

Abstract
Hippocampal sclerosis of aging (HS-Aging) is a high-morbidity brain disease in the elderly but risk factors are largely unknown. We report the first genome-wide association study (GWAS) with HS-Aging pathology as an endophenotype. In collaboration with the Alzheimer’s Disease Genetics Consortium, data were analyzed from large autopsy cohorts: (#1) National Alzheimer’s Coordinating Center (NACC); (#2) Rush University Religious Orders Study and Memory and Aging Project; (#3) Group Health Research Institute Adult Changes in Thought study; (#4) University of California at Irvine 90+ Study; and (#5) University of Kentucky Alzheimer’s Disease Center. Altogether, 363 HS-Aging cases and 2,303 controls, all pathologically confirmed, provided statistical power to test for risk alleles with large effect size. A two-tier study design included GWAS from cohorts #1–3 (Stage I) to identify promising SNP candidates, followed by focused evaluation of particular SNPs in cohorts #4–5 (Stage II). Polymorphism in the ATP-binding cassette, sub-family C member 9 (ABCC9) gene, also known as sulfonylurea receptor 2, was associated with HS-Aging pathology. In the meta-analyzed Stage I GWAS, ABCC9 polymorphisms yielded the lowest p values, and factoring in the Stage II results, the meta-analyzed risk SNP (rs704178:G) attained genome-wide statistical significance (p = 1.4 × 10−9), with odds ratio (OR) of 2.13 (recessive mode of inheritance). For SNPs previously linked to hippocampal sclerosis, meta-analyses of Stage I results show OR = 1.16 for rs5848 (GRN) and OR = 1.22 rs1990622 (TMEM106B), with the risk alleles as previously described. Sulfonylureas, a widely prescribed drug class used to treat diabetes, also modify human ABCC9 protein function. A subsample of patients from the NACC database (n = 624) were identified who were older than age 85 at death with known drug history. Controlling for important confounders such as diabetes itself, exposure to a sulfonylurea drug was associated with risk for HS-Aging pathology (p = 0.03). Thus, we describe a novel and targetable dementia risk factor.
Keywords: Oldest old, Neuropathology, KATP, CTAGE5, ADGC, Potassium channel
Introduction
In elderly individuals, hippocampal sclerosis of aging (HS-Aging) is a relatively common brain disease [41, 50]; for recent reviews, see Refs. [51, 80]. HS-Aging is defined by pathological manifestations: neuron loss, gliosis, and atrophy in the hippocampal formation, not deemed attributable to Alzheimer disease (AD)-type plaques and tangles [1, 44, 51]. The presence of HS-Aging pathology is associated with substantial cognitive impairment, independent of comorbid pathologies [48], yet this disease is generally misdiagnosed as AD in the clinical setting [9, 57].
Genetic risk factors of HS-Aging are not fully characterized. Hippocampal sclerosis pathology in AD cases has been linked to single nucleotide polymorphisms (SNPs) at genomic loci previously associated with frontotemporal lobar degeneration (FTLD), namely rs5848 (GRN) and rs1990622 (near TMEM106B) [17, 63, 76]. In contrast to AD, the risk of developing HS-Aging pathology is not associated with apolipoprotein E (APOE) alleles [9, 41, 50, 72].
There has been no prior published genome-wide association study (GWAS) focused on HS-Aging pathology. The endophenotype is deceptively complex [51, 80] and confers both potential obstacles and opportunities for a GWAS. Challenges include the requirement for reliable autopsy data on every research subject and the advanced old age of many patients with HS-Aging [50, 79]. Even archival neuropathology data must be interpreted with caution because HS-Aging has only recently been the subject of widespread attention by researchers [9, 51, 57, 62]. After accounting for these difficulties, there are characteristics of HS-Aging pathology that are conducive for GWAS. Most important is the high disease prevalence which enables analyses of diverse study cohorts including population-based samples [41, 51, 80]. HS-Aging pathology tends to co-occur with hippocampal TAR DNA-binding protein 43 (TDP-43) pathology [2, 36, 54], an important component of the pathological endophenotype [51]. Thus, a successful HS-Aging GWAS study would detect allele(s) associated with hippocampal sclerosis and TDP-43 pathologies across multiple cohorts of autopsied elderly individuals.
For the current study, genomic and pathologic data from dozens of different research centers were analyzed with the goal of identifying alleles associated with risk for HS-Aging pathology. Altogether these analyses incorporated 363 HS-Aging cases and 2,303 controls with neuropathologic evaluation. A two-tiered study design employed a GWAS to identify promising risk allele candidates, with subsequent evaluation of specific candidate alleles using separate cohorts. Our main findings are (1) a polymorphism in the ATP-binding cassette, sub-family C member 9 (ABCC9) gene, also known as sulfonylurea receptor 2 (SUR2), is associated with HS-Aging pathology, and (2) exposure to sulfonylurea drugs is associated with increased risk for HS-Aging pathology among individuals who died age 85 and older.
Materials and methods
Datasets
The Alzheimer’s Disease Genetics Consortium (ADGC) accrued genomics data from 34 different research centers (US National Institute on Aging Alzheimer’s Disease Centers, or ADCs) with multiple iterations of GWAS data (References [29, 39, 47] and see below), which were analyzed with neuropathological and clinical data obtained through the National Alzheimer’s Coordinating Center (NACC; see Reference [4]). Research using NACC data was approved by the University of Washington Human Subjects Division; other protocols were supervised by local Institutional Review Boards. Neuropathologic evaluations were performed according to center-specific protocols–including whether neuropathologists studied left, right, or bilateral hippocampi [9]—and entered into a database in standardized format. Details of HS-Aging case/control operationalization have been discussed before [9] and are described in detail in Supplemental Methods. University of Kentucky submits data to NACC, but data from that center were excluded from the Stage I cohorts. In addition to the ADGC/NACC group, autopsy series included in the Stage I phase were the Rush University Religious Orders Study and Memory and Aging Project (ROS-MAP) with recruitment in Chicago, IL, USA [7], and the Adult Changes in Thought (ACT) study from the Group Health Cooperative and University of Washington with recruitment in Seattle, WA, USA [45].
Following the GWAS on Stage I datasets, additional studies were performed analyzing data from two separate cohorts (Stage II): the University of Kentucky Alzheimer’s Disease Center (UK-ADC; included in this cohort were research subjects (n = 49) from the Georgia Centenarian Study [59] that were evaluated neuropathologically at the University of Kentucky [50]), and the University of California at Irvine 90+ Study (UCI90+). UCI90+ is a population-based longitudinal study of individuals aged 90 and older, with recruitment in Orange County, CA, USA; there is no overlap with the ADGC/NACC data. Descriptions of these autopsy cohorts were published previously [3, 15, 50, 65]. Immunohistochemical stains on hippocampal formation were performed using phosphorylated TDP-43 (P-TDP43) antibody 1D3 [53] as described previously [51]. The reason for using P-TDP43 immunostaining-based endophenotype for UK-ADC and not UCI90+ relates to a lack of analogous P-TDP43 immunohistochemical data.
For assessment of the prevalence of HS-Aging pathology in a population-based cohort, relative to other neuropathologically defined diseases, the Nun Study data [46, 74] were analyzed (n = 436 research subjects). These data were separate from the genomics analyses. Nun study neuropathologic methods were performed as previously described [50]. For the present study, neuropathologic observations were graded independently of each other: HS-Aging pathology [44, 51], AD pathology [stratified by Braak stages [8] using both a stringent criterion (Stages V/VI) and another that also incorporates milder cases (Stages IV–VI)], neocortical Lewy body pathology of a severity to indicate “high likelihood” that “the pathologic findings are associated with a DLB clinical syndrome” [43], and FTLD [42].
Statistical methods
In silico studies were performed for feasibility and power analyses. With the sample size of the Stage I cohorts (241 cases, 1,998 controls), the probability of determining a “true-positive” SNP associated with HS-Aging pathology, i.e. statistical power, was calculated. QUANTO software v.1.2.4 [25] was used to model the impact on statistical power of varying minor allele frequency (MAF) and the effect size of the risk allele. That effect size is operationalized by the odds ratio (OR) of HS-Aging pathology in risk SNP carriers versus non-carriers. Analyses were performed for an experiment that would meet criteria for genome-wide significance, pre-specified at p < 5 × 10−8 [11, 39]. This threshold corrects for multiple comparisons while also assuming that many SNPs are in linkage disequilibrium with each other and thus are not truly independent tests [20, 28]. A second model was performed relaxing the criteria for genome-wide statistical significance to p < 10−5 to reflect the requirement of a risk allele candidate that could be tested separately in Stage II cohorts.
