cells undergo apoptosis, leading to microcephaly. This effect seems to be caused by inefficient centrosome clustering in these cells and the consequent generation of multipolar mitoses, suggesting that in these cells centrosome amplification leads to tissue degeneration [69]. The implication is that efficient centrosome clustering is key for the ability of cells to proliferate and possibly to form tumours. If methods could be developed to ‘force’ centrosome clustering in this model, it would then be possible to determine if centrosome amplification can promote the development of brain tumours.
(b). Consequences of centrosome amplification in interphase
(i). Cell polarity and microtubules
Centrosomes play an important role in maintaining the organization of MT arrays in non-dividing interphase cells, which affects many aspects of cell signalling [1]. It is therefore possible that centrosome amplification could also influence tumour biology independently of generating aneuploidy by altering cell shape, polarity or motility, which could influence the architecture of tumour tissue as well as the tendency of tumours to metastasize. Indeed, increased MT nucleation capacity in cells containing extra centrosomes is correlated with high histological grade in breast cancer, independently of the degree of aneuploidy [113].
During interphase, extra centrosomes are typically clustered, but how this occurs is unclear. Clustered extra centrosomes in interphase recruit extra PCM leading to an enlarged centrosome with increased MT nucleation capacity [23,27], a kind of ‘super-centrosome’. The consequences of enlarged centrosomes for cell polarization are not known. Centrosomes play major roles in the establishment of cell polarity, in part by dictating the organization of the MTs that in turn will determine cell shape and motility [114]. The position of the centrosome, by directing MT nucleation, can determine the site of axon outgrowth in neurons, the proper secretion of lytic granules during formation of the immunological synapse, or directional migration by positioning the Golgi towards the leading edge [114]. In principle, the presence of an enlarged ‘super-centrosome’ has the potential to promote stronger polarization, but the biological consequences of this enlargement need further detailed study.
Increased MT nucleation capacity from centrosome amplification could affect cellular physiology in many ways. Focal adhesion (FA) disassembly, essential for cell migration, is regulated by MTs, which involves components of the endocytic machinery, such as dynamin and clathrin, inhibition of actomyosin-mediated contractile forces, or proteolytic cleavage of the link between FA and the actin cytoskeleton [115]. Moreover, MTs affect the activity of Rho GTPases which are known to play central roles in the regulation of invasion [116]. Depolymerization of MTs leads to RhoA activation, probably through the release of the guanine nucleotide exchange factors GEF-H1 and p190RhoGEF [117,118]. By contrast, MT polymerization can induce Rac1 activation, leading to Arp2/3-mediated actin polymerization, essential for lamelipodia formation and cell migration [119]. Hence, it is likely that centrosome amplification, through increased MT nucleation, could affect processes such as migration and invasion. Indeed, we recently reported that increased MT nucleation in cells with extra centrosomes leads to increased Rac1 activity and cell invasion in three-dimensional culture models, a process that is independent of aneuploidy (figure 4) [36]. In this model, centrosome amplification mimicked and enhanced the effects of expression of a bona fide breast cancer oncogene, ERBB2. Interestingly, in breast cancer, increased MT nucleation capacity in cells containing extra centrosomes is correlated with high histological grade, independently of aneuploidy [113]. These findings could partially explain the strong association between centrosome amplification and advanced tumour stage, in particular metastasis formation.
Figure 4.
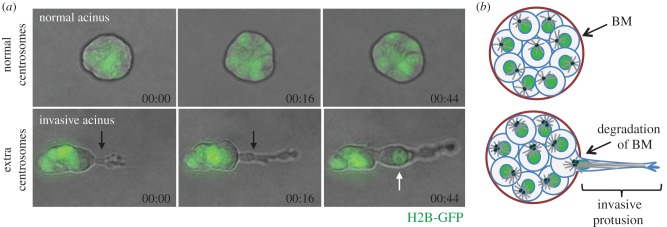
Centrosome amplification induces the formation of invasive protrusions. (a) Still images from videos of MCF10A cells plated in three-dimensional cultures for 4 days. MCF10A cells form spheroids, acini, when grown in three dimensions, whereas MCF10A cells with extra centrosomes form invasive protrusions in three-dimensional cultures (black arrow). Nuclei, labelled with H2B-GFP (green) can migrate into an invasive protrusion (white arrow). Time-scale, hour : min. (b) Scheme of normal and invasive acini. Note that invasive protrusions form at sites where the basement membrane is (BM) degraded.
(ii). Signalling
Activation of signalling pathways in cells often requires that an initial threshold of activation to be reached. One common way to cross this threshold is to concentrate signalling components locally on a structure such as the centrosome. Locally concentrating signalling molecules can also enhance signalling specificity if there is a requirement for multiple independent molecular events to occur simultaneously, a scenario often referred to as ‘coincidence detection’. The idea that centrosomes can function in this way as solid-state platforms for cellular signalling is clearly established in both budding and fission yeast [120,121].
One clear example of signalling promoted by the centrosome is the regulation of mitotic entry in fission yeast [122]. A compelling body of work suggests that the spindle pole body (SPB) functions as a platform to increase a feedback loop that amplifies cyclin B/Cdk1 activity, which is then propagated throughout the cell. This positive feedback loop involves the recruitment of the Plk1 homologue, Plo1, to the SPB. During late G2, NIMA kinase, Fin1, blocks the recruitment of the protein phosphatase 1 to the SPB, which then allows the recruitment of Plo1 to Cut12 and the SPB [123]. Increased Plo1 activation at the SPB is thought to further activate cyclin B/Cdk1 by enhancing Cdc25 (Cdk1 activator) and inhibiting Wee1 (Cdk1 inhibitor) activities [124]. Importantly, regulation of mitotic entry by the centrosome has also been observed in human cells, C. elegans and Xenopus egg extracts, suggesting a broadly conserved mechanism [125–128].
