

Abstract
The centrosome, a key microtubule organizing centre, is composed of centrioles, embedded in a protein-rich matrix. Centrosomes control the internal spatial organization of somatic cells, and as such contribute to cell division, cell polarity and migration. Upon exiting the cell cycle, most cell types in the human body convert their centrioles into basal bodies, which drive the assembly of primary cilia, involved in sensing and signal transduction at the cell surface. Centrosomal genes are targeted by mutations in numerous human developmental disorders, ranging from diseases exclusively affecting brain development, through global growth failure syndromes to diverse pathologies associated with ciliary malfunction. Despite our much-improved understanding of centrosome function in cellular processes, we know remarkably little of its role in the organismal context, especially in mammals. In this review, we examine how centrosome dysfunction impacts on complex physiological processes and speculate on the challenges we face when applying knowledge generated from in vitro and in vivo model systems to human development.
Keywords: centrosome, centriole, microcephaly, dwarfism, cilia, ciliopathy
1. Centrosomes and primary cilia
The vertebrate centrosome comprises a pair of centrioles embedded in a pericentriolar matrix (PCM), which is enriched in γ-tubulin complexes responsible for microtubule nucleation. The involvement of centrosomes in mitosis is well documented; they facilitate mitotic spindle assembly, improve the fidelity of chromosome segregation and orient mitotic spindles in relation to the cell cortex and extracellular cues. In addition, centrosomes have been implicated in cell migration, vesicle trafficking and cell polarity [1]. Centrosomes duplicate once per cell cycle in a semi-conservative manner, whereby a procentriole assembles at a perpendicular angle to each old centriole (figure 1) [2]. Centrosome duplication is tightly controlled: per centriole only one procentriole forms per cell cycle. The engagement between mother centriole and its procentriole persists until late mitosis, ensuring that they co-segregate as part of the same spindle pole in cytokinesis. Centrosomes mature prior to mitotic entry by recruitment of further PCM components and γ-tubulin complexes, thereby increasing microtubule nucleation capacity. In many cell types, cell cycle exit triggers the conversion of the mother centriole into a basal body, which nucleates the assembly of a primary cilium (figure 2) [3]. Primary cilia comprise a microtubule axoneme, built of nine doublet microtubules, surrounded by plasma membrane. Motile cilia contain an additional central pair of singlet microtubules. Once believed to be vestigial organelles, primary cilia have fundamental sensory and signalling functions that mediate a range of processes, from proliferation through polarity to differentiation. In particular, primary cilia have been implicated in Sonic Hedgehog (Shh) and Wnt signalling pathways [4]. Moreover, ciliary dysfunction has emerged as the root cause of many human disorders, collectively termed ciliopathies [5]. Below, we will discuss the role of centrosomes in development and pathologies.
Figure 1.
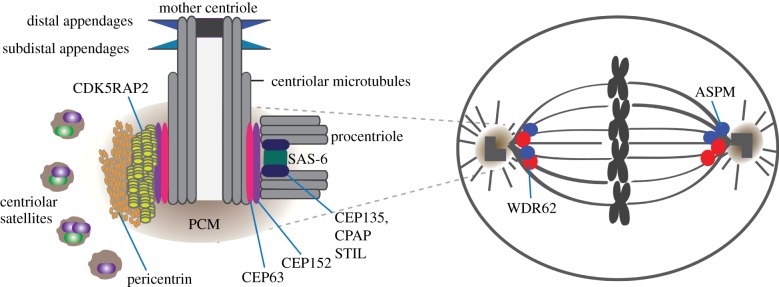
Schematic of the vertebrate centrosome and the mitotic spindle. Key structural features of centrosomes are indicated. Locations of disease-associated centrosomal proteins are shown. Mother centrioles acquire two sets of appendages during centriole maturation. Procentriole formation requires assembly of SAS-6 homodimers into a cartwheel-like structure. STIL interacts with CPAP and possibly SAS-6 in the procentriole. By binding SAS-6, CPAP and centriolar microtubules, CEP135 could stabilize centrioles. CDK5RAP2 and pericentrin are scaffolding proteins located in the PCM, surrounded by centriolar satellites. Centrosomes are positioned at the mitotic spindle poles where spindle microtubules focus and nucleate astral microtubules. ASPM and WDR62 localize to the mitotic spindle poles. (Online version in colour.)
Figure 2.
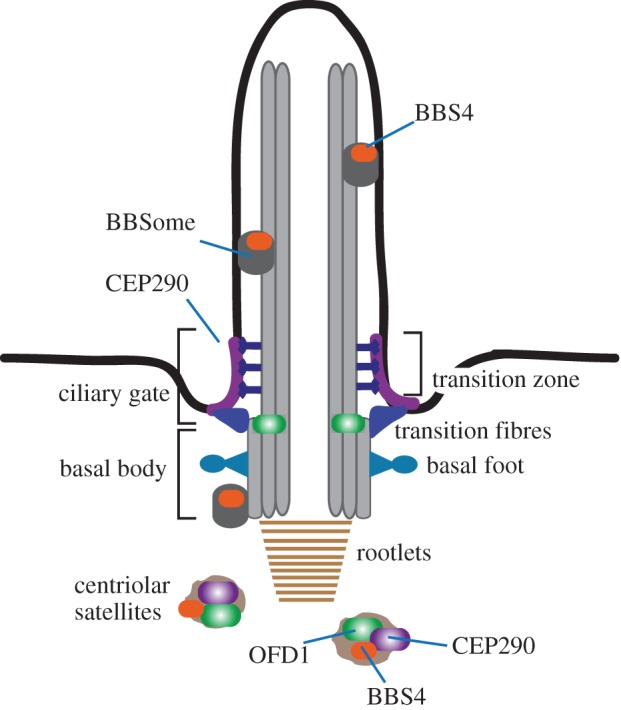
Schematic of the primary cilium. Key structural features of cilia are indicated. The basal body is attached to the plasma membrane via transition fibres derived from the distal appendages. Structural stability is provided by the ciliary rootlet. During ciliogenesis, centriolar satellites deliver proteins like CEP290, BBS4 and OFD1 to the cilium. CEP290 localizes to the transition zone and functions as the ciliary gate. BBS4 assembles into the BBSome at the ciliary base and is transported inside the cilium. OFD1 is localized to the distal end of the basal body. (Online version in colour.)
2. Centrosomes in brain development
(a). Autosomal primary recessive microcephaly: a disorder of neurogenesis?
Autosomal primary recessive microcephaly (MCPH) is a genetically heterogeneous neurodevelopmental disorder caused by mutations in at least nine genes. Its main characteristics are non-progressive mental retardation and reduction in head circumference at birth primarily affecting cerebral cortex size [6].
Development of the cerebral cortex begins with the rapid expansion of neuroepithelial progenitor (NEP) cells leading to the thickening of the ectoderm and formation of the neural plate. Normal cortical development relies on neurogenesis, a process responsible for the production of neurons through coordinated neural stem cell proliferation and differentiation. NEP and NEP-derived radial glial (RG) cells reside in the ventricular and sub-ventricular zones (SVZs) and are collectively referred to as apical progenitors (APs) (figure 3). APs are polarized cells with characteristic cytoplasmic extensions: the long basal process extends to the pial surface, whereas the centrosome-containing short apical process contacts the lumen of the ventricle. Initially, most APs undergo symmetric cell divisions to expand the progenitor pool, but the balance subsequently shifts towards asymmetric neurogenic divisions yielding an AP and an immature neuron [7]. Inheritance or loss of the apical and/or basal processes seems important for cell fate decisions [8–11]. Post-mitotic neurons generated by neurogenic divisions migrate radially to the pial surface, where they differentiate and form the six-layered neocortex. Therefore, in addition to balancing self-renewing and neurogenic divisions, the formation of the cortex requires neuronal migration. Although MCPH is widely believed to arise from abnormal neurogenesis, simplified gyral patterns have been reported in some cases, raising the possibility that neuronal migration defects occur in MCPH [12–16].
Figure 3.