Stage I (GWAS) and Stage II (focused on individual SNP) studies
GWAS data access was coordinated through ADGC, providing quality-controlled genotypes using imputation to the Phase I v.3 1000 Genomes Project March 2012 release with imputation as previously described [39]. Additional quality control (QC) was applied within each Stage I dataset. To reduce the potential biases associated with population stratification, principal components (PCs) were iteratively calculated with normalized SNP data, and subjects with outlying values were removed after each of six iterations using SVS™ software. An additive mode of inheritance (MOI) was assumed in each cohort from the Stage I stage to balance statistical power with concerns of multiple testing, and logistic regression was performed for each SNP with correction for PCs. The number of PCs chosen for adjustment was based on examination of scree plots (Supplemental Fig. 1) for each Stage I cohort: four PCs for NACC and ROS-MAP; three PCs for ACT. For assessment of rs5848 and rs1990622 SNPs, for which prior published studies derived from Mayo Clinic data [57, 61, 63], a dataset lacking that institution’s participants was used for analyses of those alleles. Results across the three Stage I datasets were meta-analyzed using an inverse-variance-based statistic [70]. LocusZoom plots [60] reflect regional meta-analyzed p values from all the Stage I datasets.
In the Stage II cohorts, SNPs were characterized using Life Technologies’ TaqMan-based SNP assays providing direct assessment of genotype. All tests for genotype/HS-Aging association were two-tailed. Meta-analyses were performed for the two Stage II cohorts, and then data were combined across all Stage I and Stage II cohorts, using an inverse-variance-based statistic as above. Forest plots displaying cohort-specific and aggregated ORs and confidence intervals were created reflecting these meta-analyzed results using R v.3.0.2 programming language [71].
Genomic ABCC9 sequencing from HS-Aging cases and controls
Gene-specific genomic sequencing was performed on DNA from six individuals with pathologic and genomic information (three rs704178 G/G HS-Aging cases, three elderly rs704178 C/C controls). Data pertaining to all six individuals are provided in Supplemental Methods, as are details of the sequencing protocol. Briefly, ABCC9 sequences were captured using the HaloPlex Target Enrichment System (Agilent Technologies) to amplify fragments spanning the human genomic region chr12:21950273-22094386 (GRCh37/hg19 genomic assembly). The six resulting fragment libraries were run in a single flow cell on a HiSeq 2500 (Illumina). The variant files were inspected both manually and with custom Perl scripts.
Western blots on hippocampal proteins from HS-Aging cases and controls
Sequential biochemical fractionation and immunoblot studies employed techniques modified from previous protocols [51, 54, 64] as described in detail in Supplemental Methods. Briefly, hippocampal tissue (CA1 and subiculum) was dissected from a convenience sample of postmortem brain specimens (n = 9) including subjects with or without the ABCC9 polymorphism, and with or without HS-Aging pathology, which were extracted into separate low salt-, 1 % Triton-X100-, 1 % Sarkosyl+heat-, and 7M urea-extractable fractions. Equal amounts of protein were loaded to a denaturing SDS polyacrylamide gel prior to immunoblot analyses. Antibodies used for immunoblots were as follows: anti-SUR2B (C-15): sc-5793; anti-SUR2A (T-19): sc-32461; and anti-SUR2 (H-80): sc-25684, all three from Santa Cruz Biotechnology, and anti-SUR2B (N323A/31) and anti-SUR2A (N319A/14) from EMD Millipore. Control immunoblots using the same samples were performed using anti-PGRN/GRN (R&D Cat # AF 2420) and anti-β-Actin (Rockland Code 600-401-886) antibodies; see Supplemental Methods.
NACC neuropathology and drug data
The initial data source for these analyses was research volunteers evaluated after 2009 at an ADC with both reported clinical (including drug information) and autopsy data. Participants were excluded if they were younger than 85 at death to best match all participants to HS-Aging age range. Methods and rationale for the UDS clinical examination have been previously published [4, 5]. All participants received an initial in-person clinical evaluation and up to seven follow-up evaluations; data were collected on an approximately annual basis. In this analysis, HS-Aging pathology was defined as present if the neuropathologist recorded a “primary or contributing pathologic diagnosis of hippocampal sclerosis”. Assessment of TDP-43 pathology was not collected on the NACC Neuropathology form, but allowed as a write-in; for individuals without FTLD, where some sort of medial temporal lobe TDP-43 pathology was noted, these cases were considered to be HS-Aging (n = 4).
For statistical analyses of the NACC data, longitudinal self-reported medication histories were reviewed for the 624 included subjects. Medication use was determined by asking subjects to provide a list of all medications taken within the last 2 weeks prior to their UDS assessment. Individuals who reported taking any sulfonylurea at any time during follow-up (September 2005 through April 2013) were coded as positive for use. Crude and adjusted ORs for the presence of HS-Aging at autopsy were calculated separately. Adjusted ORs were obtained via logistic regression, controlling for age at death (centered at 90) and year of death (centered at 2011). Possible group differences in mean age at death and interval between last evaluation and death were assessed using linear regression. Sensitivity analyses were conducted restricting the medication history to the last UDS assessment. Analyses were conducted using SAS/STAT 9.3®. Two-tailed tests were used to evaluate the hypothesis that use of sulfonylureas promotes HS-Aging pathology (α = 0.05).
Results
Endophenotype for GWAS and subsequent studies
The endophenotype used for correlation with genomic information was HS-Aging pathology [9, 51]. Photomicrographs (Fig. 1) demonstrate features of HS-Aging pathology discernible after staining human hippocampal sections with hematoxylin and eosin (H&E) and using P-TDP-43 immunohistochemistry. A three-tier (0–2) semiquantitative scale of P-TDP-43 immunoreactive pathology was used in the UK-ADC dataset. Data about the prevalence of HS-Aging pathology, from the Nun Study, are shown in Fig. 2. Nun Study research volunteers were followed as a population-based cohort, mostly from normal status, and with high autopsy rate [46, 74]; this group lacked some of the recruitment biases of dementia clinic cohorts [6, 35, 46, 50]. Even when younger Nun Study research volunteers were included from the sample studied neuropathologically at University of Kentucky (total n = 526), no FTLD-TDP cases were observed (data not shown). By contrast, HS-Aging pathology is relatively prevalent in population-based cohorts, which are key sources of data in the current study (ROS-MAP, ACT, and UCI90+ studies).
Fig. 1.
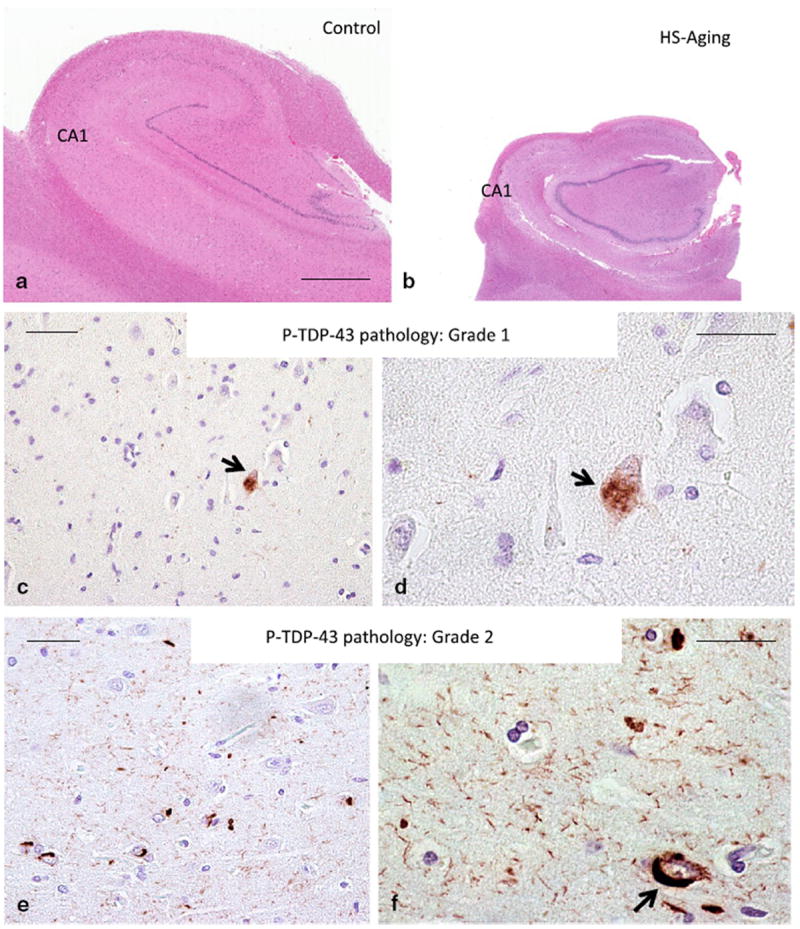
Hippocampal sclerosis of aging (HS-Aging) pathology is a complex endophenotype for genomic studies. Pathology defines the disease, illustrated in photomicrographs of hematoxylin and eosinstained sections to compare an aged control hippocampus (a) with a shrunken HS-Aging hippocampus (b); same scale bar 2 mm, CA1 subfield indicated. For grading P-TDP43 pathology in CA1 and subiculum of the hippocampal formation, a three-tier semiquantitative system was employed with “Grade 0” indicating no P-TDP43 pathology, “Grade 1” (c, d) indicating very sparse isolated pathology (P-TDP43 positive neuronal inclusion at arrow) and “Grade 2” (e, f) indicating any more severe pathology, usually with intranuclear, intracytoplasmic (arrow), and neuritic P-TDP-43 pathology. Scale bar 60 microns (c, e) and 25 microns (d, f)
Fig. 2.