In mammals, proteomic analysis of purified centrosomes has shown that components of many signalling pathways can associate with the centrosomes, if only transiently [129,130]. Members of the Wnt, NF-κB and integrin signalling that are known to contribute to tumorigenesis can localize to the centrosomes [131–133]. For example, the centrosomal localization of Diversin, an inhibitor of the Wnt pathway, seems to be important for its function. Diversin mutants defective for centrosome localization failed to antagonize Wnt signalling [132]. The centrosome is also a focal point for ubiquitin-mediated proteolysis [134]. In response to bone morphogenic protein (BMP) signalling, phosphorylated and polyubiquitinated Smad1 is targeted to the centrosome. Inhibition of the proteasome leads to a greater accumulation of phospho-Smad1 at the centrosome suggesting the possibility that centrosome might function as a site for proteasome-mediated degradation [135]. This supports earlier observations showing that the size of the centrosome can change in response to proteasome inhibition and increased levels of misfolded proteins [134]. Thus, whether by promoting phosphorylation, degradation or simply by sequestering proteins from the cytoplasm, the centrosome could exert either positive or negative regulatory effects on intracellular signalling, similar to what has been described during mitotic entry. Thus we can envision a model where by increasing the centrosome size, centrosome amplification could affect the balance of many of these pathways that are typically deregulated in cancer.
Alterations of signalling pathways mediated by centrosome amplification do not necessarily need to be mediated directly by the centrosomes. Centrosomes can moonlight as cilia, which are themselves signalling hubs and control several pathways, such as the sonic hedgehog (Shh) pathway [136]. Activation of Shh involves the transport of the transmembrane protein smoothened to the cilium, thereby allowing the transcription of Gli-responsive genes. Recently, it was shown that centrosome amplification can generate extra cilia that, somewhat surprisingly, diminished Shh signalling and induced defects in tissue architecture [137]. The finding that more cilia produce less Shh signalling may be reconciled by the observation that extra cilia seem to dilute some signalling molecules. Defects in cilia signalling are associated with several ciliopathies and brain developmental defects, such as primary recessive microcephaly. Interestingly, the presence of centrosome amplification has been recently described in some patients with cilia-related diseases [138]. However, it is unclear whether the extra centrosomes are cause or consequence in these diseases. It is also not known whether the extra centrosomes reported in these diseases result in extra cilia. At least in a mouse model, the induction of centrosome amplification appears to induce microcephaly independently of cilia. Instead, gross aneuploidy that leads to cell death appears to be the basis for the observed disease phenotype [69].
6. Targeting cells with extra centrosomes for cancer therapy
Selective cancer therapies rely on the identification and targeting of features that are unique to cancer cells. Inhibition of centrosome clustering by depletion of the kinesin HSET/KIFC1 or the ILK selectively kills cancer cells containing extra centrosomes [92,94]. Notably, flies without Ncd are viable [101] and human cells also have minimal requirement for HSET during normal somatic mitosis [102,139]. These observations form the basis for new therapeutic strategies that target cells carrying extra centrosomes. The recent development of inhibitors that compromise centrosome clustering could be the beginning of the validation of this idea in vivo.
Several inhibitors have been developed that induce multipolar mitosis preferentially in cancer cells, such as a phenanthrene-derived PARP inhibitor, GF-15, a derivative of griseofulvin and taxol [140–142]. There is interest in the clinical use of these compounds, such as for example GF-15 which decreases tumour growth in xenograft mouse models [141]. Although cells with extra centrosomes have increased sensitivity to such inhibitors, whether this is specifically due to centrosome amplification in tumour cells is not firmly established. An alternative possibility is that indirect effects on MTs could, at least in part, explain the induced spindle multipolarity. Griseofluvin shares many properties with taxol, an MT-stabilizing drug well known to induce multipolar spindles [142]. Griseofulvin promotes MT stabilization at low concentrations and the tubulin binding of griseofulvin overlaps with the taxol binding site, thus suggesting a common mechanism for the induction of multipolar spindles [143].
More recently, a screen to identify small molecules that inhibit centrosome clustering resulted in the identification of 14 chemical compounds that specifically induced multipolar spindles in a breast cancer cell line containing extra centrosomes, BT-549. One of the tested compounds significantly reduced the viability of cancer cells while sparing normal mammary epithelial cells and also bone marrow haematopoietic progenitors, which are frequently affected by conventional chemotherapies [144]. The first HSET/KIFC1 inhibitor described, AZ82, was shown to specifically induce multipolar spindles in BT-549 cells, but not in cancer cells with normal centrosome number, such as HeLa [145]. A novel allosteric HSET/KIFC1 inhibitor has also recently been described [146]. As predicted, this inhibitor, CW069, induces multipolar spindles in cells with extra centrosomes (e.g. N1E-115, MDA-231 and BT-549) without compromising the ability of cells with normal centrosome number (NHDF, MCF-7 and HeLa) to form a bipolar spindle. The formation of multipolar spindles led to multipolar cell divisions in cells with centrosome amplification, as previously shown for depletion of HSET/KIFC1 [92]. Importantly, CW069 significantly decreased the viability of cells with extra centrosomes without compromising normal cells. However, this drug also compromised the viability of the MCF-7 breast cancer cell line without inducing multipolar mitoses [146]. The basis for the sensitivity of MCF-7 cells to CW069 is not clear and whether this effect is direct or indirect, and whether it is ‘on target’ or off target’ is likewise not known.
In addition to the requirement of HSET to cluster extra centrosomes, some cancer cells appear to have fragile spindle poles and thus upon HSET inhibition generate acentrosomal spindles [147]. Depletion of HSET/KIFC1 in BT-549 cells caused not only centrosome declustering but also induced the formation of multipolar spindles with acentrosomal poles [147]. These observations demonstrate that HSET has a dual role in promoting bipolar spindle formation in cancer cells: promoting the clustering of extra centrosomes and the coalescence of acentrosomal poles that are aberrantly generated in BT-549 cells [147]. This study suggested that DNA damage signalling, which is commonly activated in cancer cells and is induced by many chemotherapeutic agents, contributes to the generation of acentrosomal spindle poles that need to be clustered to achieve a bipolar mitosis. Thus, in addition to centrosome amplification, other cancer-specific pathologies may sensitize cancer cells to HSET/KIFC1 inhibitors.