Neurogenesis in the rodent brain. NEPs and RG are collectively termed APs that reside in the ventricular zone (VZ). NEPs first expand through rapid symmetric divisions, but after the onset of neurogenesis they give rise to RG cells, which can divide symmetrically into two RGs or asymmetrically into an RG and a nascent neuron. APs undergoing symmetric proliferative divisions display a vertical cleavage plane relative to the apical surface, whereas those undergoing neurogenic divisions show oblique or horizontal cleavage planes. In the case of symmetric divisions, the cleavage furrow bisects the apical membrane and adjacent adherens junctions. In asymmetric divisions, the RG inherits the apical plasma membrane, whereas the newborn neuron migrates through the SVZ towards the cortical plate (CP) where it matures. Loss-of-function of CDK5RAP2 and ASPM in mouse models compromises symmetric AP divisions, similarly to CDK5RAP2-deficient human brain organoids. Individuals with WDR62 mutations exhibit cortical malformations and neuronal migration defects. (Online version in colour.)
(b). Autosomal primary recessive microcephaly genes: the centrosome connection
Centrosomes have been implicated in multiple processes during brain development, including neurogenesis, neuronal migration and polarity, although their precise contributions are not well established [17]. To date, MCPH represents the best candidate for a bona fide genetic disorder of the centrosome. Of the nine genes implicated in MCPH, five encode core centrosomal components (CPAP, CEP152, CEP135, STIL and CDK5RAP2) and two code for proteins associated with mitotic spindle poles (WDR62 and ASPM) (table 1) [18]. The remaining two proteins exhibit no obvious functional overlap; CASC5/Blinkin is a centromere-associated protein involved in the spindle assembly checkpoint [19,20], whereas MCPH1, a nuclear and centrosomal protein, controls cell cycle checkpoint transitions, DNA damage response and repair [21,22].
Table 1.
Summary of centrosomal genes implicated in genetic disorders. (NP, neural progenitors.)
| gene | disease locus (disease) | major clinical features | protein localization | protein function | phenotype in mouse models |
|---|---|---|---|---|---|
| ASPM | MCPH5 | microcephaly, mild to severe cognitive impairment, seizures and short stature | mitotic spindle pole and midbody | mitotic spindle orientation and organization, mitotic progression, interkinetic nuclear migration and cytokinesis | microcephaly, reduced proliferation of NPs, neuronal migration defects, abnormal cortical layering, germ cell defects and abnormal Wnt signalling |
| CDK5RAP2 | MCPH3 | microcephaly and short stature | centrosome | centrosome organization and maturation, microtubule organization, centrosome cohesion, centriole engagement and centrosome-spindle pole connection | microcephaly, reduced proliferation of NPs, mitotic progression defects and apoptosis in NPs, supernumerary centrosomes and mild macrocytic anaemia |
| CEP63 | SCKL6 (Seckel) | microcephaly and short stature | centrosome | centriole duplication | |
| CEP135 | MCPH8 | microcephaly | centrosome | centriole biogenesis, centrosome cohesion and microtubule organization | |
| CEP152 | MCPH9 | microcephaly, moderate cognitive impairment and simplified gyri | centrosome | centriole biogenesis | |
| SCKL5 (Seckel) | proportionate short stature, severe microcephaly, mental retardation and simplified gyri | ||||
| CPAP/CENPJ | MCPH6 | microcephaly, mild to severe cognitive impairment and facial dysmorphism | centrosome | centriole biogenesis and scaffolding PCM assembly | dwarf, microcephaly, increased apoptosis, abnormal centrosome numbers, aneuploidy, memory impairment and skeletal anomalies |
| SCKL4 (Seckel) | Proportionate short stature, microcephaly, skeletal anomalies | ||||
| MCPH1 | MCPH1 | microcephaly, short stature, premature chromosome condensation and mild to severe cognitive impairment | centrosome and nucleus | DNA damage response and repair and cell cycle checkpoints | growth retardation, microcephaly, reduced proliferation of NPs and radiation sensitivity |
| ORC1 | MGS1 (Meier-Gorlin) | proportionate short stature, microcephaly, knee and ear abnormalities and skeletal abnormalities | centrosome and nucleus | DNA replication, centrosome cycle and centriole copy number regulation | |
| PCNT | MOPD2 (microcephalic osteodysplastic PD II) | extreme but proportionate short stature, bone abnormalities, microcephaly and near normal intelligence | centrosome | centrosome maturation and mitotic spindle organization | perinatal lethality, microcephaly, reduced olfactory bulb, defective olfactory cilia and neuronal migration defects |
| STIL | MCPH7 | microcephaly, ataxia and short stature | centrosome | centriole biogenesis, mitotic entry | embryonic lethality, defects in neural tube closure and heart development |
| WDR62 | MCPH2 | microcephaly, pachygyria, hypoplesia of corpus callosum, cortical thickening, lissencephaly, polymicrogyria and facial dysmorphism | mitotic spindle pole | mitotic spindle assembly and orientation, and centrosome-spindle pole connection | premature cell cycle exit and reduced proliferation of NPs (in utero siRNA only) |
| BBS4 | BBS4 (Bardet–Biedl) | retinal dystrophy, obesity, polydactyly, renal malformation, hypogenitalism and male infertiliy | centriolar satellites, centrosome and cilium | centriolar satellite function, microtubule anchorage and cilium formation | partial embryonic lethality, low birth weight and small size, obesity, defects in airway cilia function, retinal degeneration and sperm lacks flagellum |
| CEP290 | BBS14 (Bardet–Biedl) | retinal dystrophy, obesity, polydactyly, cognitive impairments, hypogenitalism and renal malformation | centriolar satellites, centrosome, cilia (transition zone) and nucleus | microtubule organization, cilium assembly and ciliary gatekeeper | retinal degeneration, olfactory dysfunction and defective cerebellar midline fusion |
| JBTS5 (Joubert) | cerebellar malfunction, ataxia, retinal degeneration and nephronepthisis | ||||
| MKS4 (Meckel) | occipital encephalocele (neural tube defect), polydactyly and dysplastic kidneys | ||||
| SLSN6 (Senior–Loken) | retinal dystrophy and nephronepthisis | ||||
| OFD1 | OFD type I | male embryonic lethality; abnormalities of face, oral cavity and digits, polycystic kidney disease, mental retardation and macrocephaly | centriolar satellites and centrosome | regulation of centriole length, control of distal appendage formation and cilium assembly | male mice: early gestation lethal and neural tube closure defects; female mice: die at birth, left-right axis randomization, facial and oral abnormalities, and polydactyly |
| Joubert syndrome 10 | mental retardation, recurrent infections, developmental delay, cerebellar abnormalities and retinitis pigmentosa | ||||
| SGBS2 | perinatal male lethality, macrocephaly, craniofacial anomalies and respiratory problems |
Despite not being core centrosomal components, both ASPM and WDR62 localize to the mitotic spindle poles and are involved in mitotic spindle assembly and normal mitotic progression [23–25]. Centriole biogenesis requires a small core set of proteins: PLK4, SAS-6, STIL/Ana2/SAS-5, CPAP/CENPJ/SAS-4, CEP135/Bld10 and CEP152/Asl [2]. Strikingly, four of these carry MCPH-linked mutations [26–28]. CPAP/MCPH6 appears to be a key node of the MCPH protein network, because it binds STIL/MCPH7, CEP152/MCPH9 and CEP135/MCPH8, although probably not simultaneously [29–32]. A missense MCPH mutation weakens the ability of CPAP to bind STIL and impairs centriole production [32–35]. By contrast, MCPH-causing mutations in STIL do not interfere with centriole formation, but prevent cell cycle-dependent degradation of the protein, thereby triggering centriole amplification [36]. Thus, MCPH mutations seem to target essential components of the centriole biogenesis pathway. By perturbing the precise control of centriole duplication, these mutations are able to deregulate centrosome numbers in cells. Indeed, although not required for centriole formation per se, CDK5RAP2 has also been implicated in controlling centrosome numbers, in addition to roles in centrosomal microtubule nucleation and organization, mitotic spindle orientation and ciliogenesis [37].