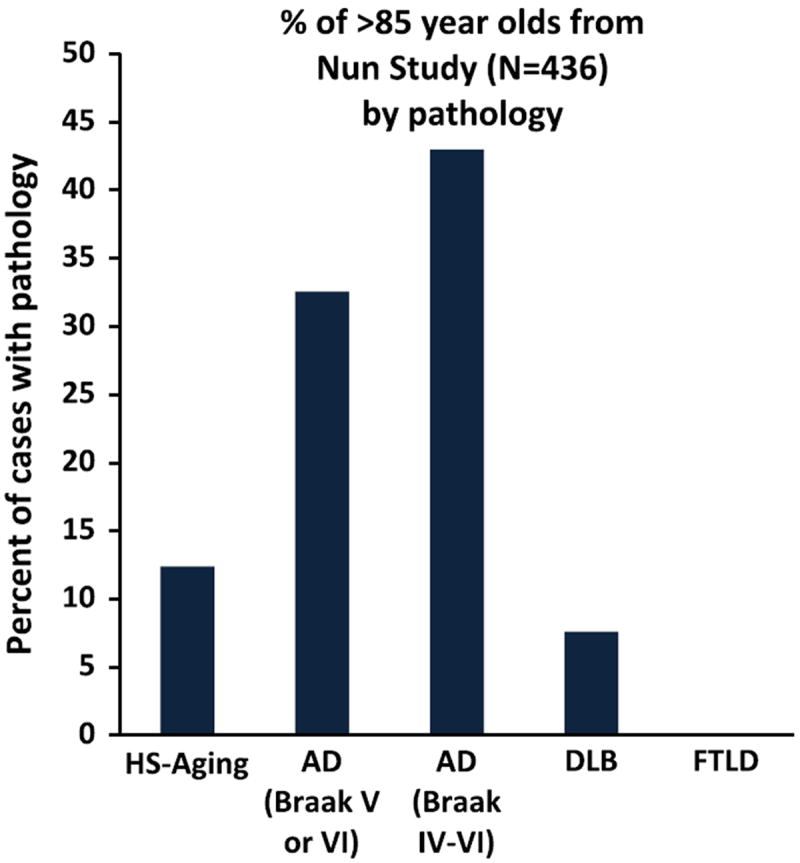
Prevalence of brain pathology subtypes in the Nun Study. The prevalence of HS-Aging pathology is germane to a study that incorporates different autopsy cohort types. Here, we show data about the prevalence of brain pathology subtypes in the Nun Study, a population-based cohort as previously described in detail [50]. As in most community-based or population-based cohorts (as opposed to cohorts connected with memory disorder clinics), frontotemporal lobar degeneration (FTLD) pathology is rare [19, 69, 73]
Statistical and feasibility considerations
GWAS in Stage I cohorts was used to identify risk SNP candidates, followed by separate analyses of individual SNPs in Stage II cohorts (Fig. 3). This study design involved three key assumptions:
Assumption #1 (central hypothesis being tested in the study)> As there has been no prior published HS-Aging GWAS, polymorphisms remain to be described with strongly increased odds of HS-Aging pathology in risk allele carriers versus non-carriers, i.e. OR >1.5 for the risk allele;
Assumption #2> The Stage I cohort GWAS comprises adequate sample sizes to yield a limited number of credible candidate risk SNPs for evaluation in the Stage II cohorts; and
Assumption #3> The Stage II cohorts have sufficient numbers of HS-Aging cases and controls to adequately test for an HS-Aging risk SNP candidate.
Fig. 3.
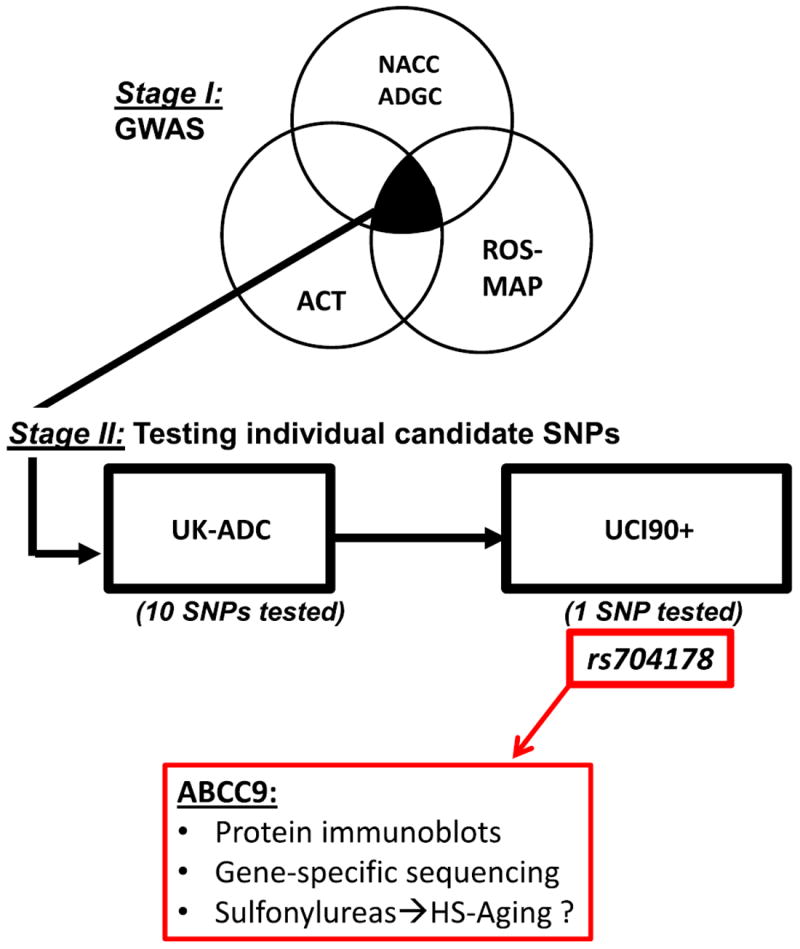
Study design for elucidating a large effect size human genomic polymorphism that is associated with HS-Aging pathology. The GWAS (Stage I) incorporated genomic data from Alzheimer’s Disease Genetics Consortium (ADGC) and pathologic data from National Alzheimer’s Coordinating Center (NACC) derived from 34 different US Alzheimer’s Disease Centers; Rush University Religious Orders Study and Memory and Aging Project (ROS-MAP); and the Group Health Research Institute Adult Changes in Thought study (ACT). Subsequent evaluation of particular risk alleles was then performed (Stage II) using data from two additional cohorts: University of Kentucky ADC (UK-ADC), and University of California at Irvine 90+ Study (UCI90+). Post-hoc studies were performed on the ABCC9 allele (rs704178) showing the most robust association with HS-Aging pathology
Power analyses were performed to test whether Assumptions #2 and #3 were valid if the large effect size SNP (Assumption #1) exists. The effect size strongly influences statistical considerations (Fig. 4). Given sample sizes, the likelihood of uncovering an HS-Aging risk allele with genome-wide statistical significance (p < 5 × 10−8) in the Stage I GWAS was only modest, unless the effect size was quite large (Fig. 4a). However, the Stage I cohort was adequately powered for the purposes of identifying risk SNP candidates for downstream evaluation. In the presence of a strong HS-Aging risk allele (OR >1.5), there was a high chance that the risk allele would be identified in the Stage I cohort GWAS with p < 10−5 (Fig. 4b). Definitive testing of the candidate risk SNPs would require downstream validation in independent cohorts. Stage II would be most effective in the case of a SNP with high MAF and with studies of individual SNPs performed serially so that the final cohort’s analyses require no adjustment for multiple comparisons.
Fig. 4.