Although inhibitors that specifically target cells with extra centrosomes are appealing because of their potential cancer selectivity, it will be essential to assess the tolerance of normal polyploid tissues containing extra centrosomes to such inhibitors, in particular hepatocytes that still retain a capacity to divide [148]. In addition, because most tumours are a heterogeneous mixture of cells with and without centrosome amplification, it is unclear how depletion of the population containing extra centrosomes will affect tumour progression. Because extra centrosomes are correlated with both aneuploidy and increased tumour grade, elimination of these cells may have a beneficial effect for patients, but this remains to be tested.
The utility of these newly developed inhibitors in the clinic is hindered by our ability to identify the patients who will respond to such therapy. To date, the best method to identify tumours containing extra centrosomes is by immunostaining for centriolar components, which is cumbersome and gives results that are difficult to quantify. One alternative would be to identify a gene signature that predicts the presence of centrosome amplification in tumour samples. However, this too is expected to face challenges of specificity, separating centrosome-specific effects from the general mitotic signature common in aneuploid tumours [149], and also sensitivity, given that cells with extra centrosomes often comprise only a fraction of the tumour cells. Perhaps most realistically, it would be better to define the genetic changes that lead to centrosome amplification and then stratify patients based on mutational status. Irrespective of the challenges, it is clear that the clinical utility of agents that target centrosome amplification will be highly dependent on our ability to identify the relevant patients rapidly and inexpensively.
7. Future perspectives
Almost 100 years after the pioneering work of Boveri, Hansemann and Galeotti, centrosomes are once again centre stage for cancer biology. Although we are still far from having a complete picture of the role of centrosome amplification in tumorigenesis, it is becoming clear that extra centrosomes are more than simple bystanders in tumour progression. Whether through the generation of aneuploidy [80,104], effects on signalling, such as through Rho-type GTPases [36], centrosome amplification is likely to affect cancer cells in multiple ways. Whatever the potential for a positive contribution to tumour development, centrosome amplification has known detrimental effects for cell proliferation. It is therefore expected that the net effect of centrosome amplification will be context-dependent. In this sense, centrosome amplification can be viewed as being similar to aneuploidy, which in different models can be cause, consequence or a neutral bystander to the process of tumour development [106].
Remarkably, most tumours consist of a heterogeneous population of cells with normal and abnormal centrosomes, suggesting that centrosome defects are a source of tumour heterogeneity. The dynamics of centrosome duplication within tumours, how the fraction of cells with centrosome amplification develops and is maintained are important and unresolved questions. Whether the heterogeneity that comes from having a fraction of cells with extra centrosomes contributes to tumorigenesis is also unknown. This is an interesting possibility because tumour heterogeneity, whether defined by genetic alterations or influenced by the tumour microenvironment, plays major roles in tumour evolution and response to therapy [150].
Taken together, much progress has been made over the past 20 years in understanding the centrosome—its parts and the complex controls of its duplication. Even with a nearly complete parts list, we are still discovering unexpected things about how these parts fit together. For example, the application of super-resolution imaging technology has only recently enabled the discovery of previously unanticipated structure of the PCM [151]. Together with these imaging approaches, much of the forefront for the basic understanding of centrosome organization is coming from structural studies and in vitro reconstitution [54,55]. By contrast, progress in understanding how centrosomes impact cellular signalling has been slower. This is in essence a problem of defining the local function of proteins, and may benefit from new approaches using light to control protein activity at different locations within cells. We are also beginning to obtain a better understanding of how the process of centrosome biogenesis goes awry in cancer. However, there is much to be done to understand both causes and consequences of centrosome defects in cancer. In this review, we have tried to identify key unanswered questions. Although there has been important work done in tissue culture cells, an important direction for the field will be in newly developed mouse models, more physiologically relevant three-dimensional culture systems and ultimately human patient samples.
Acknowledgements
We apologize for to the many authors whose work we were unable to cite due to space limitations. We thank to Gayathri Chandrasekaran and Fanni Gergely for providing the image displayed in figure 1.
Funding statement
D.P. is supported by Howard Hughes Medical Institute and awards from the National Institutes of Health.