Thus far, there is little evidence to suggest that centriole biogenesis is impaired in MCPH patient-derived cells, although ultrastructural studies will be needed to confirm if this is indeed the case [38]. Conversely, the presence of normal centrosomes in patient-derived adult somatic cells does not necessarily rule out defective centriole biogenesis during development, because cell division cycles in the embryo must be subject to stricter temporal control. Centrioles are involved in recruiting and organizing the PCM, and centriole length correlates with the amount of PCM in the centrosome [39–41]. CPAP, an important node in the MCPH protein network, is a key regulator of centriolar microtubule elongation, and thus centriole length may be impaired by MCPH-linked mutations in CPAP and its interactors [41–43]. We speculate that if centrioles are too short in MCPH cells, this could impair PCM formation and centrosome maturation, which in turn may interfere with centriole duplication [44]. Moreover, short centrioles may not be able to acquire appendages, structures implicated in centriole maturation as well as basal body function and ciliogenesis.
Below we describe the cellular pathways most susceptible to centrosome dysfunction and their potential contribution to neurogenesis.
(c). Linking cellular phenotypes of autosomal primary recessive microcephaly mutations to neurogenesis
(i). Cell cycle progression, centrosome duplication and mitosis
The cell cycle machinery faces an immense challenge during development; in addition to its crucial task of maintaining genome integrity, it needs to ensure that cell division occurs in a temporally controlled fashion. Changes in cell cycle length have been reported during the development of the mammalian cortex. In particular, G1 phase lengthens and S phase shortens as cells transition from proliferative to neurogenic divisions [45–47]. Thus, cell cycle duration and cell fate determination may be linked in the neuroepithelium. Loss of centrosome integrity in cultured cells has been reported to induce senescence or p38 mitogen-activated protein kinase-dependent G1 arrest but whether these occur in cells with MCPH mutations remains to be seen [48–50].
Cells with abnormal centrosome numbers take longer to form a bipolar spindle and consequently show a mitotic delay [51–55]. In fact, mitotic delay is the major phenotype seen after depletion of WDR62, ASPM and STIL in developing zebrafish retinal neuroepithelium [56]. Mutations in CDK5RAP2 also cause mitotic delay in the mouse brain [57]. A recent study suggests that cells sense the duration of mitosis; in particular, prolonged prometaphase in the mother cell causes daughter cells to exit the cell cycle, even if mitosis is completed normally [58]. Therefore, mitotic delay triggered by aberrant centrosomes could obliterate long-term proliferative capacity of progenitors leading to MCPH. In support of this scenario, reduced proliferation is the shared phenotype of CDK5RAP2 mutation and WDR62- or ASPM-depletion in the mouse neocortex [24,57,59]. Premature entry into mitosis has also been implicated in MCPH [60].
Cells containing too few or too many centrosomes exhibit transient anastral, mono- or multipolar spindles and frequent chromosome missegregation [53,61,62]. Chromosomal instability generates aneuploidy, which has a strong anti-proliferative effect owing to imbalanced gene expression [63]. Remarkably, centrosome amplification induced by PLK4 overexpression in the mouse brain causes mitotic delay, aneuploidy and apoptosis in neural progenitors, collectively leading to microcephaly [64]. Of these, mitotic delay and aneuploidy are more likely to account for the proliferative impairment than apoptosis, because p53 disruption does not restore normal brain size in PLK4-overexpressing mice, and elevated apoptosis is not consistently observed in mouse models of MCPH. Multipolar spindles are also seen in CDK5RAP2 mutant mouse neuroepithelium, but a link to aneuploidy is yet to be established in this system [57].
An example for the physiological effects of aneuploidy is provided by the rare genetic disease, mosaic variegated aneuploidy syndrome [65–67]. Whole chromosome aneuploidy occurs in greater than 25% of cells of affected individuals. Consistent with a deleterious effect of aneuploidy on brain development, microcephaly is frequent in this syndrome, and additional structural brain defects and seizures are also seen in some patients [66]. The severity of these defects may depend on the extent of aneuploidy acquired during prenatal brain development. We hypothesize that the aneuploidy triggered by centrosome dysfunction in MCPH is not as profound as in mosaic variegated aneuploidy, thus explaining the overall milder brain phenotype of MCPH.
(ii). Mitotic spindle orientation
Balancing the number of progenitors and differentiated cells is crucial for normal brain development. Cell fate decisions frequently depend on the orientation of the cell division axis, which controls partitioning of fate determinants into daughter cells [7]. APs undergoing symmetric proliferative divisions display vertical cleavage plane (i.e. perpendicular to the apical surface), whereas those undergoing neurogenic divisions show oblique or horizontal cleavage planes (figure 3) [7]. Only if the plane is vertical to the apical surface, do both daughter cells inherit a region of the apical membrane, important for maintenance of stem cell fate [68,69]. Mitotic spindle orientation is governed by centrosome-nucleated astral microtubules that are captured by cortically tethered microtubule motor complexes [70]. For centrosomes to orient the mitotic spindle, they must nucleate astral microtubules and maintain stable association with the mitotic spindle poles, processes impaired in CDK5RAP2- and WDR62-deficient cells [24,37,71,72]. Mitotic spindle orientation defects have been described in cells depleted of WDR62, CPAP and STIL, and a premature shift to oblique orientation occurs in developing mouse brains depleted of ASPM, MCPH1 and CDK5RAP2 [24,57,73–75]. Correct positioning of the cleavage furrow and accurate execution of cytokinesis are also crucial for regulating the cell division plane. So far, ASPM is the only MCPH protein directly implicated in cytokinesis [76–78]. Although spindle orientation defects are likely to contribute to the aetiology of MCPH, they probably account only for part of the cell loss, because randomized spindle orientation in the mouse neocortex does not preclude normal neuron production rates [8].
(iii). Centrosome asymmetry and ciliogenesis
By nature of the centrosome duplication cycle, the two centrosomes contain differentially aged centrioles: a fully mature mother centriole and an immature daughter centriole produced in the preceding cell cycle. In the mouse neocortex, inheritance of the mother centriole maintains stemness; interfering with this process causes premature depletion of progenitors from the proliferative zone [79]. Why is this so? Recent work suggests that centrosome asymmetry determines the speed of cilia assembly in daughter cells following mitosis. The cell in possession of the mother centriole assembles a primary cilium faster [80]. Cilia transmit Shh and Wnt signals, both involved in neurogenesis, and rapid access to these could impact on cell fate decisions [81–83]. If MCPH mutations impaired centriole maturation, this could impede speed of cilium formation, causing abnormal signalling and premature neural differentiation.
Centrosomes are also considered relevant to interkinetic nuclear migration (INM), a process characteristic of APs. INM involves the movement of nuclei between the apical and basal surfaces in phase with the cell cycle [84]. Although its precise role in neurogenesis is not clear, INM exposes nuclei to signalling gradients, thereby potentially influencing cell fate decisions [85]. It will be important to establish whether INM is affected in MCPH mouse models, especially since ASPM in Drosophila contributes to this process [86].
(d). In vivo autosomal primary recessive microcephaly models: rodent or human?
Our current understanding of MCPH, and much of neurogenesis, is based on studies of murine brain. However, the human cortex is increased in size compared with most other species, partly owing to the protracted proliferative activity of progenitors in the outer SVZ, a zone unique to gyrencephalic brains. Unlike APs in the rodent cortex, these progenitors display basal but not apical processes, yet undergo self-renewing and neurogenic divisions [9–11]. Thus, gyrencephalic cortical development requires cell behaviour atypical of the rodent brain. A recent breakthrough in brain research has been the generation of human cerebral organoids from induced pluripotent stem cells [16]. These recapitulate many of the complex features of embryonic brain development and could prove invaluable in deciphering disease mechanisms of MCPH. Remarkably, organoids derived from CDK5RAP2-deficient MCPH patient cells display premature neural differentiation and spindle orientation defects. Premature differentiation could reduce the number of early progenitors (i.e. founder cells), causing an overall collapse in neuron numbers.