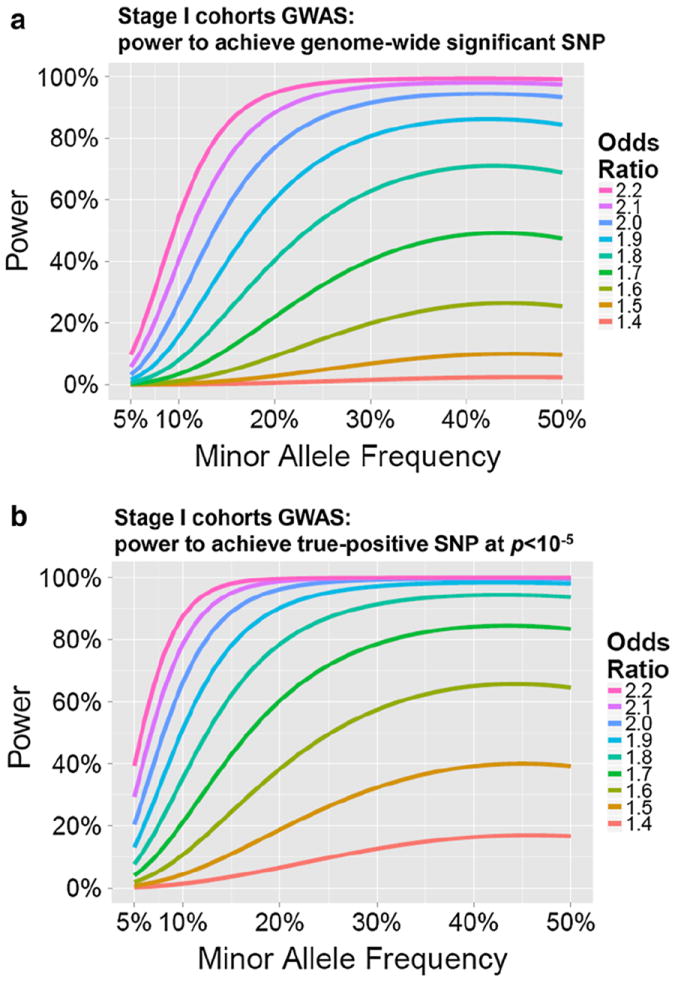
Statistical power analyses. With the sample size of the Stage I cohorts (241 cases, 1,998 controls), the probability of elucidating a “true-positive” SNP associated with HS-Aging pathology was modeled. Two different genome-wide significance thresholds were modeled, each with varying minor allele frequency and risk allele effect sizes operationalized by the odds ratio of HS-Aging pathology in risk SNP carriers versus non-carriers. To reflect the study design, the p values were set at p < 5 × 10−8 (a) and p < 10−5 (b)
GWAS
As a positive control, a GWAS from AD pathology was performed initially on a sample from the ADGC cohort (1,443 AD cases and 99 controls) and as expected, only SNPs near the APOE gene reached genome-wide significance (p = 4.2 × 10−13; Supplemental Tables 1 and 2; Supplemental Fig. 2). We then performed a HS-Aging GWAS using data from the three Stage I datasets: ADGC/NACC, ROS-MAP, and ACT (Table 1 and Supplemental Table 3). The number of nominal SNPs that passed QC and were included in the GWAS was ~4.9 million (Table 2). HS-Aging cases and controls were classified according to pathology irrespective of AD or other prevalent pathologies; see Supplemental Methods for details. Manhattan plot and Q–Q plot for the HS-Aging GWAS are shown in Fig. 5; no Stage II cohort data were factored into these analyses.
Table 1.
Research subjects (n = 2,666 research subjects total) and the five autopsy cohorts analyzed in the current study
| Autopsy cohort | Pathologic phenotype evaluated | HS cases | Avg age (years ± st dev) | Non-HS controls | Avg age (years ± st dev) | Total | Role in study |
|---|---|---|---|---|---|---|---|
| ADGC | HS pathology | 186 | 84.1 ± 8.2 | 1,450 | 81.5 ± 8.7 | 1,636 | Stage I (GWAS) |
| ROS-MAP | HS pathology | 29 | 93.3 ± 5.4 | 398 | 90.8 ± 4.1 | 427 | |
| ACT | HS pathology | 26 | 87.6 ± 4.2 | 150 | 88.4 ± 2.2 | 176 | |
| UK-ADC | Hippocampal TDP-43 pathology | 94 | 92.3 ± 6.9 | 161 | 93.3 ± 6.4 | 255 | Stage II #1 |
| UCI90+ | HS pathology | 28 | 94.1 ± 2.3 | 144 | 92.3 ± 4.5 | 172 | Stage II #2 |
| Total | 363 | 2,303 | 2,666 |
GWAS was performed on the three Stage I datasets, from Alzheimer’s Disease Genetics Consortium (ADGC, which are based mostly on dementia research clinics), Rush Religious Order Study and Memory and Aging Project (ROS-MAP, which is an epidemiologic cohort), and Adult Changes in Though (ACT, which is a community-based autopsy cohort). First Stage II was performed at University of Kentucky Alzheimer’s Disease Center (UK-ADC), and second Stage II at the University of California Irvine 90+ Study (UCI90+). None of the cases used in the assessment of HS-Aging overlapped between cohorts
HS Hippocampal sclerosis
Table 2.
Genotyping quality control (QC) for GWAS: Stage I datasets
| Cohort (ADCs, via ADGC) | Level 0 QC | Level 1 QC | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Number of SNPs above imputation threshold | Number of SNPs be low initial call rate threshold (0.85) | Number of SNPs be low initial MAF threshold (0.01) | Number of SNPs out of Hardy–Weinberg equilibrium (p < 1E–5) | Number of SNPs post-level 1 QC | Number of subjects with NACC and ADGC data | |
| ADC1 | 36,642,556 | 832,579 | 28,964,437 | 75,832 | 6,886,357 | 1,484 |
| ADC2 | 36,274,850 | 643,766 | 28,678,144 | 36,123 | 6,964,483 | 210 |
| ADC3 | 36,552,919 | 540,092 | 28,804,571 | 48,516 | 7,222,900 | 529 |
| ADC4 | 36,644,873 | 848,668 | 29,326,492 | 119,205 | 6,594,201 | 127 |
| ADC5 | 36,644,873 | 893,321 | 29,350,835 | 30,593 | 6,581,733 | 103 |
| ADC6 | 36,644,873 | 849,334 | 29,616,694 | 24,304 | 6,345,693 | 56 |
|
| ||||||
| Cohort | Level 2 QC
|
|||||
| Union of SNPs passing level 1 QC | Number of SNPs below call rate threshold (0.95) | Number of SNPs below MAF threshold (0.05) | Number of SNPs out of Hardy–Weinberg equilibrium (p < 1E–5) | Number of SNPs post-level 2 QC | Number of subjects | |
|
| ||||||
| ADCs | 7,948,061 | 2,771,882 | 2,604,515 | 3,142 | 4,271,710 | 2,509 |
|
| ||||||
| Cohort | Number of SNPs above imputation threshold | Number of SNPs below call rate threshold (0.95) | Number of SNPs below MAF threshold (0.05) | Number of SNPs out of Hardy–Weinberg equilibrium (p < 1E–5) | Number of SNPs post-QC | Number of subjects |
|
| ||||||
| ACT | 35,550,922 | 1,831,347 | 30,043,290 | 24,473 | 4,407,882 | 176 |
| ROS-MAP | 35,606,835 | 2,242,313 | 30,159,840 | 36,529 | 4,059,683 | 427 |
Genotypes were imputed to the Phase I v.3 1000 Genomes Project March 2012 release by the ADGC. ADCs were imputed in six iterations (ADC 1–6). Resulting SNPs within each dataset were screened for call rate, minor allele frequency (MAF) and Hardy–Weinberg equilibrium. QC filters were applied in two separate stages for the ADGC dataset which comprises six subsets (ADC 1–6). Note that a SNP can fail more than one of the QC criteria. Overall the number of SNPs that passed QC and were evaluated in the GWAS, i.e., the union of SNPs passing QC across the ADC, ACT and ROS-MAP cohorts, was 4,913,579
Fig. 5.