References
- 1.Bettencourt-Dias M, Glover DM. 2007. Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 8, 451–463. ( 10.1038/nrm2180) [DOI] [PubMed] [Google Scholar]
- 2.Kim S, Dynlacht BD. 2013. Assembling a primary cilium. Curr. Opin. Cell Biol. 25, 506–511. ( 10.1016/j.ceb.2013.04.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nigg EA. 2007. Centrosome duplication: of rules and licenses. Trends Cell Biol. 17, 215–221. ( 10.1016/j.tcb.2007.03.003) [DOI] [PubMed] [Google Scholar]
- 4.Bettencourt-Dias M, et al. 2005. SAK/PLK4 is required for centriole duplication and flagella development. Curr. Biol. 15, 2199–2207. ( 10.1016/j.cub.2005.11.042) [DOI] [PubMed] [Google Scholar]
- 5.Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW. 2006. Flies without centrioles. Cell 125, 1375–1386. ( 10.1016/j.cell.2006.05.025) [DOI] [PubMed] [Google Scholar]
- 6.Khodjakov A, Cole RW, Oakley BR, Rieder CL. 2000. Centrosome-independent mitotic spindle formation in vertebrates. Curr. Biol. 10, 59–67. ( 10.1016/S0960-9822(99)00276-6) [DOI] [PubMed] [Google Scholar]
- 7.Debec A. 1978. Haploid cell cultures of Drosophila melanogaster. Nature 274, 255–256. ( 10.1038/274255a0) [DOI] [PubMed] [Google Scholar]
- 8.Sir JH, Putz M, Daly O, Morrison CG, Dunning M, Kilmartin JV, Gergely F. 2013. Loss of centrioles causes chromosomal instability in vertebrate somatic cells. J. Cell Biol. 203, 747–756. ( 10.1083/jcb.201309038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boveri T. 2008. Concerning the origin of malignant tumours by Theodor Boveri. [Transl. and annotated by Henry Harris] J. Cell Sci. 121(Suppl. 1), 1–84. ( 10.1242/jcs.025742) [DOI] [PubMed] [Google Scholar]
- 10.Boveri T. 1887. Uber die Befruchtung der Eier von Ascaris megalocephala. SitzBer. Ges. Morph. Phys. Munchen. 3, 71–80. [Google Scholar]
- 11.Boveri T. 1888. Zellen-Studien 2: Die Befruchtung und Teilung des Eies von Ascaris megalocephala. Jenaische Zeitschr. Med. Naturw. 22, 685–882. [Google Scholar]
- 12.Galeotti G. 1893. Beitrag zum Studium des Chromatins in den Epithelzellen der Carcinome. Beitr. Pathol. Anat. Allg. Pathol. 14, 249–271. [Google Scholar]
- 13.Hansemann D. 1890. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 119, 299–326. [Google Scholar]
- 14.Hardy PA, Zacharias H. 2005. Reappraisal of the Hansemann–Boveri hypothesis on the origin of tumors. Cell Biol. Int. 29, 983–992. ( 10.1016/j.cellbi.2005.10.001) [DOI] [PubMed] [Google Scholar]
- 15.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. 1996. Abnormal centrosome amplification in the absence of p53. Science 271, 1744–1747. ( 10.1126/science.271.5256.1744) [DOI] [PubMed] [Google Scholar]
- 16.Chan JY. 2011. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 7, 1122–1144. ( 10.7150/ijbs.7.1122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lingle WL, Lutz WH, Ingle JN, Maihle NJ, Salisbury JL. 1998. Centrosome hypertrophy in human breast tumors: implications for genomic stability and cell polarity. Proc. Natl Acad. Sci. USA 95, 2950–2955. ( 10.1073/pnas.95.6.2950) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pihan GA, et al. 1998. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 58, 3974–3985. [PubMed] [Google Scholar]
- 19.Hsu LC, Kapali M, DeLoia JA, Gallion HH. 2005. Centrosome abnormalities in ovarian cancer. Int. J. Cancer 113, 746–751. ( 10.1002/ijc.20633) [DOI] [PubMed] [Google Scholar]
- 20.Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, Ogawa T, Tanaka M. 1999. Centrosome abnormalities in pancreatic ductal carcinoma. Clin. Cancer Res. 5, 963–970. [PubMed] [Google Scholar]
- 21.Kramer A, Neben K, Ho AD. 2005. Centrosome aberrations in hematological malignancies. Cell Biol. Int. 29, 375–383. ( 10.1016/j.cellbi.2005.03.004) [DOI] [PubMed] [Google Scholar]
- 22.Giehl M, Fabarius A, Frank O, Hochhaus A, Hafner M, Hehlmann R, Seifarth W. 2005. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 19, 1192–1197. ( 10.1038/sj.leu.2403779) [DOI] [PubMed] [Google Scholar]
- 23.Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL. 2002. Centrosome amplification drives chromosomal instability in breast tumor development. Proc. Natl Acad. Sci. USA 99, 1978–1983. ( 10.1073/pnas.032479999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pihan GA, Wallace J, Zhou Y, Doxsey SJ. 2003. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 63, 1398–1404. [PubMed] [Google Scholar]
- 25.Segat D, et al. 2010. Pericentriolar material analyses in normal esophageal mucosa, Barrett's metaplasia and adenocarcinoma. Histol. Histopathol. 25, 551–560. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto Y, et al. 2004. Centrosome hyperamplification predicts progression and tumor recurrence in bladder cancer. Clin. Cancer Res. 10, 6449–6455. ( 10.1158/1078-0432.CCR-04-0773) [DOI] [PubMed] [Google Scholar]
- 27.D'Assoro AB, et al. 2002. Amplified centrosomes in breast cancer: a potential indicator of tumor aggressiveness. Breast Cancer Res. Treat. 75, 25–34. ( 10.1023/A:1016550619925) [DOI] [PubMed] [Google Scholar]
- 28.Pihan GA, Purohit A, Wallace J, Malhotra R, Liotta L, Doxsey SJ. 2001. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 61, 2212–2219. [PubMed] [Google Scholar]
- 29.Reiter R, et al. 2009. Centrosome abnormalities in head and neck squamous cell carcinoma (HNSCC). Acta Otolaryngol. 129, 205–213. ( 10.1080/00016480802165767) [DOI] [PubMed] [Google Scholar]
- 30.Nigg EA. 2006. Origins and consequences of centrosome aberrations in human cancers. Int. J. Cancer 119, 2717–2723. ( 10.1002/ijc.22245) [DOI] [PubMed] [Google Scholar]
- 31.Kirkham M, Muller-Reichert T, Oegema K, Grill S, Hyman AA. 2003. SAS-4 is a C. elegans centriolar protein that controls centrosome size. Cell 112, 575–587. ( 10.1016/S0092-8674(03)00117-X) [DOI] [PubMed] [Google Scholar]
- 32.Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Gonczy P. 2009. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 19, 1012–1018. ( 10.1016/j.cub.2009.05.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, Nigg EA. 2009. Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011. ( 10.1016/j.cub.2009.05.016) [DOI] [PubMed] [Google Scholar]