3. Centrosomes and body size
(a). Primordial dwarfism syndromes
Microcephalic primordial dwarfism (PD) defines a group of autosomal recessive human genetic disorders that show pre- and postnatal growth failure accompanied by microcephaly [87]. Of these, the most prominent diseases are Seckel syndrome, microcephalic osteodysplastic PD (MOPD) types I and II, and Meier-Gorlin syndrome. At least 14 different genes have been implicated in PD so far. These encode proteins involved in DNA replication such as subunits of the origin recognition complex (ORC), in DNA damage response like ATR kinase, ATR-interacting protein (ATRIP) [88] and CtIP [89], or in centrosome function like CEP152, CPAP, pericentrin (PCNT) and CEP63 [90–94] (table 1). In line with the consensus that PD represents a type of hypocellular dwarfism, PD proteins are all implicated in the cell cycle. Below, we will discuss how centrosome-related pathways could contribute to the profound cell loss during fetal development in PD.
(b). Cellular pathways disrupted by mutations in centrosomal primordial dwarfism genes and their putative link to organismal growth
(i). DNA damage response and replicative stress
The first Seckel allele identified was a mutation in ATR (ATM- and RAD3-related), a kinase that orchestrates cellular response to single-stranded DNA resulting from stalled replication forks or DNA repair intermediates [95,96]. Subsequent discoveries of Seckel mutations in proteins implicated in ATR activation, ATRIP and CtIP, further strengthen the case for impaired ATR signalling being the cause of Seckel syndrome [88,89]. ATR, and its substrate, the checkpoint kinase CHK1, coordinate the response to replicative stress, accumulation of which can disrupt development and tissue homoeostasis [97]. Mutations in the ORC proteins, involved in DNA replication, cause Meier-Gorlin syndrome [90,92]. Cells derived from MOPDII patients with PCNT mutations exhibit defective ATR signalling and a marked reduction in the centrosomal pool of CHK1 [60,93]. Although the precise role of this pool remains controversial, reduced centrosomal CHK1 levels correlate with premature activation of cyclin B-CDK1 in pericentrin-deficient cells [60,98,99]. Thus, anti-proliferative effects of replicative stress and premature CDK1 activation could contribute to the global growth failure seen in PD patients. However, impaired ATR signalling may not be universal across all PD complementation groups. CHK1 kinase function is normal in CEP152-Seckel fibroblasts, and instead these show activation of ataxia telangiectasia-mutated (ATM) kinase, which recognizes double-stranded DNA breaks [100]. By contrast, DNA damage response is intact in a mouse model of CPAP-Seckel, which recapitulates features of Seckel syndrome including intrauterine growth retardation and small brain size [101,102]. Therefore, dwarfism caused by mutations in centrosomal genes may not be directly linked with their capacity to activate DNA damage response or DNA repair.
(ii). Centrosome amplification and aneuploidy
Centrosome amplification has been observed in fibroblasts with disease-associated mutations in pericentrin, CEP152, CPAP and ORC1 [91,96,100]. It is thus possible that centrosomal PD proteins prevent centriole overduplication, for instance by promoting engagement between the mother centriole and its procentriole [38,103,104]. Recent studies suggest that ORC1 exercises centrosome copy number control by suppressing CDK2-cyclin E-dependent reduplication of centrioles, a function specifically disrupted by Meier-Gorlin mutations in ORC1 [91]. Cells with centrosome amplification must inactivate excess centrosomes or cluster these into two poles [105]. In the absence of such mechanisms, centrosome amplification results in multipolar cytokinesis and cell death [106]. If inactivation or clustering is efficient, cells can survive but at a cost; centrosome clustering causes not only mitotic delay, but also chromosomal instability [55,61]. Importantly, genome instability has been documented in CPAP-Seckel mouse and in Seckel patient-derived cells; CEP152-Seckel lymphocytes exhibit aneuploidy in addition to abnormal mitotic spindle morphologies [100,101].
(iii). Ciliogenesis
Recent studies implicate cilia dysfunction in the aetiology of PD. Both ORC1 and pericentrin mutant cells show defective ciliary recruitment of the Shh receptor, Smoothened and consequently abnormal Shh signalling with potentially wide-ranging effects on development [107]. As the primary cilium assembles on a basal body, a structure derived from the mother centriole, CEP152 and CPAP, along with the MCPH proteins, STIL and CEP135, are required not only for centriole biogenesis but also for ciliogenesis [108–112]. Given the link between centriole biogenesis and primary cilia assembly, it is puzzling that patients carrying MCPH- or PD-associated mutations in these essential regulators do not exhibit clinical signs of defective ciliary function. We speculate that in the context of a tissue, cells with numerical or structural centrosome abnormalities might be eliminated and/or prevented from assembling aberrant cilia [64].
4. Ciliopathies
(a). Primary cilia formation and centriolar satellites
Most human cell types are ciliated and therefore defects in ciliary function or structure cause pleiotropic genetic disorders, termed ciliopathies. These share many clinical features, with kidney, eye, liver and brain being the most affected organs [113]. Over the last decade, cilia have emerged as vital cellular compartments for transducing mechanical and extracellular signals, regulating organogenesis, planar polarity, proliferation, DNA damage response and autophagy [4,114,115]. Indeed, impaired signalling is considered a key underlying factor in ciliopathies.
Mutations in over 30 proteins have been identified in ciliopathies (see [113] for extensive review). Despite many of these localizing to centrosomes and basal bodies, only a handful have known functions both in centrosomes and ciliogenesis, and these include CEP290 and the oral-facial-digital syndrome 1 (OFD1) protein. CEP290, OFD1 and a third ciliopathy protein, Bardet–Biedl syndrome (BBS4), also associate with centriolar satellites, electrondense granules concentrated around the PCM [116,117]. Through a detailed discussion of these factors, we will highlight key themes of ciliary research and aim to provide insight into the complex nature of ciliopathies.
The first step in ciliogenesis is the transformation of the mother centriole into a basal body, which then docks at the plasma membrane and nucleates the assembly of axonemal microtubules (figure 2). Golgi-derived vesicles fuse with the plasma membrane to extend the membrane protrusion around the growing axoneme, a process dependent on the GTPase, Rab8a, and its guanine nucleotide exchange factor, Rabin8. Cilium assembly and maintenance require a selective transport system for ciliary proteins called intraflagellar transport (IFT) [3]. Centriolar satellites have been implicated in centrosomal protein transport and ciliogenesis [118]. In addition to providing a platform for pro-ciliogenesis proteins, satellites suppress untimely cilia formation by sequestering key ciliogenesis factors [115,119–122]. Thus, certain ciliopathy phenotypes could arise from ill-timed ciliogenesis and signalling.
In cycling cells, CEP290 mediates interphase microtubule organization [123]. In ciliogenesis, CEP290 promotes vesicle trafficking to the ciliary membrane and ciliary outgrowth; it facilitates ciliary targeting of Rab8a, but it also promotes ciliary translocation of BBS4, thereby completing the assembly of the BBSome, a multiprotein complex that binds Rabin8 and triggers Rab8a activation [122–124]. CEP290 may also act as a ciliary gatekeeper; in Chlamydomonas reinhardtii, CEP290 is found at the transition zone, a region that restricts entry of soluble proteins into the ciliary compartment [125]. By contrast, OFD1 acts at the distal ends of centrioles to control distal appendage formation and centriole length [126]. It is involved in targeting the IFT protein, IFT88, to the distal end of the centriole and possibly to the basal body [127]. Through recruitment of CEP164, a distal appendage protein mutated in cystic kidney disease, OFD1 also promotes basal body docking to the plasma membrane [114,128].
(b). Phenotypic spectra of ciliopathies caused by mutations in centriolar satellite components
Animal models underscore the importance of OFD1, CEP290 and BBS4 in ciliogenesis. Zebrafish depleted of Ofd1 show typical signs of defective cilia: body curvature and hydrocephalus. Laterality defects are also present owing to shorter motile primary cilia in the Kupffer vesicle, an embryonic organ required for left–right asymmetry [129]. Such defects also occur in CEP290- and BBS4-depleted zebrafish, with the former exhibiting additional retinal anomalies [130,131].