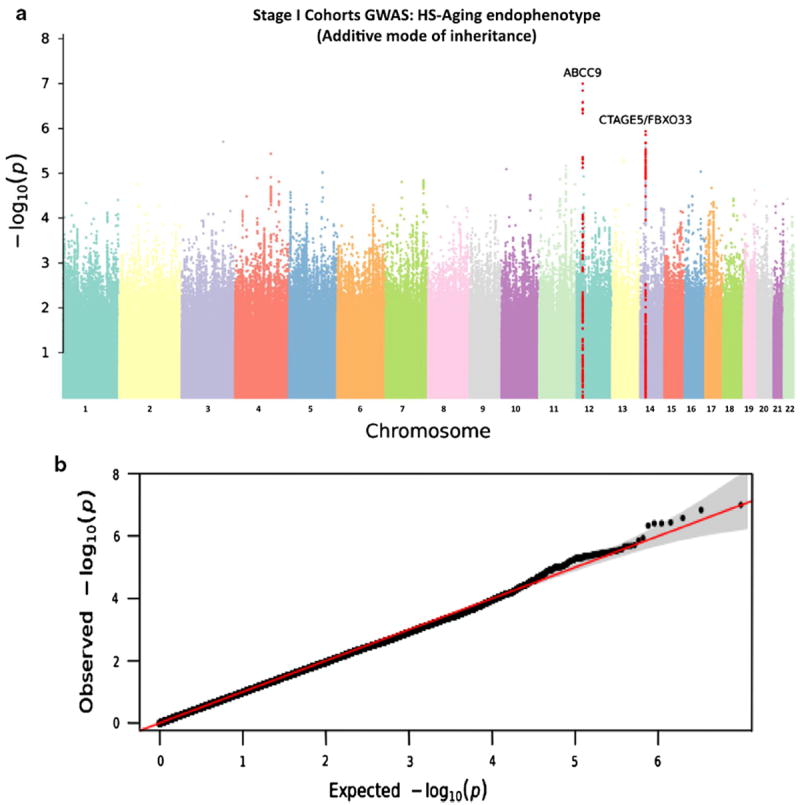
HS-Aging GWAS using data meta-analyzed from the three Stage I cohorts. a Manhattan plot showing p values for HS-Aging GWAS with meta-analyzed data from the three Stage I cohorts. No SNP was found to reach genome-wide statistical significance (p < 5 × 10−8 to correct for multiple comparisons) in this GWAS. Two loci with the lowest p values SNPs in the Stage I GWAS are highlighted in red: ABCC9 (Chr 12p) and CTAGE5/FBXO33 (Chr 14q). b QQ Plot for HS-Aging GWAS using ADGC data. Observed (log-transformed) p values for each SNP are plotted against their corresponding expected values under the null hypothesis of no association with HS-Aging
The GWAS included individual SNPs previously linked to HS-type pathology [57], specifically, rs5848 [61] and rs1990622 [63]. For analyses of these alleles, a separate GWAS was performed using Stage I cohort cases and controls that did not overlap with prior studies of rs5848 and rs1990622. The SNPs previously linked to HS-Aging pathology can be tested with less stringent p value cutoffs due to targeted testing (α = 0.05; two-tailed tests are reported without adjustment for the performance of two tests). Results are shown in Table 3 and Fig. 6. Meta-analyses of Stage I cohorts results show OR = 1.16 (95 % CI 0.92–1.46) for rs5848, OR = 1.22 (95 % CI 1.00–1.49) for rs1990622, with all cohorts tested demonstrating the same risk alleles (T allele for both SNPs) as previously described [61, 63].
Table 3.
Results from Stage I datasets for polymorphisms (rs5848 and rs1990622) linked previously to hippocampal sclerosis pathology in aged individuals
| Dataset | Major allele | Minor allele | Odds ratio | 95 % Confidence interval | Risk allele | p value* |
|---|---|---|---|---|---|---|
| rs5848 (GRN) | ||||||
| ADGC | C | T | 1.08 | 0.84–1.40 | T | 0.541 |
| ACT | C | T | 1.90 | 0.95–3.83 | T | 0.073 |
| ROS-MAP | C | T | 1.12 | 0.53–2.37 | T | 0.774 |
| Meta-analysis results | 1.16 | 0.92–1.46 | T | 0.218 | ||
| rs1990622 (TMEM106B) | ||||||
| ADGC | T | C | 1.19 | 0.95–1.49 | T | 0.132 |
| ACT | T | C | 1.24 | 0.63–2.44 | T | 0.528 |
| ROS-MAP | T | C | 1.43 | 0.82–2.50 | T | 0.198 |
| Meta-analysis results | 1.22 | 1.00–1.49 | T | 0.049 | ||
p values shown here are assuming a two-tailed test
Fig. 6.
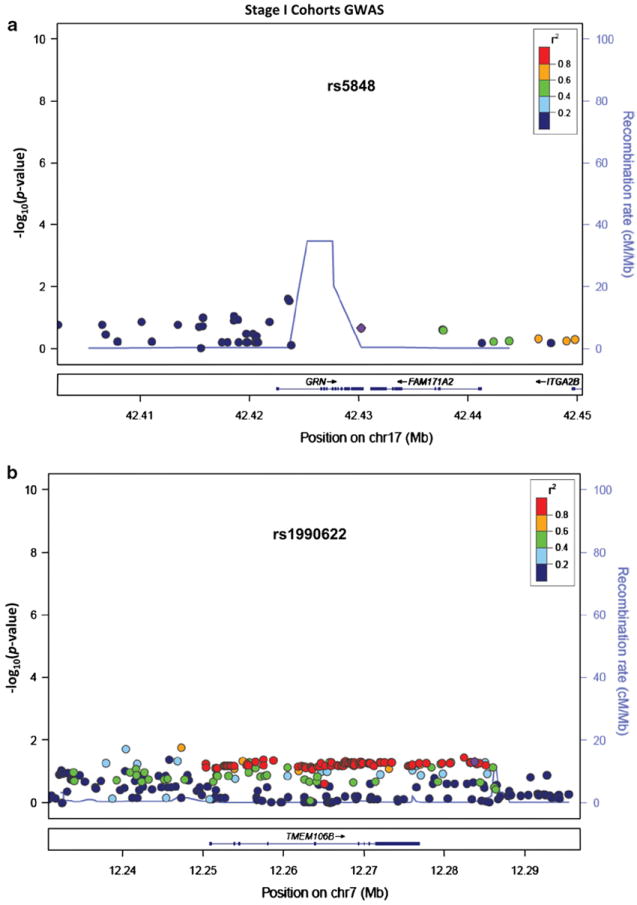
LocusZoom plots of HS-Aging GWAS data. Results of HS-Aging GWAS meta-analyzed data from Stage I datasets are shown. The y axis shows p values describing the results of association study with HS-Aging phenotype centered on a GRN locus (rs5848) on Chromosome 17 and b TMEM106B locus (rs1990622) on Chromosome 7. Note that the SNPs being evaluated have been published previously [17, 63] and this is a sample of cases and controls that is independent from prior published studies of these alleles. Since this part of our study is a replication experiment, and focused on the two particular alleles, p < 0.05 findings would constitute a successful replication
GWAS from meta-analyzed Stage I datasets detected no SNP associated with HS-Aging pathology so strongly as to achieve genome-wide statistical significance. The genomic locus bearing SNPs with the lowest p values in the GWAS data were in the ABCC9 gene on chromosome 12p. The individual SNP that yielded the lowest p value in the GWAS itself was rs7966849 (p = 1.0 × 10−7, uncorrected, see Supplemental Fig. 3) in an intron of ABCC9. SNPs from only nine different genomic regions showed p < 10−5 in the GWAS (Table 4, Supplemental Table 4), including ABCC9 (15 nominal SNPs) and SNPs in and near CTAGE5/FBOXO33 on chromosome 14 (50 nominal SNPs; Supplemental Fig. 4).
Table 4.
GWAS from Stage I cohorts with p < 10−5: genomic loci, including the genes (if any) and individual SNP at each locus with the lowest p value
| Gene/locus area | # SNPS at locus with p < 10−5 | SNP at locus with lowest p value | Chromosome: genomic positiona | Minor allele | Major allele | MAF cases | MAF controls | p value (additive MOI) |
|---|---|---|---|---|---|---|---|---|
| ABCC9 | 15 | rs7966849 | 12: 22004199 | G | A | 0.38 | 0.51 | 1.01E–07 |
| CTAGE5/FBXO 33 | 50 | rs8018486 | 14: 39818617 | G | A | 0.11 | 0.21 | 1.15E–06 |
| Intergenic | 1 | rs114816986 | 3: 155035752 | A | G | 0.30 | 0.08 | 1.97E–06 |
| Intergenic | 1 | rs62324179 | 4: 127661386 | T | C | 0.26 | 0.18 | 3.65E–06 |
| Intergenic | 3 | rs80155385 | 13: 59845469 | C | G | 0.22 | 0.05 | 4.95E–06 |
| Intergenic | 2 | rs3018573 | 11: 98516757 | G | A | 0.52 | 0.41 | 6.81E–06 |
| FRMD4A | 1 | rs72769570 | 10: 13810428 | A | G | 0.19 | 0.05 | 8.09E–06 |
| WWOX | 1 | rs55751884 | 16: 78231495 | C | T | 0.26 | 0.12 | 9.19E–06 |
| Intergenic | 12 | rs12514988 | 5: 123179750 | T | C | 0.16 | 0.04 | 9.48E–06 |
GWAS from Stage I cohorts yielded 86 different nominal SNPs with p < 10−5. These all derived from nine genomic regions or loci, which are shown here with each genomic locus represented by the SNP with the lowest p value from each
Genomic position refers to GRCh37/hg19 assembly
Stage II analyses
The initial criteria for deciding which candidate HS-Aging risk alleles to test in the Stage II cohorts were that the same alleles would yield p < 0.05 in all three Stage I cohorts. This led us to consider ABCC9 putative risk SNPs including rs704178, a SNP with MAF ~0.45, in strong linkage disequilibrium with rs7966849 (R2 = 0.97 and D′ = 1.0) [23]. Genotype frequencies for rs704178 in HS-Aging cases and controls are displayed in Table 5. After accumulation of additional data, the rs704178 allele became the study’s focal-point (Figs. 7, 8, 9). Primary data from ABCC9 risk SNP rs704178 for HS-Aging cases and controls from the Stage II cohorts are presented in Table 6.