- 34.Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. 2009. CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell Biol. 11, 825–831. ( 10.1038/ncb1889) [DOI] [PubMed] [Google Scholar]
- 35.Guo HQ, Gao M, Ma J, Xiao T, Zhao LL, Gao Y, Pan Q. 2007. Analysis of the cellular centrosome in fine-needle aspirations of the breast. Breast Cancer Res. 9, R48 ( 10.1186/bcr1752) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godinho SA, et al. 2014. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 510, 167–171. ( 10.1038/nature13277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Godinho SA, Kwon M, Pellman D. 2009. Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 28, 85–98. ( 10.1007/s10555-008-9163-6) [DOI] [PubMed] [Google Scholar]
- 38.Fukasawa K. 2007. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 7, 911–924. ( 10.1038/nrc2249) [DOI] [PubMed] [Google Scholar]
- 39.Brownlee CW, Rogers GC. 2013. Show me your license, please: deregulation of centriole duplication mechanisms that promote amplification. Cell. Mol. Life Sci. 70, 1021–1034. ( 10.1007/s00018-012-1102-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nigg EA, Stearns T. 2011. The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat. Cell Biol. 13, 1154–1160. ( 10.1038/ncb2345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holland AJ, Lan W, Cleveland DW. 2010. Centriole duplication: a lesson in self-control. Cell Cycle 9, 2731–2736. ( 10.4161/cc.9.14.12184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. 2005. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 7, 1140–1146. ( 10.1038/ncb1320) [DOI] [PubMed] [Google Scholar]
- 43.Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. 2007. Plk4-induced centriole biogenesis in human cells. Dev. Cell 13, 190–202. ( 10.1016/j.devcel.2007.07.002) [DOI] [PubMed] [Google Scholar]
- 44.O'Connell KF, Caron C, Kopish KR, Hurd DD, Kemphues KJ, Li Y, White JG. 2001. The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell 105, 547–558. ( 10.1016/S0092-8674(01)00338-5) [DOI] [PubMed] [Google Scholar]
- 45.Cunha-Ferreira I, Rodrigues-Martins A, Bento I, Riparbelli M, Zhang W, Laue E, Callaini G, Glover DM, Bettencourt-Dias M. 2009. The SCF/Slimb ubiquitin ligase limits centrosome amplification through degradation of SAK/PLK4. Curr. Biol. 19, 43–49. ( 10.1016/j.cub.2008.11.037) [DOI] [PubMed] [Google Scholar]
- 46.Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW. 2010. Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188, 191–198. ( 10.1083/jcb.200911102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers GC, Rusan NM, Roberts DM, Peifer M, Rogers SL. 2009. The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184, 225–239. ( 10.1083/jcb.200808049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sillibourne JE, Tack F, Vloemans N, Boeckx A, Thambirajah S, Bonnet P, Grand-Perret T. 2010. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 21, 547–561. ( 10.1091/mbc.E09-06-0505) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guderian G, Westendorf J, Uldschmid A, Nigg EA. 2010. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J. Cell Sci. 123, 2163–2169. ( 10.1242/jcs.068502) [DOI] [PubMed] [Google Scholar]
- 50.Brownlee CW, Klebba JE, Buster DW, Rogers GC. 2011. The Protein Phosphatase 2A regulatory subunit Twins stabilizes Plk4 to induce centriole amplification. J. Cell Biol. 195, 231–243. ( 10.1083/jcb.201107086) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leidel S, Delattre M, Cerutti L, Baumer K, Gonczy P. 2005. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell Biol. 7, 115–125. ( 10.1038/ncb1220) [DOI] [PubMed] [Google Scholar]
- 52.Dammermann A, Muller-Reichert T, Pelletier L, Habermann B, Desai A, Oegema K. 2004. Centriole assembly requires both centriolar and pericentriolar material proteins. Dev. Cell 7, 815–829. ( 10.1016/j.devcel.2004.10.015) [DOI] [PubMed] [Google Scholar]
- 53.Rodrigues-Martins A, Bettencourt-Dias M, Riparbelli M, Ferreira C, Ferreira I, Callaini G, Glover DM. 2007. DSAS-6 organizes a tube-like centriole precursor, and its absence suggests modularity in centriole assembly. Curr. Biol. 17, 1465–1472. ( 10.1016/j.cub.2007.07.034) [DOI] [PubMed] [Google Scholar]
- 54.Kitagawa D, et al. 2011. Structural basis of the 9-fold symmetry of centrioles. Cell 144, 364–375. ( 10.1016/j.cell.2011.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Breugel M, et al. 2011. Structures of SAS-6 suggest its organization in centrioles. Science 331, 1196–1199. ( 10.1126/science.1199325) [DOI] [PubMed] [Google Scholar]
- 56.Strnad P, Leidel S, Vinogradova T, Euteneuer U, Khodjakov A, Gonczy P. 2007. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 13, 203–213. ( 10.1016/j.devcel.2007.07.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Puklowski A, et al. 2011. The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat. Cell Biol. 13, 1004–1009. ( 10.1038/ncb2282) [DOI] [PubMed] [Google Scholar]
- 58.Leidel S, Gonczy P. 2003. SAS-4 is essential for centrosome duplication in C. elegans and is recruited to daughter centrioles once per cell cycle. Dev. Cell 4, 431–439. ( 10.1016/S1534-5807(03)00062-5) [DOI] [PubMed] [Google Scholar]
- 59.Spektor A, Tsang WY, Khoo D, Dynlacht BD. 2007. Cep97 and CP110 suppress a cilia assembly program. Cell 130, 678–690. ( 10.1016/j.cell.2007.06.027) [DOI] [PubMed] [Google Scholar]