Ofd1 is located on the X chromosome both in mice and humans. Loss-of-function studies in mice show a gender-dependent effect of Ofd1 inactivation; hemizygous males die during gestation probably as a result of neural tube closure failure, whereas heterozygous females die at birth exhibiting abnormalities in oral and facial structures (table 1) [132]. On the cellular level, ciliary axoneme elongation is defective in the developing forebrain of mutant females despite basal bodies being able to dock normally to the plasma membrane [133]. These models recapitulate many of the phenotypes of human oral-facial-digital syndrome type 1, which also exhibits an X-linked dominant male-lethal trait [134]. Male lethality occurs in early pregnancy, whereas affected females exhibit polycystic kidney disease and neurodevelopmental abnormalities. There is considerable phenotypic variability even within the same family, most likely owing to cellular mosaicism created by X-inactivation. Mutations in OFD1 have also been uncovered in other ciliopathies such as Joubert syndrome and Simpsons–Golabi–Behmel syndrome type 2 (SGBS2) [135]. While mental retardation is common to both, Joubert syndrome involves cerebellar malfunction and ataxia, whereas SGBS2 is characterized by macrocephaly.
BBS4 null mice display partial embryonic lethality. Newborns show low body weight before weaning, yet subsequently develop obesity and retinal degeneration [136]. By contrast, CEP290 mutant mice are viable. Hypomorphic alleles cause retinal degeneration and impaired olfaction [137,138], whereas null animals exhibit a defective cerebellar midline fusion [139]. In humans, BBS4 mutations cause BBS characterized by retinal dystrophy, obesity, cognitive impairments and renal malformation [140]. BBS4 mutations are also present in the embryonically lethal Meckel syndrome [141]. Likewise, CEP290 mutations have been linked to both BBS and Meckel, in addition to Senior–Loken and Joubert syndromes [142].
How can mutations in a single gene lead to so many disease phenotypes and disorders? First, evidence suggests that hypomorphic mutations target specific aspects of protein function [126]. Second, CEP290 and OFD1 participate in multiple protein complexes during ciliogenesis, which could be differentially affected by mutations [122,123,126,143]. Last, patients often carry mutations in multiple ciliary genes [116]. As normal ciliary function is implicated in a growing number of fundamental signalling pathways, mutations causing suboptimal function will have wide-ranging effects in development and homoeostasis. Further work will be necessary to shed light on how disease-linked perturbations of cilia function and signalling pathways generate these complex organ-specific phenotypes.
5. Perspectives
Remarkably, CPAP and CEP152 are mutated in both MCPH and Seckel syndrome, suggesting that the two disorders might represent different ends of a disease continuum [26,100,102,144]. Indeed, recent reports uncovered mutations in the Seckel genes ATR and CtIP that cause primary microcephaly without PD, whereas short stature has been noted in some individuals with mutations in the MCPH gene, CDK5RAP2 [16,89,145]. Thus, MCPH and Seckel must also share common cellular mechanisms. We speculate that mitotic defects, especially mitotic delay and chromosome segregation errors, are probable common causes of MCPH and PD (figure 4). Over 30 billion neurons are produced during human fetal brain development with little proliferation later in life, rendering the brain particularly susceptible to mitotic defects and aneuploidy. Indeed, microcephaly is frequent in mice and humans with trisomies. Moreover, CEP63 and CEP152 form a complex with CEP57, a protein mutated in mosaic variegated aneuploidy syndrome, a genetic disorder associated with severe microcephaly and short stature [67,146]. Genome instability disorders such as Fanconi anaemia, Nijmegen breakage or Bloom syndromes also manifest with microcephaly and short stature, arguing for a vital role of genome integrity in neurogenesis and normal body size [147]. A striking difference between these disorders and MCPH or PD is the lack of cancer predisposition in the latter, indicative of a robust anti-proliferative effect of abnormal centrosomes. Validated in vivo models that mimic disease-associated mutations will be needed to tease apart the cellular mechanisms and address the physiological consequences of centrosome dysfunction.
Figure 4.

Putative cellular mechanisms implicated in MCPH and PD. (Online version in colour.)
Acknowledgements
We thank members of the Gergely group and the reviewers for helpful comments.
Funding statement
We thank Cancer Research UK for the generous core funding of our laboratory.
References
- 1.Bornens M. 2012. The centrosome in cells and organisms. Science 335, 422–426. ( 10.1126/science.1209037) [DOI] [PubMed] [Google Scholar]
- 2.Gonczy P. 2012. Towards a molecular architecture of centriole assembly. Nat. Rev. Mol. Cell Biol. 13, 425–435. ( 10.1038/nrm3373) [DOI] [PubMed] [Google Scholar]
- 3.Ishikawa H, Marshall WF. 2011. Ciliogenesis: building the cell's antenna. Nat. Rev. Mol. Cell Biol. 12, 222–234. ( 10.1038/nrm3085) [DOI] [PubMed] [Google Scholar]
- 4.Singla V, Reiter JF. 2006. The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629–633. ( 10.1126/science.1124534) [DOI] [PubMed] [Google Scholar]
- 5.Fliegauf M, Benzing T, Omran H. 2007. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893. ( 10.1038/nrm2278) [DOI] [PubMed] [Google Scholar]
- 6.Mahmood S, Ahmad W, Hassan MJ. 2011. Autosomal recessive primary microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J. Rare Dis. 6, 39 ( 10.1186/1750-1172-6-39) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gotz M, Huttner WB. 2005. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788. ( 10.1038/nrm1739) [DOI] [PubMed] [Google Scholar]
- 8.Konno D, Shioi G, Shitamukai A, Mori A, Kiyonari H, Miyata T, Matsuzaki F. 2008. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat. Cell Biol. 10, 93–101. ( 10.1038/ncb1673) [DOI] [PubMed] [Google Scholar]
- 9.Shitamukai A, Konno D, Matsuzaki F. 2011. Oblique radial glial divisions in the developing mouse neocortex induce self-renewing progenitors outside the germinal zone that resemble primate outer subventricular zone progenitors. J. Neurosci. 31, 3683–3695. ( 10.1523/JNEUROSCI.4773-10.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen DV, Lui JH, Parker PR, Kriegstein AR. 2010. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. ( 10.1038/nature08845) [DOI] [PubMed] [Google Scholar]
- 11.Fietz SA, et al. 2010. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 13, 690–699. ( 10.1038/nn.2553) [DOI] [PubMed] [Google Scholar]
- 12.Thornton GK, Woods CG. 2009. Primary microcephaly: do all roads lead to Rome? Trends Genet. 25, 501–510. ( 10.1016/j.tig.2009.09.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bilguvar K, et al. 2010. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. ( 10.1038/nature09327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murdock DR, Clark GD, Bainbridge MN, Newsham I, Wu YQ, Muzny DM, Cheung SW, Gibbs RA, Ramocki MB. 2011. Whole-exome sequencing identifies compound heterozygous mutations in WDR62 in siblings with recurrent polymicrogyria. Am. J. Med. Genet. A 155, 2071–2077. ( 10.1002/ajmg.a.34165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desir J, Cassart M, David P, Van Bogaert P, Abramowicz M. 2008. Primary microcephaly with ASPM mutation shows simplified cortical gyration with antero-posterior gradient pre- and post-natally. Am. J. Med. Genet. A 146A, 1439–1443. ( 10.1002/ajmg.a.32312) [DOI] [PubMed] [Google Scholar]
- 16.Lancaster MA, et al. 2013. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. ( 10.1038/nature12517) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuijpers M, Hoogenraad CC. 2011. Centrosomes, microtubules and neuronal development. Mol. Cell Neurosci. 48, 349–358. ( 10.1016/j.mcn.2011.05.004) [DOI] [PubMed] [Google Scholar]
- 18.Gilmore EC, Walsh CA. 2013. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2, 461–478. ( 10.1002/wdev.89) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genin A, et al. 2012. Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum. Mol. Genet. 21, 5306–5317. ( 10.1093/hmg/dds386) [DOI] [PubMed] [Google Scholar]
- 20.Kiyomitsu T, Obuse C, Yanagida M. 2007. Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell 13, 663–676. ( 10.1016/j.devcel.2007.09.005) [DOI] [PubMed] [Google Scholar]