Table 5.
Genotype frequencies by HS-Aging status from Stage I cohorts
| HS-Aging | rs704178 genotypes
|
||
|---|---|---|---|
| CC | GC | GG | |
| Cases | 0.164 | 0.441 | 0.395 |
| Controls | 0.259 | 0.500 | 0.242 |
| Total | 0.249 | 0.493 | 0.258 |
The three Stage I cohorts (ADGC/NACC, ROS-MAP, and ACT) are aggregated to compute genotype frequencies for the ABCC9 putative risk SNP, rs704178
Fig. 7.
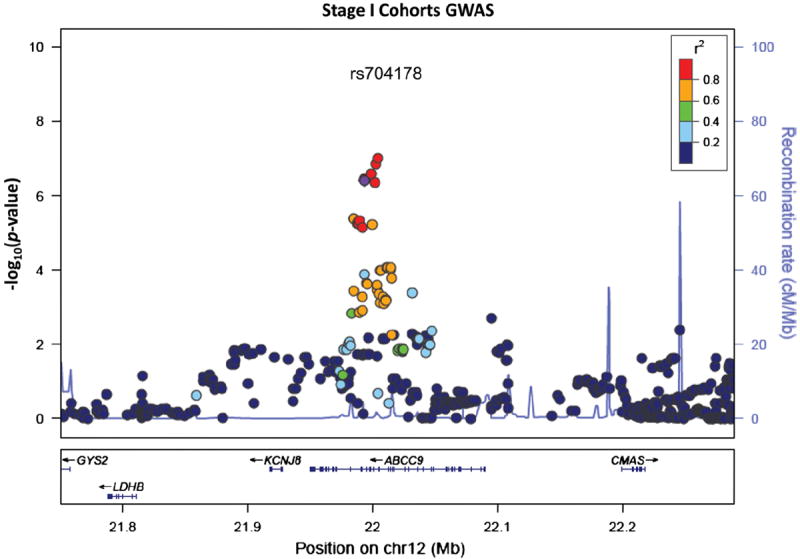
LocusZoom plots of HS-Aging GWAS data. Results of HS-Aging GWAS meta-analyzed data from Stage I datasets are shown. The y axis shows p values describing the results of association study with HS-Aging phenotype centered on the ABCC9 rs704178 risk allele that was the subject of further study. The data shown here are independent of the subsequent studies in two additional Stage II cohorts
Fig. 8.
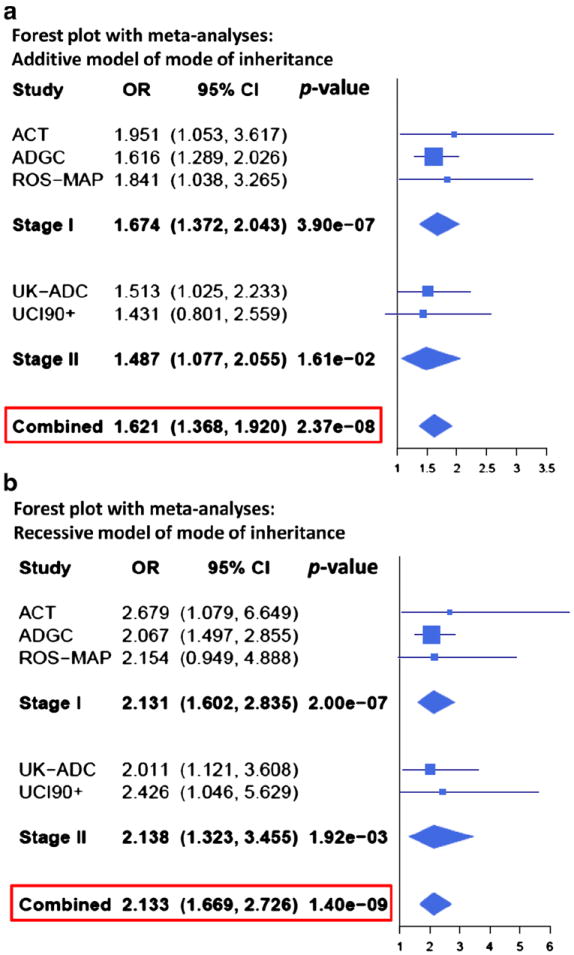
The ABCC9 risk allele (rs704178) association with HS-Aging pathology. Analyses were performed for each individual autopsy cohort, and then combined using meta-analyses across all five Stage I and Stage II cohorts, using inverse-variance-based statistic [70]. Forest plots displaying cohort-specific and aggregated odds ratios (OR) and 95 % confidence intervals (CIs) were created reflecting these meta-analyzed results. Lines represent 95 % CIs for the ORs, and the size of the square (cohort) or trapezoid (meta-analyzed group) reflects the cohort size. Shown here are the results when assuming an additive mode of inheritance (a) or recessive mode of inheritance (b)
Fig. 9.
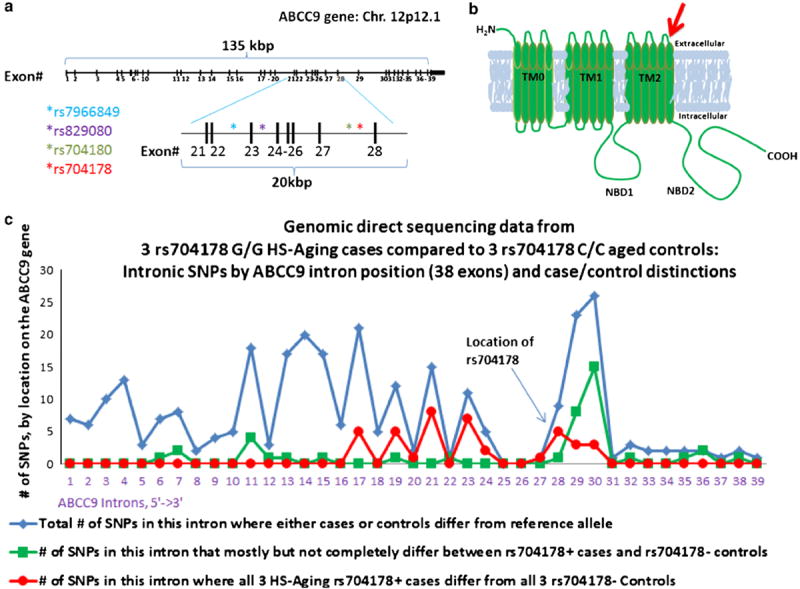
ABCC9 gene and protein organization in relation to the HS-Aging risk alleles. a There are at least 38 exons in the ABCC9 gene, and the SNPs that are most strongly linked to HS-Aging are present within a 20-kilobase region. b The approximate location of the exons on the corresponding ABCC9 polypeptide is shown with a red arrow. TM transmembrane, NBD nucleotide-binding domains. c Direct genomic sequencing of the ABCC9 gene was performed for six cases (3 rs704178 G/G HS-Aging cases, 3 elderly controls with rs704178 C/C genotype). Intron-by-intron analyses indicate that the intronic polymorphisms linked to HS-Aging pathology span more than ten exons
Table 6.