- 60.Li J, D'Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, Campos EI, Pagano M, Dynlacht BD. 2013. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495, 255–259. ( 10.1038/nature11941) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hannak E, Kirkham M, Hyman AA, Oegema K. 2001. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J. Cell Biol. 155, 1109–1116. ( 10.1083/jcb.200108051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lane HA, Nigg EA. 1996. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol. 135, 1701–1713. ( 10.1083/jcb.135.6.1701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsou MF, Wang WJ, George KA, Uryu K, Stearns T, Jallepalli PV. 2009. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev. Cell 17, 344–354. ( 10.1016/j.devcel.2009.07.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guardavaccaro D, et al. 2003. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev. Cell 4, 799–812. ( 10.1016/S1534-5807(03)00154-0) [DOI] [PubMed] [Google Scholar]
- 65.Wojcik EJ, Glover DM, Hays TS. 2000. The SCF ubiquitin ligase protein slimb regulates centrosome duplication in Drosophila. Curr. Biol. 10, 1131–1134. ( 10.1016/S0960-9822(00)00703-X) [DOI] [PubMed] [Google Scholar]
- 66.Duensing A, Spardy N, Chatterjee P, Zheng L, Parry J, Cuevas R, Korzeniewski N, Duensing S. 2009. Centrosome overduplication, chromosomal instability, and human papillomavirus oncoproteins. Environ. Mol. Mutagenesis 50, 741–747. ( 10.1002/em.20478) [DOI] [PubMed] [Google Scholar]
- 67.Korzeniewski N, Treat B, Duensing S. 2011. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer 10, 61 ( 10.1186/1476-4598-10-61) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J, Tan M, Li L, Pamarthy D, Lawrence TS, Sun Y. 2005. SAK, a new polo-like kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. Neoplasia 7, 312–323. ( 10.1593/neo.04325) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R. 2013. Centrosome amplification causes microcephaly. Nat. Cell Biol. 15, 731–740. ( 10.1038/ncb2746) [DOI] [PubMed] [Google Scholar]
- 70.Meraldi P, Honda R, Nigg EA. 2002. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53-/- cells. EMBO J. 21, 483–492. ( 10.1093/emboj/21.4.483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Borel F, Lohez OD, Lacroix FB, Margolis RL. 2002. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl Acad. Sci. USA 99, 9819–9824. ( 10.1073/pnas.152205299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Loncarek J, Hergert P, Magidson V, Khodjakov A. 2008. Control of daughter centriole formation by the pericentriolar material. Nat. Cell Biol. 10, 322–328. ( 10.1038/ncb1694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD. 2004. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol. Cell Biol. 24, 8457–8466. ( 10.1128/MCB.24.19.8457-8466.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loncarek J, Hergert P, Khodjakov A. 2010. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr. Biol. 20, 1277–1282. ( 10.1016/j.cub.2010.05.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ganem NJ, Storchova Z, Pellman D. 2007. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 17, 157–162. ( 10.1016/j.gde.2007.02.011) [DOI] [PubMed] [Google Scholar]
- 76.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047. ( 10.1038/nature04217) [DOI] [PubMed] [Google Scholar]
- 77.Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. 2007. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 17, 431–437. ( 10.1016/j.cub.2007.01.049) [DOI] [PubMed] [Google Scholar]
- 78.Davoli T, de Lange T. 2012. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 21, 765–776. ( 10.1016/j.ccr.2012.03.044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krzywicka-Racka A, Sluder G. 2011. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J. Cell Biol. 194, 199–207. ( 10.1083/jcb.201101073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ganem NJ, Godinho SA, Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. ( 10.1038/nature08136) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holland AJ, Fachinetti D, Zhu Q, Bauer M, Verma IM, Nigg EA, Cleveland DW. 2012. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 26, 2684–2689. ( 10.1101/gad.207027.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thompson SL, Compton DA. 2010. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 188, 369–381. ( 10.1083/jcb.200905057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brinkley BR. 2001. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 11, 18–21. ( 10.1016/S0962-8924(00)01872-9) [DOI] [PubMed] [Google Scholar]
- 84.Timonen S, Therman E. 1950. The changes in the mitotic mechanism of human cancer cells. Cancer Res. 10, 431–439. [PubMed] [Google Scholar]
- 85.Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. 2008. Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032–1042. ( 10.1016/j.cell.2008.05.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iwao Y, Murakawa T, Yamaguchi J, Yamashita M. 2002. Localization of gamma-tubulin and cyclin B during early cleavage in physiologically polyspermic newt eggs. Dev. Growth Diff. 44, 489–499. ( 10.1046/j.1440-169X.2002.00661.x) [DOI] [PubMed] [Google Scholar]
- 87.Mikeladze-Dvali T, von Tobel L, Strnad P, Knott G, Leonhardt H, Schermelleh L, Gonczy P. 2012. Analysis of centriole elimination during C. elegans oogenesis. Development 139, 1670–1679. ( 10.1242/dev.075440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brinkley BR, Cox SM, Pepper DA, Wible L, Brenner SL, Pardue RL. 1981. Tubulin assembly sites and the organization of cytoplasmic microtubules in cultured mammalian cells. J. Cell Biol. 90, 554–562. ( 10.1083/jcb.90.3.554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ring D, Hubble R, Kirschner M. 1982. Mitosis in a cell with multiple centrioles. J. Cell Biol. 94, 549–556. ( 10.1083/jcb.94.3.549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marthiens V, Piel M, Basto R. 2012. Never tear us apart--the importance of centrosome clustering. J. Cell Sci. 125, 3281–3292. ( 10.1242/jcs.094797) [DOI] [PubMed] [Google Scholar]
- 91.Kramer A, Maier B, Bartek J. 2011. Centrosome clustering and chromosomal (in)stability: a matter of life and death. Mol. Oncol. 5, 324–335. ( 10.1016/j.molonc.2011.05.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. 2008. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203. ( 10.1101/gad.1700908) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. 2005. Spindle multipolarity is prevented by centrosomal clustering. Science 307, 127–129. ( 10.1126/science.1104905) [DOI] [PubMed] [Google Scholar]