- 21.Lin SY, Rai R, Li K, Xu ZX, Elledge SJ. 2005. BRIT1/MCPH1 is a DNA damage responsive protein that regulates the Brca1-Chk1 pathway, implicating checkpoint dysfunction in microcephaly. Proc. Natl Acad. Sci. USA 102, 15 105–15 109. ( 10.1073/pnas.0507722102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jackson AP, et al. 1998. Primary autosomal recessive microcephaly (MCPH1) maps to chromosome 8p22-pter. Am. J. Hum. Genet. 63, 541–546. ( 10.1086/301966) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouprina N, et al. 2005. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum. Mol. Genet. 14, 2155–2165. ( 10.1093/hmg/ddi220) [DOI] [PubMed] [Google Scholar]
- 24.Bogoyevitch MA, Yeap YY, Qu Z, Ngoei KR, Yip YY, Zhao TT, Heng JI, Ng DCH. 2012. WD40-repeat protein 62 is a JNK-phosphorylated spindle pole protein required for spindle maintenance and timely mitotic progression. J. Cell Sci. 125, 5096–5109. ( 10.1242/jcs.107326) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholas AK, et al. 2010. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat. Genet. 42, 1010–1014. ( 10.1038/ng.682) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bond J, et al. 2005. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet. 37, 353–355. ( 10.1038/ng1539) [DOI] [PubMed] [Google Scholar]
- 27.Kumar A, Girimaji SC, Duvvari MR, Blanton SH. 2009. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am. J. Hum. Genet. 84, 286–290. ( 10.1016/j.ajhg.2009.01.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussain MS, et al. 2012. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function. Am. J. Hum. Genet. 90, 871–878. ( 10.1016/j.ajhg.2012.03.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin YC, Chang CW, Hsu WB, Tang CJ, Lin YN, Chou EJ, Wu C-T, Tang TK. 2013. Human microcephaly protein CEP135 binds to hSAS-6 and CPAP, and is required for centriole assembly. EMBO J. 32, 1141–1154. ( 10.1038/emboj.2013.56) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cizmecioglu O, Arnold M, Bahtz R, Settele F, Ehret L, Haselmann-Weiss U, Antony C, Hoffmann I. 2010. Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J. Cell Biol. 191, 731–739. ( 10.1083/jcb.201007107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dzhindzhev NS, et al. 2010. Asterless is a scaffold for the onset of centriole assembly. Nature 467, 714–718. ( 10.1038/nature09445) [DOI] [PubMed] [Google Scholar]
- 32.Tang CJ, Lin SY, Hsu WB, Lin YN, Wu CT, Lin YC, Chang C-W, Wu K-S, Tang TK. 2011. The human microcephaly protein STIL interacts with CPAP and is required for procentriole formation. EMBO J. 30, 4790–4804. ( 10.1038/emboj.2011.378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cottee MA, et al. 2013. Crystal structures of the CPAP/STIL complex reveal its role in centriole assembly and human microcephaly. Elife 2, e01071 ( 10.7554/eLife.01071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hatzopoulos GN, Erat MC, Cutts E, Rogala KB, Slater LM, Stansfeld PJ, Vakonakis I. 2013. Structural analysis of the G-box domain of the microcephaly protein CPAP suggests a role in centriole architecture. Structure 21, 2069–2077. ( 10.1016/j.str.2013.08.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X, et al. 2014. Conserved TCP domain of Sas-4/CPAP is essential for pericentriolar material tethering during centrosome biogenesis. Proc. Natl Acad. Sci. USA 111, E354–E363. ( 10.1073/pnas.1317535111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arquint C, Nigg EA. 2014. STIL microcephaly mutations interfere with APC/C-mediated degradation and cause centriole amplification. Curr. Biol. 24, 351–360. ( 10.1016/j.cub.2013.12.016) [DOI] [PubMed] [Google Scholar]
- 37.Megraw TL, Sharkey JT, Nowakowski RS. 2011. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 21, 470–480. ( 10.1016/j.tcb.2011.04.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sir JH, et al. 2011. A primary microcephaly protein complex forms a ring around parental centrioles. Nat. Genet. 43, 1147–1153. ( 10.1038/ng.971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gopalakrishnan J, et al. 2011. Sas-4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat. Commun. 2, 359 ( 10.1038/ncomms1367) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conduit PT, Brunk K, Dobbelaere J, Dix CI, Lucas EP, Raff JW. 2010. Centrioles regulate centrosome size by controlling the rate of Cnn incorporation into the PCM. Curr. Biol. 20, 2178–2186. ( 10.1016/j.cub.2010.11.011) [DOI] [PubMed] [Google Scholar]
- 41.Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Dynlacht BD, Khodjakov A, Gönczy P. 2009. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 19, 1012–1018. ( 10.1016/j.cub.2009.05.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK. 2009. CPAP is a cell-cycle regulated protein that controls centriole length. Nat. Cell Biol. 11, 825–831. ( 10.1038/ncb1889) [DOI] [PubMed] [Google Scholar]
- 43.Schmidt TI, Kleylein-Sohn J, Westendorf J, Le Clech M, Lavoie SB, Stierhof YD, Nigg EA. 2009. Control of centriole length by CPAP and CP110. Curr. Biol. 19, 1005–1011. ( 10.1016/j.cub.2009.05.016) [DOI] [PubMed] [Google Scholar]
- 44.Loncarek J, Hergert P, Magidson V, Khodjakov A. 2008. Control of daughter centriole formation by the pericentriolar material. Nat. Cell Biol. 10, 322–328. ( 10.1038/ncb1694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takahashi T, Nowakowski RS, Caviness versus 1995. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J. Neurosci. 15, 6046–6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB. 2011. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat. Commun. 2, 154 ( 10.1038/ncomms1155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Calegari F, Haubensak W, Haffner C, Huttner WB. 2005. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J. Neurosci. 25, 6533–6538. ( 10.1523/JNEUROSCI.0778-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srsen V, Gnadt N, Dammermann A, Merdes A. 2006. Inhibition of centrosome protein assembly leads to p53-dependent exit from the cell cycle. J. Cell Biol. 174, 625–630. ( 10.1083/jcb.200606051) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. 2007. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 9, 160–170. ( 10.1038/ncb1529) [DOI] [PubMed] [Google Scholar]
- 50.Uetake Y, Loncarek J, Nordberg JJ, English CN, La Terra S, Khodjakov A, Sluder G. 2007. Cell cycle progression and de novo centriole assembly after centrosomal removal in untransformed human cells. J. Cell Biol. 176, 173–182. ( 10.1083/jcb.200607073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW. 2006. Flies without centrioles. Cell 125, 1375–1386. ( 10.1016/j.cell.2006.05.025) [DOI] [PubMed] [Google Scholar]
- 52.Yang Z, Loncarek J, Khodjakov A, Rieder CL. 2008. Extra centrosomes and/or chromosomes prolong mitosis in human cells. Nat. Cell Biol. 10, 748–751. ( 10.1038/ncb1738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sir JH, Putz M, Daly O, Morrison CG, Dunning M, Kilmartin JV, Gergely F. 2013. Loss of centrioles causes chromosomal instability in vertebrate somatic cells. J. Cell Biol. 203, 747–756. ( 10.1083/jcb.201309038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. 2008. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 22, 2189–2203. ( 10.1101/gad.1700908) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silkworth WT, Nardi IK, Scholl LM, Cimini D. 2009. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 4, e6564 ( 10.1371/journal.pone.0006564) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Novorol C, et al. 2013. Microcephaly models in the developing zebrafish retinal neuroepithelium point to an underlying defect in metaphase progression. Open Biol. 3, 130065 ( 10.1098/rsob.130065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lizarraga SB, et al. 2010. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137, 1907–1917. ( 10.1242/dev.040410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uetake Y, Sluder G. 2010. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr. Biol. 20, 1666–1671. ( 10.1016/j.cub.2010.08.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchman JJ, Durak O, Tsai LH. 2011. ASPM regulates Wnt signaling pathway activity in the developing brain. Genes Dev. 25, 1909–1914. ( 10.1101/gad.16830211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tibelius A, et al. 2009. Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. J. Cell Biol. 185, 1149–1157. ( 10.1083/jcb.200810159) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ganem NJ, Godinho SA, Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. ( 10.1038/nature08136) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silkworth WT, Nardi IK, Paul R, Mogilner A, Cimini D. 2012. Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 23, 401–411. ( 10.1091/mbc.E11-02-0095) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheltzer JM, Amon A. 2011. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet. 27, 446–453. ( 10.1016/j.tig.2011.07.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R. 2013. Centrosome amplification causes microcephaly. Nat. Cell Biol. 15, 731–740. ( 10.1038/ncb2746) [DOI] [PubMed] [Google Scholar]