Number of research subjects in each Stage II cohort arranged by ABCC9 genotype status (risk allele is G) and neuropathology [P-TDP-43 or hippocampal sclerosis (HS)]
| Cohort | Neuropathology | rs704178 genotypes
|
||
|---|---|---|---|---|
| CC | GC | GG | ||
| UK-ADC | 0a | 35 | 75 | 35 |
| 1a | 7 | 11 | 7 | |
| 2a | 8 | 24 | 25 | |
| UCI90+ | HS− | 34 | 76 | 34 |
| HS+ | 7 | 9 | 12 | |
Stage II cohorts used autopsy series independent of the Stage I datasets, with phospho-TDP-43 (P-TDP43) pathology in the UK-ADC as the endophenotype. Different strategies and models could be applied for statistical testing; for results of statistical analyses, see Fig. 8 and Supplemental Fig. 5
In the UK-ADC, severity of aberrant P-TDP-43 (0–2 scale) was the pathologic readout
The UK-ADC was the first-stage Stage II cohort for evaluating HS-Aging candidate risk SNPs. For this cohort, the primary criterion for a positive “case” status was aberrant P-TDP-43 immunoreactivity in the hippocampus and subiculum, which we hypothesize provides (in the context of elderly individuals without FTLD) a sensitive and specific readout of HS-Aging pathology [2, 51]. From this convenience sample of 227 patients, 82 (36.1 %) were positive for P-TDP43 pathology. Ten initial SNP risk allele candidates from the Stage I datasets were evaluated in the UK-ADC cohort: rs7206273, rs1538842, rs4869959, rs5848, rs201825, rs2274567, rs6656401, rs2287910, rs4144545, rs704179, and rs704178. Of these, rs704178 was the only SNP showing positive association with the pathologic endophenotype.
Data from the UCI90+ study [15, 16], a population-based sample of 144 controls and 28 HS-Aging cases, was the second Stage II cohort. Here, H&E-based observation of hippocampal sclerosis pathology was used as the readout, since not all the cases were stained with the same P-TDP43 methods. The risk alleles (rs704178:G) trended toward positive association with HS pathology; no other SNP was tested in this dataset except rs704180, which was in complete linkage disequilibrium with rs704178 as expected (not shown). This trend was only statistically significant if the recessive MOI model was applied (p < 0.03 using two-tailed test); however, the effect size was still large at OR = 1.43 (95 % CI 0.80–2.6) even with additive MOI model and two-tailed test.
Combined meta-analyses of all Stage I and Stage II cohorts yielded a p value of 2.4 × 10−8 with an additive MOI model and a p value of 1.4 × 10−9 with recessive MOI (Fig. 8). Dominant MOI model had far weaker association as is shown in Supplemental Fig. 5. Having found the same ABCC9 gene polymorphism is associated with hippocampal P-TDP43 pathology or HS-Aging pathology in two Stage II cohorts in addition to HS-Aging pathology in the Stage I cohorts, we focused on ABCC9 in subsequent studies.
Post-hoc studies related to ABCC9
To assess preliminarily whether the ABCC9 risk alleles are proxies for as-yet uncharacterized genomic feature(s), ABCC9 gene-specific sequencing was performed using genomic DNA (Supplemental Methods). For these studies, DNA was isolated from hippocampi of three autopsy-confirmed HS-Aging cases homozygous for ABCC9 risk SNPs (rs704178: G/G), and the results were compared to three individuals over 95 at death (rs704178: C/C) and lacking HS-Aging pathology (Supplemental Methods). No previously uncharacterized genomic feature (including exonic SNP) was discovered for which rs704178 would be a proxy. As expected, SNPs in known linkage disequilibrium with rs704178 span a region over 10 exons (Fig. 9).
Western blots were performed on protein samples from snap-frozen human hippocampi to address whether differences in ABCC9 polypeptides can be observed in correlation with particular HS-Aging risk alleles or pathologic status. These experiments were designed to test for a qualitative change in ABCC9 protein such as insoluble aggregates, proteolytic fragments, or complete loss of expression. The convenience subsample from the UK-ADC comprised nine human hippocampi including HS-Aging and controls, and different homozygous ABCC9 genotypes. Different protein extraction methods were utilized and five different commercially available anti-ABCC9 antibodies were applied. These experiments discovered no evidence of a specific qualitative change in hippocampal ABCC9 protein products, with caveats related to the small number of cases evaluated and to the inconsistency in the protein blotting characteristics observed for these antibodies (Supplemental Fig. 6). The imperfect antigenic specificity for the five putative ABCC9 antibodies also rendered immunohistochemistry technically unsatisfactory, although this was attempted (not shown).
The ABCC9 gene is also known as sulfonylurea receptor 2, SUR2. Sulfonylureas are a widely prescribed class of drugs used to treat type II diabetes mellitus; these drugs antagonize the function of the ABCC9 protein product [10, 13, 33, 68]. We tested the hypothesis that exposure to sulfonylurea drugs is associated with altered risk for HS-Aging pathology (Table 7). Data were obtained from NACC database with both longitudinal drug history and postmortem data from individuals dying age 85 and above with autopsies between 2010 and 2013. HS-Aging pathology was observed in 17.3 % (108/624) of all autopsied cases in the sample, 30.6 % of sulfonylurea users and 16.5 % of non-users. Based on a two-tailed test and α = 0.05, the crude OR for autopsy-confirmed HS-Aging pathology after use of any sulfonylurea versus no sulfonylurea use was 2.23 [95 % CI: 1.06–4.68, p = 0.03; age-adjusted OR = 2.19 (95 % CI: 1.04–4.63, p = 0.04)]. Use of any sulfonylurea was reported by 11 HS-Aging and 25 non HS-Aging subjects. When the analysis was restricted to the medication history given at the last assessment before death, the crude OR for use of any sulfonylurea was 2.52 [95 % CI: 1.10–5.76, p = 0.02; age-adjusted OR = 2.49 (95 % CI: 1.08–5.75, p = 0.03)]. There was neither positive nor negative association between sulfonylurea use and AD pathology, operationalized as AD-positive if Braak stage V or VI and AD-negative otherwise (not shown). These data are compatible with the hypothesis that sulfonylurea exposure is associated with HS-Aging pathology in this sample.
Table 7.
Sulfonylurea use and HS-Aging risk in persons age 85 years and older at death: data from the National Alzheimer’s Coordinating Center (NACC) database
| Sulfonylurea use (n = 36)
|
No sulfonylurea use (n = 588)
|
|||
|---|---|---|---|---|
| HS-Aging pathology (n = 11) | No HS-Aging pathology (n = 25) | HS-Aging Pathology (n = 97) | No HS-Aging pathology (n = 491) | |
| Age at death, years | 88.7 ± 4.8 | 90.8 ± 4.3 | 91.2 ± 4.3 | 90.9 ± 4.4 |
| Number of longitudinal evaluations | 4.5 ± 1.1 | 4.4 ± 1.7 | 3.9 ± 1.8 | 3.9 ± 1.7 |
| Last evaluation to death, months | 13.8 ± 10.0 | 15.3 ± 15.3 | 18.6 ± 19.7 | 15.1 ± 15.1 |
| Estimated sulfonylurea exposure, years | 3.4 (range 0.5–6.2) | 3.5 (range 0.5–6.8) | N/A | N/A |
| Taking sulfonylurea at last evaluation, % | 81.8 % | 72.0 % | N/A | N/A |
Summary statistics for data used to test of the hypothesis that sulfonylurea use is correlated with increased risk for HS-Aging pathology in the NACC database. Results presented are mean ± SD unless otherwise stated. These analyses involved 624 patients who died after age 85, between 2010 and 2013, with detailed longitudinal drug history and autopsy data. “Sulfonylurea use” indicated self-reported use of any sulfonylurea drug at any longitudinal visit. Crude OR for use of any sulfonylurea was 2.23 [95 % CI: 1.06–4.68, p = 0.03; age-adjusted OR = 2.19 (95 % CI: 1.04–4.63, p = 0.04)]. Mean age at death was not different for subjects stratified by the presence of HS-Aging and use of any sulfonylurea (F3,620 = 1.10, p = 0.35) or was mean time between last evaluation and date of death (F3,620 = 1.33, p = 0.26). The average estimated drug exposure and % of patients taking a sulfonylurea at last visit was not different in persons with and without eventual HS-Aging pathology
N/A not applicable
Potential confounders include the clinical indication for sulfonylurea use, namely type II diabetes. Of subjects who reported taking any anti-diabetic agent (62/624), most reported use of a sulfonylurea (36/62). The association with HS-Aging pathology was not dependent upon diabetes itself because, in the NACC database overall, history of diabetes had a null association with HS-Aging pathology [52]. In this subsample, the odds of observing HS-Aging pathology were not increased for anti-diabetic agent users as a whole [age-adjusted OR = 1.43 (95 % CI: 0.75–2.71, p = 0.28)], for anti-diabetic agent users who never reported using a sulfonylurea [age-adjusted OR = 0.60 (95 % CI: 0.17–2.04, p = 0.41)], or for self-reported diabetes [age-adjusted OR = 1.08 (95 % CI: 0.58–2.03, p = 0.81)].