- 94.Fielding AB, Lim S, Montgomery K, Dobreva I, Dedhar S. 2011. A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 30, 521–534. ( 10.1038/onc.2010.431) [DOI] [PubMed] [Google Scholar]
- 95.Yang Z, Loncarek J, Khodjakov A, Rieder CL. 2008. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat. Cell Biol. 10, 748–751. ( 10.1038/ncb1738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Leber B, et al. 2010. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2, 33ra8 ( 10.1126/scitranslmed.3000915) [DOI] [PubMed] [Google Scholar]
- 97.Breuer M, Kolano A, Kwon M, Li CC, Tsai TF, Pellman D, Brunet S, Verlhac M-H. 2010. HURP permits MTOC sorting for robust meiotic spindle bipolarity, similar to extra centrosome clustering in cancer cells. J. Cell Biol. 191, 1251–1260. ( 10.1083/jcb.201005065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Logarinho E, Maffini S, Barisic M, Marques A, Toso A, Meraldi P, Meraldi P, Maiato H. 2012. CLASPs prevent irreversible multipolarity by ensuring spindle-pole resistance to traction forces during chromosome alignment. Nat. Cell Biol. 14, 295–303. ( 10.1038/ncb2423) [DOI] [PubMed] [Google Scholar]
- 99.Levesque AA, Howard L, Gordon MB, Compton DA. 2003. A functional relationship between NuMA and kid is involved in both spindle organization and chromosome alignment in vertebrate cells. Mol. Biol. Cell 14, 3541–3552. ( 10.1091/mbc.E03-02-0082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Thery M, Racine V, Pepin A, Piel M, Chen Y, Sibarita JB, Bornens M. 2005. The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 7, 947–953. ( 10.1038/ncb1307) [DOI] [PubMed] [Google Scholar]
- 101.Endow SA, Komma DJ. 1998. Assembly and dynamics of an anastral:astral spindle: the meiosis II spindle of Drosophila oocytes. J. Cell Sci. 111, 2487–2495. [DOI] [PubMed] [Google Scholar]
- 102.Mountain V, Simerly C, Howard L, Ando A, Schatten G, Compton DA. 1999. The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle. J. Cell Biol. 147, 351–366. ( 10.1083/jcb.147.2.351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zyss D, Gergely F. 2009. Centrosome function in cancer: guilty or innocent? Trends Cell Biol. 19, 334–346. ( 10.1016/j.tcb.2009.04.001) [DOI] [PubMed] [Google Scholar]
- 104.Silkworth WT, Nardi IK, Scholl LM, Cimini D. 2009. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 4, e6564 ( 10.1371/journal.pone.0006564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cimini D. 2008. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim. Biophys. Acta 1786, 32–40. [DOI] [PubMed] [Google Scholar]
- 106.Schvartzman JM, Sotillo R, Benezra R. 2010. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev Cancer 10, 102–115. ( 10.1038/nrc2781) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. 2011. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333, 1895–1898. ( 10.1126/science.1210214) [DOI] [PubMed] [Google Scholar]
- 108.Crasta K, et al. 2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58. ( 10.1038/nature10802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. 2013. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. ( 10.1016/j.cell.2013.06.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stephens PJ, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. ( 10.1016/j.cell.2010.11.055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Castellanos E, Dominguez P, Gonzalez C. 2008. Centrosome dysfunction in Drosophila neural stem cells causes tumors that are not due to genome instability. Curr. Biol. 18, 1209–1214. ( 10.1016/j.cub.2008.07.029) [DOI] [PubMed] [Google Scholar]
- 112.Caussinus E, Gonzalez C. 2005. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat. Genet. 37, 1125–1129. ( 10.1038/ng1632) [DOI] [PubMed] [Google Scholar]
- 113.Salisbury JL, D'Assoro AB, Lingle WL. 2004. Centrosome amplification and the origin of chromosomal instability in breast cancer. J. Mammary Gland Biol. Neoplasia 9, 275–283. ( 10.1023/B:JOMG.0000048774.27697.30) [DOI] [PubMed] [Google Scholar]
- 114.Tang N, Marshall WF. 2012. Centrosome positioning in vertebrate development. J. Cell Sci. 125(Pt 21), 4951–4961. ( 10.1242/jcs.038083) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stehbens S, Wittmann T. 2012. Targeting and transport: how microtubules control focal adhesion dynamics. J. Cell Biol. 198, 481–489. ( 10.1083/jcb.201206050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lozano E, Betson M, Braga VM. 2003. Tumor progression: small GTPases and loss of cell-cell adhesion. Bioessays 25, 452–463. ( 10.1002/bies.10262) [DOI] [PubMed] [Google Scholar]
- 117.Chang YC, Nalbant P, Birkenfeld J, Chang ZF, Bokoch GM. 2008. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol. Biol. Cell 19, 2147–2153. ( 10.1091/mbc.E07-12-1269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van Horck FP, Ahmadian MR, Haeusler LC, Moolenaar WH, Kranenburg O. 2001. Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 276, 4948–4956. ( 10.1074/jbc.M003839200) [DOI] [PubMed] [Google Scholar]
- 119.Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED. 1999. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1, 45–50. ( 10.1038/9018) [DOI] [PubMed] [Google Scholar]
- 120.Jaspersen SL, Winey M. 2004. The budding yeast spindle pole body: structure, duplication, and function. Annu. Rev. Cell Dev. Biol. 20, 1–28. ( 10.1146/annurev.cellbio.20.022003.114106) [DOI] [PubMed] [Google Scholar]
- 121.Bardin AJ, Amon A. 2001. Men and sin: what's the difference? Nat. Rev. Mol. Cell Biol. 2, 815–826. ( 10.1038/35099020) [DOI] [PubMed] [Google Scholar]
- 122.Hagan IM, Grallert A. 2013. Spatial control of mitotic commitment in fission yeast. Biochem. Soc. Trans. 41, 1766–1771. [DOI] [PubMed] [Google Scholar]