- 65.Hanks S, et al. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat. Genet. 36, 1159–1161. ( 10.1038/ng1449) [DOI] [PubMed] [Google Scholar]
- 66.Kawame H, Sugio Y, Fuyama Y, Hayashi Y, Suzuki H, Kurosawa K, Maekawa K. 1999. Syndrome of microcephaly, Dandy-Walker malformation, and Wilms tumor caused by mosaic variegated aneuploidy with premature centromere division (PCD): report of a new case and review of the literature. J. Hum. Genet. 44, 219–224. ( 10.1007/s100380050147) [DOI] [PubMed] [Google Scholar]
- 67.Snape K, et al. 2011. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat. Genet. 43, 527–529. ( 10.1038/ng.822) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chenn A, Walsh CA. 2002. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369. ( 10.1126/science.1074192) [DOI] [PubMed] [Google Scholar]
- 69.Chae TH, Kim S, Marz KE, Hanson PI, Walsh CA. 2004. The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat. Genet. 36, 264–270. ( 10.1038/ng1302) [DOI] [PubMed] [Google Scholar]
- 70.Kotak S, Gonczy P. 2013. Mechanisms of spindle positioning: cortical force generators in the limelight. Curr. Opin. Cell Biol. 25, 741–748. ( 10.1016/j.ceb.2013.07.008) [DOI] [PubMed] [Google Scholar]
- 71.Lesage B, Gutierrez I, Marti E, Gonzalez C. 2010. Neural stem cells: the need for a proper orientation. Curr. Opin. Genet. Dev. 20, 438–442. ( 10.1016/j.gde.2010.04.013) [DOI] [PubMed] [Google Scholar]
- 72.Barr AR, Kilmartin JV, Gergely F. 2010. CDK5RAP2 functions in centrosome to spindle pole attachment and DNA damage response. J. Cell Biol. 189, 23–39. ( 10.1083/jcb.200912163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. 2006. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl Acad. Sci. USA 103, 10 438–10 443. ( 10.1073/pnas.0604066103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitagawa D, Kohlmaier G, Keller D, Strnad P, Balestra FR, Fluckiger I, Gonczy P. 2011. Spindle positioning in human cells relies on proper centriole formation and on the microcephaly proteins CPAP and STIL. J. Cell Sci. 124, 3884–3893. ( 10.1242/jcs.089888) [DOI] [PubMed] [Google Scholar]
- 75.Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ. 2011. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway. Nat. Cell Biol. 13, 1325–1334. ( 10.1038/ncb2342) [DOI] [PubMed] [Google Scholar]
- 76.Paramasivam M, Chang YJ, LoTurco JJ. 2007. ASPM and citron kinase co-localize to the midbody ring during cytokinesis. Cell Cycle 6, 1605–1612. ( 10.4161/cc.6.13.4356) [DOI] [PubMed] [Google Scholar]
- 77.Higgins J, et al. 2010. Human ASPM participates in spindle organisation, spindle orientation and cytokinesis. BMC Cell Biol. 11, 85 ( 10.1186/1471-2121-11-85) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pulvers JN, et al. 2010. Mutations in mouse ASPM (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc. Natl Acad. Sci. USA 107, 16 595–16 600. ( 10.1073/pnas.1010494107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spalding KL, et al. 2013. Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. ( 10.1016/j.cell.2013.05.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Anderson CT, Stearns T. 2009. Centriole age underlies asynchronous primary cilium growth in mammalian cells. Curr. Biol. 19, 1498–1502. ( 10.1016/j.cub.2009.07.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paridaen JT, Wilsch-Brauninger M, Huttner WB. 2013. Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 155, 333–344. ( 10.1016/j.cell.2013.08.060) [DOI] [PubMed] [Google Scholar]
- 82.Wallingford JB, Mitchell B. 2011. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 25, 201–213. ( 10.1101/gad.2008011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tasouri E, Tucker KL. 2011. Primary cilia and organogenesis: is Hedgehog the only sculptor? Cell Tissue Res. 345, 21–40. ( 10.1007/s00441-011-1192-8) [DOI] [PubMed] [Google Scholar]
- 84.Kosodo Y. 2012. Interkinetic nuclear migration: beyond a hallmark of neurogenesis. Cell. Mol. Life Sci. 69, 2727–2738. ( 10.1007/s00018-012-0952-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Del Bene F, Wehman AM, Link BA, Baier H. 2008. Regulation of neurogenesis by interkinetic nuclear migration through an apical-basal notch gradient. Cell 134, 1055–1065. ( 10.1016/j.cell.2008.07.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rujano MA, Sanchez-Pulido L, Pennetier C, le Dez G, Basto R. 2013. The microcephaly protein ASP regulates neuroepithelium morphogenesis by controlling the spatial distribution of myosin II. Nat. Cell Biol. 15, 1294–1306. ( 10.1038/ncb2858) [DOI] [PubMed] [Google Scholar]
- 87.Klingseisen A, Jackson AP. 2011. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 25, 2011–2024. ( 10.1101/gad.169037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ogi T, et al. 2012. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel syndrome. PLoS Genet. 8, e1002945 ( 10.1371/journal.pgen.1002945) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qvist P, Huertas P, Jimeno S, Nyegaard M, Hassan MJ, Jackson SP, Børglum AD. 2011. CtIP mutations cause Seckel and Jawad syndromes. PLoS Genet. 7, e1002310 ( 10.1371/journal.pgen.1002310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bicknell LS, et al. 2011. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat. Genet. 43, 350–355. ( 10.1038/ng.776) [DOI] [PubMed] [Google Scholar]
- 91.Hossain M, Stillman B. 2012. Meier-Gorlin syndrome mutations disrupt an Orc1 CDK inhibitory domain and cause centrosome reduplication. Genes Dev. 26, 1797–1810. ( 10.1101/gad.197178.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guernsey DL, et al. 2011. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. 43, 360–364. ( 10.1038/ng.777) [DOI] [PubMed] [Google Scholar]
- 93.Griffith E, et al. 2008. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 40, 232–236. ( 10.1038/ng.2007.80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rauch A, et al. 2008. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 319, 816–819. ( 10.1126/science.1151174) [DOI] [PubMed] [Google Scholar]
- 95.O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501. ( 10.1038/ng1129) [DOI] [PubMed] [Google Scholar]
- 96.Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O'Driscoll M. 2004. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 13, 3127–3138. ( 10.1093/hmg/ddh335) [DOI] [PubMed] [Google Scholar]
- 97.Ruzankina Y, et al. 2007. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126. ( 10.1016/j.stem.2007.03.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matsuyama M, Goto H, Kasahara K, Kawakami Y, Nakanishi M, Kiyono T, Goshima N, Inagaki M. 2011. Nuclear Chk1 prevents premature mitotic entry. J. Cell Sci. 124, 2113–2119. ( 10.1242/jcs.086488) [DOI] [PubMed] [Google Scholar]
- 99.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J. 2004. Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6, 884–891. ( 10.1038/ncb1165) [DOI] [PubMed] [Google Scholar]
- 100.Kalay E, et al. 2011. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat. Genet. 43, 23–26. ( 10.1038/ng.725) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McIntyre RE, et al. 2012. Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS Genet. 8, e1003022 ( 10.1371/journal.pgen.1003022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. 2010. Novel CENPJ mutation causes Seckel syndrome. J. Med. Genet. 47, 411–414. ( 10.1136/jmg.2009.076646) [DOI] [PubMed] [Google Scholar]
- 103.Stevens NR, Roque H, Raff JW. 2010. DSas-6 and Ana2 coassemble into tubules to promote centriole duplication and engagement. Dev. Cell 19, 913–919. ( 10.1016/j.devcel.2010.11.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Loncarek J, Hergert P, Khodjakov A. 2010. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr. Biol. 20, 1277–1282. ( 10.1016/j.cub.2010.05.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. 2005. Spindle multipolarity is prevented by centrosomal clustering. Science 307, 127–129. ( 10.1126/science.1104905) [DOI] [PubMed] [Google Scholar]