- 123.Grallert A, et al. 2013. Removal of centrosomal PP1 by NIMA kinase unlocks the MPF feedback loop to promote mitotic commitment in S. pombe. Curr. Biol. 23, 213–222. ( 10.1016/j.cub.2012.12.039) [DOI] [PubMed] [Google Scholar]
- 124.Hagan IM. 2008. The spindle pole body plays a key role in controlling mitotic commitment in the fission yeast Schizosaccharomyces pombe. Biochem. Soc. Trans. 36(Pt 5), 1097–1101. ( 10.1042/BST0361097) [DOI] [PubMed] [Google Scholar]
- 125.Jackman M, Lindon C, Nigg EA, Pines J. 2003. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 5, 143–148. ( 10.1038/ncb918) [DOI] [PubMed] [Google Scholar]
- 126.Perez-Mongiovi D, Beckhelling C, Chang P, Ford CC, Houliston E. 2000. Nuclei and microtubule asters stimulate maturation/M phase promoting factor (MPF) activation in Xenopus eggs and egg cytoplasmic extracts. J. Cell Biol. 150, 963–974. ( 10.1083/jcb.150.5.963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hachet V, Canard C, Gonczy P. 2007. Centrosomes promote timely mitotic entry in C. elegans embryos. Dev. Cell 12, 531–541. ( 10.1016/j.devcel.2007.02.015) [DOI] [PubMed] [Google Scholar]
- 128.Portier N, Audhya A, Maddox PS, Green RA, Dammermann A, Desai A, Oegema K. 2007. A microtubule-independent role for centrosomes and Aurora A in nuclear envelope breakdown. Dev. Cell 12, 515–529. ( 10.1016/j.devcel.2007.01.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. 2003. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574. ( 10.1038/nature02166) [DOI] [PubMed] [Google Scholar]
- 130.Jakobsen L, et al. 2011. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 30, 1520–1535. ( 10.1038/emboj.2011.63) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kfoury Y, et al. 2008. Ubiquitylated Tax targets and binds the IKK signalosome at the centrosome. Oncogene 27, 1665–1676. ( 10.1038/sj.onc.1210804) [DOI] [PubMed] [Google Scholar]
- 132.Itoh K, Jenny A, Mlodzik M, Sokol SY. 2009. Centrosomal localization of Diversin and its relevance to Wnt signaling. J. Cell Sci. 122(Pt 20), 3791–3798. ( 10.1242/jcs.057067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fielding AB, Dobreva I, McDonald PC, Foster LJ, Dedhar S. 2008. Integrin-linked kinase localizes to the centrosome and regulates mitotic spindle organization. J. Cell Biol. 180, 681–689. ( 10.1083/jcb.200710074) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wigley WC, et al. 1999. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 145, 481–490. ( 10.1083/jcb.145.3.481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fuentealba LC, Eivers E, Ikeda A, Hurtado C, Kuroda H, Pera EM, De Robertis EM. 2007. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 131, 980–993. ( 10.1016/j.cell.2007.09.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Goetz SC, Anderson KV. 2010. The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344. ( 10.1038/nrg2774) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mahjoub MR, Stearns T. 2012. Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr. Biol. 22, 1628–1634. ( 10.1016/j.cub.2012.06.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mahjoub MR. 2013. The importance of a single primary cilium. Organogenesis 9, 61–69. ( 10.4161/org.25144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cai S, Weaver LN, Ems-McClung SC, Walczak CE. 2009. Kinesin-14 family proteins HSET/XCTK2 control spindle length by cross-linking and sliding microtubules. Mol. Biol. Cell 20, 1348–1359. ( 10.1091/mbc.E08-09-0971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Castiel A, Visochek L, Mittelman L, Dantzer F, Izraeli S, Cohen-Armon M. 2011. A phenanthrene derived PARP inhibitor is an extra-centrosomes de-clustering agent exclusively eradicating human cancer cells. BMC Cancer 11, 412 ( 10.1186/1471-2407-11-412) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Raab MS, et al. 2012. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 72, 5374–5385. ( 10.1158/0008-5472.CAN-12-2026) [DOI] [PubMed] [Google Scholar]
- 142.Chen JG, Horwitz SB. 2002. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res. 62, 1935–1938. [PubMed] [Google Scholar]
- 143.Rathinasamy K, Jindal B, Asthana J, Singh P, Balaji PV, Panda D. 2010. Griseofulvin stabilizes microtubule dynamics, activates p53 and inhibits the proliferation of MCF-7 cells synergistically with vinblastine. BMC Cancer 10, 213 ( 10.1186/1471-2407-10-213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kawamura E, Fielding AB, Kannan N, Balgi A, Eaves CJ, Roberge M, Dedhar S. 2013. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 4, 1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu J, et al. 2013. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem. Biol. 8, 2201–2208. ( 10.1021/cb400186w) [DOI] [PubMed] [Google Scholar]
- 146.Watts CA, et al. 2013. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem. Biol. 20, 1399–1410. ( 10.1016/j.chembiol.2013.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kleylein-Sohn J, Pollinger B, Ohmer M, Hofmann F, Nigg EA, Hemmings BA, Wartmann M. 2012. Acentrosomal spindle organization renders cancer cells dependent on the kinesin HSET. J. Cell Sci. 125, 5391–5402. ( 10.1242/jcs.107474) [DOI] [PubMed] [Google Scholar]
- 148.Gentric G, Desdouets C, Celton-Morizur S. 2012. Hepatocytes polyploidization and cell cycle control in liver physiopathology. Int. J. Hepatol. 2012, 282430 ( 10.1155/2012/282430) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. 2006. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048. ( 10.1038/ng1861) [DOI] [PubMed] [Google Scholar]
- 150.De Sousa EMF, Vermeulen L, Fessler E, Medema JP. 2013. Cancer heterogeneity—a multifaceted view. EMBO Rep. 14, 686–695. ( 10.1038/embor.2013.92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mennella V, Agard DA, Huang B, Pelletier L. 2014. Amorphous no more: subdiffraction view of the pericentriolar material architecture. Trends Cell Biol. 24, 188–197. ( 10.1016/j.tcb.2013.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]