- 106.Gergely F, Basto R. 2008. Multiple centrosomes: together they stand, divided they fall. Genes Dev. 22, 2291–2296. ( 10.1101/gad.1715208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stiff T, Alagoz M, Alcantara D, Outwin E, Brunner HG, Bongers EM, O'Driscoll M, Jeggo PA, Dutcher SK. 2013. Deficiency in origin licensing proteins impairs cilia formation: implications for the aetiology of Meier-Gorlin syndrome. PLoS Genet. 9, e1003360 ( 10.1371/journal.pgen.1003360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu KS, Tang TK. 2012. CPAP is required for cilia formation in neuronal cells. Biol. Open 1, 559–565. ( 10.1242/bio.20121388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blachon S, Gopalakrishnan J, Omori Y, Polyanovsky A, Church A, Nicastro D, Malicki J, Avidor-Reiss T. 2008. Drosophila asterless and vertebrate Cep152. Are orthologs essential for centriole duplication. Genetics 180, 2081–2094. ( 10.1534/genetics.108.095141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA. 2007. Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 179, 321–330. ( 10.1083/jcb.200707181) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, Kuehn MR. 1999. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature 399, 691–694. ( 10.1038/21429) [DOI] [PubMed] [Google Scholar]
- 112.Carvalho-Santos Z, et al. 2012. BLD10/CEP135 is a microtubule-associated protein that controls the formation of the flagellum central microtubule pair. Dev. Cell 23, 412–424. ( 10.1016/j.devcel.2012.06.001) [DOI] [PubMed] [Google Scholar]
- 113.Waters AM, Beales PL. 2011. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 26, 1039–1056. ( 10.1007/s00467-010-1731-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chaki M, et al. 2012. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 150, 533–548. ( 10.1016/j.cell.2012.06.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pampliega O, et al. 2013. Functional interaction between autophagy and ciliogenesis. Nature 502, 194–200. ( 10.1038/nature12639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tobin JL, Beales PL. 2009. The nonmotile ciliopathies. Genet. Med. 11, 386–402. ( 10.1097/GIM.0b013e3181a02882) [DOI] [PubMed] [Google Scholar]
- 117.Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. 2011. Centrosomes and cilia in human disease. Trends Genet. 27, 307–315. ( 10.1016/j.tig.2011.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dammermann A, Merdes A. 2002. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 159, 255–266. ( 10.1083/jcb.200204023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lopes CA, Prosser SL, Romio L, Hirst RA, O'Callaghan C, Woolf AS, Fry AM. 2011. Centriolar satellites are assembly points for proteins implicated in human ciliopathies, including oral-facial-digital syndrome 1. J. Cell Sci. 124, 600–612. ( 10.1242/jcs.077156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Villumsen BH, et al. 2013. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 32, 3029–3040. ( 10.1038/emboj.2013.223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. 2013. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502, 254–257. ( 10.1038/nature12606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stowe TR, Wilkinson CJ, Iqbal A, Stearns T. 2012. The centriolar satellite proteins Cep72 and Cep290 interact and are required for recruitment of BBS proteins to the cilium. Mol. Biol. Cell 23, 3322–3335. ( 10.1091/mbc.E12-02-0134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim J, Krishnaswami SR, Gleeson JG. 2008. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum. Mol. Genet. 17, 3796–3805. ( 10.1093/hmg/ddn277) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nachury MV, et al. 2007. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. ( 10.1016/j.cell.2007.03.053) [DOI] [PubMed] [Google Scholar]
- 125.Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. 2010. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J. Cell Biol. 190, 927–940. ( 10.1083/jcb.201006105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. 2010. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 18, 410–424. ( 10.1016/j.devcel.2009.12.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Reiter JF. 2008. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 10, 70–76. ( 10.1038/ncb1670) [DOI] [PubMed] [Google Scholar]
- 128.Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou M-FB. 2013. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 27, 163–168. ( 10.1101/gad.207043.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ferrante MI, Romio L, Castro S, Collins JE, Goulding DA, Stemple DL, Woolf AS, Wilson SW. 2009. Convergent extension movements and ciliary function are mediated by ofd1, a zebrafish orthologue of the human oral-facial-digital type 1 syndrome gene. Hum. Mol. Genet. 18, 289–303. ( 10.1093/hmg/ddn356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tayeh MK, Yen HJ, Beck JS, Searby CC, Westfall TA, Griesbach H, Sheffield VC, Slusarski DC. 2008. Genetic interaction between Bardet–Biedl syndrome genes and implications for limb patterning. Hum. Mol. Genet. 17, 1956–1967. ( 10.1093/hmg/ddn093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baye LM, Patrinostro X, Swaminathan S, Beck JS, Zhang Y, Stone EM, Sheffield VC, Slusarski DC. 2011. The N-terminal region of centrosomal protein 290 (CEP290) restores vision in a zebrafish model of human blindness. Hum. Mol. Genet. 20, 1467–1477. ( 10.1093/hmg/ddr025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dollé P, Franco B. 2006. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 38, 112–117. ( 10.1038/ng1684) [DOI] [PubMed] [Google Scholar]
- 133.D'Angelo A, De Angelis A, Avallone B, Piscopo I, Tammaro R, Studer M, Franco B, Zhang X. 2012. Ofd1 controls dorso-ventral patterning and axoneme elongation during embryonic brain development. PLoS ONE 7, e52937 ( 10.1371/journal.pone.0052937) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Macca M, Franco B. 2009. The molecular basis of oral-facial-digital syndrome, type 1. Am. J. Med. Genet. C Semin. Med. Genet. 151C, 318–325. ( 10.1002/ajmg.c.30224) [DOI] [PubMed] [Google Scholar]
- 135.Coene KL, et al. 2009. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 85, 465–481. ( 10.1016/j.ajhg.2009.09.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kulaga HM, et al. 2004. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 36, 994–998. ( 10.1038/ng1418) [DOI] [PubMed] [Google Scholar]
- 137.Chang B, et al. 2006. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 15, 1847–1857. ( 10.1093/hmg/ddl107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McEwen DP, Koenekoop RK, Khanna H, Jenkins PM, Lopez I, Swaroop A, Martens JR. 2007. Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons. Proc. Natl Acad. Sci. USA 104, 15 917–15 922. ( 10.1073/pnas.0704140104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lancaster MA, et al. 2011. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med. 17, 726–731. ( 10.1038/nm.2380) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Forsythe E, Beales PL. 2013. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 21, 8–13. ( 10.1038/ejhg.2012.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Karmous-Benailly H, et al. 2005. Antenatal presentation of Bardet–Biedl syndrome may mimic Meckel syndrome. Am. J. Hum. Genet. 76, 493–504. ( 10.1086/428679) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Coppieters F, Lefever S, Leroy BP, De Baere E. 2010. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum. Mutat. 31, 1097–1108. ( 10.1002/humu.21337) [DOI] [PubMed] [Google Scholar]
- 143.Tsang WY, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, Dynlacht BD. 2008. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 15, 187–197. ( 10.1016/j.devcel.2008.07.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guernsey DL, et al. 2010. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am. J. Hum. Genet. 87, 40–51. ( 10.1016/j.ajhg.2010.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mokrani-Benhelli H, et al. 2013. Primary microcephaly, impaired DNA replication, and genomic instability caused by compound heterozygous ATR mutations. Hum. Mutat. 34, 374–384. ( 10.1002/humu.22245) [DOI] [PubMed] [Google Scholar]
- 146.Lukinavicius G, Lavogina D, Orpinell M, Umezawa K, Reymond L, Garin N, Gönczy P, Johnsson K. 2013. Selective chemical crosslinking reveals a Cep57-Cep63-Cep152 centrosomal complex. Curr. Biol. 23, 265–270. ( 10.1016/j.cub.2012.12.030) [DOI] [PubMed] [Google Scholar]
- 147.McKinnon PJ. 2013. Maintaining genome stability in the nervous system. Nat. Neurosci. 16, 1523–1529. ( 10.1038/nn.3537) [DOI] [PMC free article] [PubMed] [Google Scholar]