For a slightly younger group of research subjects, those dying age 80–84 (n = 261), among whom HS-Aging histopathology was relatively uncommon [9], post hoc sensitivity analyses indicated no association between sulfonylurea use and HS-Aging pathology [age-adjusted OR = 0.59 (95 % CI: 0.13–2.68)]. Effect estimates in this group are similar for use of any anti-diabetic medication [age-adjusted OR = 0.33 (95 % CI: 0.07–1.42)] and self-reported diabetes [age-adjusted OR = 0.43 (95 % CI: 0.15–1.29)]. Results in this slightly younger group of subjects, which included only 17 sulfonylurea users, did not support the hypothesis that sulfonylurea use is associated with HS-Aging pathology in this age group.
Discussion
In the large human autopsy cohorts studied, the risk of developing HS-Aging pathology was associated with polymorphism in the ABCC9 gene. The effect size was relatively large (OR = 1.4–2.0 with additive MOI, 2.0–2.7 with recessive MOI) in all three Stage I cohorts and also in both Stage II cohorts. In the final Stage II cohort, only a single polymorphism was queried so statistical correction for multiple testing was obviated. The HS-Aging risk allele may be directly relevant to public health because aged human subjects in the NACC database with history of sulfonylurea exposure have increased risk for HS-Aging pathology.
There are potential pitfalls inherent to the study design. The Stage I GWAS did not produce a SNP that reached genome-wide statistical significance. Bias was thus inserted into the study by selecting which SNPs would be analyzed in the Stage II cohorts. Among the candidate SNPs from GWAS, ABCC9 polymorphisms ultimately yielded the lowest p values by an order of magnitude, so that locus was an obvious choice to evaluate. Testing the association between sulfonylurea drug exposure and HS-Aging pathology in the NACC dataset is complicated by confounders and limitations. For example, the accuracy of medication self-report is imperfect since current medication use refers to the 2-week period prior to the clinical assessment, drug dosages were unknown, and some of the patients presumably used and discontinued sulfonylurea drugs prior to study enrollment as well as between annual study visits. Patient age was also a relevant factor; we found that HS-Aging pathology is relatively unusual among individuals younger than age 85 [9, 49-51] and found no evidence for correlation between the sulfonylurea drugs and HS-Aging pathologically in the post hoc analysis of younger patients. These analyses were also performed blind to patients’ genotypes so the distribution of ABCC9 SNPs versus sulfonylurea use is unknown. For these reasons, the data on HS-Aging linked to drug exposure should be interpreted with caution.
The present study was not designed to evaluate polymorphisms that show weak associations with HS-Aging pathology. There were, however, some alleles from the GWAS that appear interesting despite failing to reach genome-wide statistical significance. For example, SNPs within and near the cutaneous T-cell lymphoma-associated antigen 5 (CTAGE5) gene yielded p ~ 10−6 in the GWAS, and CTAGE5 polymorphisms have been linked to familial idiopathic basal ganglia calcification (Fahr’s disease) [40, 56]. Meta-analyses of Stage I cohort data also indicated that alleles linked previously to hippocampal sclerosis pathology—rs5848 [17] and rs1990662 SNPs [63]—trend toward association with HS-Aging pathology as operationalized in the current study, with the same risk alleles as previously described [57, 61, 63], and with OR ~1.2 (meta-analyzed data, additive MOI).
The main discovery of the present study is the association between HS-Aging pathology and polymorphism in the ABCC9 gene. Meta-analyzed data indicate an overall OR = 1.62 with the additive MOI model—a relatively large effect size for a common allele [11, 24, 81]. Notably, the chromosomal region in which ABCC9 resides (12p) has been linked to clinical late-onset AD risk [31, 66], which may be at least partially explained by ABCC9, because HS-Aging tends to be misdiagnosed clinically as AD [9, 57].
We can begin to develop hypotheses about mechanism(s) underlying the genomic associations. The presence of hippocampal TDP-43 pathology is associated with additive cognitive impairment [37, 48], and “unilateral” HS-Aging pathology usually shows TDP-43 pathology on the contralateral side [50]; the present study affirms that hippocampal TDP-43 pathology is linked to HS-Aging pathology, since risk for both is associated with the same allele, rs704178. The strength of the association between rs704178 and HS-Aging pathology with recessive model of MOI (OR = 2.13) indicates that the “non-risk alleles” may exert an adaptive function, even in the heterozygous state (“dominant” gain-of-function pattern).
The association between ABCC9 and HS-Aging provides a novel insight into neurodegenerative diseases. Prior GWAS found that different polymorphisms of the ABCC9 gene were associated with cardiac problems and altered sleep patterns [32, 34, 38, 55, 58], which could theoretically be pathogenetic factors “upstream” from HS-Aging. Alternatively, or in addition, ABCC9 polymorphisms may exert influence through blood vessel pathology. The ABCC9 polypeptide product (SUR2) is physiologically active in arteriolar smooth muscle [22, 55], and pathologically confirmed arteriolosclerosis is relatively severe in brains that also harbor HS-Aging pathology [52] (and see Ref. [18]). Stop-codon mutations in ABCC9 lead to Cantú syndrome [26, 75], a rare condition with mostly uncharacterized neuropathology. Intriguingly, Cantú syndrome patients have demonstrated vascular anomalies, including in the brain [21, 27, 67].
What about the function of the HS-Aging risk SNPs themselves? These intronic SNPs span at least ten ABCC9 exons. Some mRNA variants have been described with splice junctions near the HS-Aging risk SNPs [12, 78]. In the genotype-tissue expression project (GTEx) database [14] of expression quantitative trait loci, SNPs linked to HS-Aging pathology (particularly rs704192 which has D′ > 0.9 with rs704178) also correlate with altered ABCC9 expression specifically in both human hippocampus and amygdala (both p < 0.05) but not cerebellum. Using SNPExp web tool [30] where SNPs are correlated with gene expression using transformed lymphoblastoid cell lines, SNPs linked to HS-Aging pathology (particularly rs829080, with D′ = 1.0 with rs704178) correlate strongly with ABCC9 expression (p < 0.0001). Despite these interesting observations, it is not yet proven that the HS-Aging risk SNP variability leads directly to changes in gene splicing, mRNA expression, or polypeptide function in human brain.
Whatever the mechanism(s), ABCC9 polymorphism in a relatively large number of human cases is associated with HS-Aging pathology. The differing neuropathologic findings and differing genetic vulnerabilities of HS-Aging and AD make it likely that prevention and treatment of HS-Aging, when achieved, will be different from prevention and treatment for AD. ABCC9 function is sensitive to pharmacologic manipulation, and we here report that in subjects dying after age 85, sulfonylurea drug exposure was associated with HS-Aging pathology. This study may herald progress of clinical relevance, because ABCC9 agonists have been described, and may be neuroprotective [77, 82]. We conclude that the present study establishes a pathogenetic association between ABCC9 and HS-Aging pathology––and perhaps the basis for a novel future therapeutic strategy.
Supplementary Material
Acknowledgments
We are deeply grateful to all of the study participants and all the clinical workers, who make this research possible. We thank Ms. Sonya Anderson and Ms. Ela Patel for technical support. Thanks also to Drs. Julia Kofler and M. Ilyas Kamboh for collaborative assistance in this project. 1D3 antibody was a generous gift from Dr. Manuela Neumann. Funding included National Institutes of Health (NIH) grants for ADGC (U01 AG032984), NACC (U01 AG016976), the National Cell Repository for AD (NCRAD; U24 AG21886), K25 AG043546, UL1TR000117, and the UK-ADC P30 AG028383 from the National Institute on Aging (NIA). ACT enrollment and clinical data are from NIA U01 AG 06781 (E Larson, PI). Genetic data for ACT are from Human Genome Research Institute U01 HG006375 (E Larson, PI). Rush Religious Order Study and Memory and Aging Project (D Bennett PI) is supported by NIH grants P30AG10161, R01AG15819, R01AG17917, R01AG42210, and the Translational Genomics Research Institute. University of California Irvine work was supported by NIH grants for the UCI ADC (P50AG16573) and 90+ Study (R01AG21055; C Kawas, PI). For additional acknowledgment and funding support, please see Supplemental material.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1282-2) contains supplementary material, which is available to authorized users.
Contributor Information
Peter T. Nelson, Email: pnels2@email.uky.edu, peter.nelson@uky.edu, Department of Pathology, Division of Neuropathology, Rm 311, Sanders-Brown Center on Aging, University of Kentucky, 800 S. Limestone Avenue, Lexington, KY 40536-0230, USA.
Steven Estus, University of Kentucky, Lexington, KY, USA.
Erin L. Abner, University of Kentucky, Lexington, KY, USA
Ishita Parikh, University of Kentucky, Lexington, KY, USA.
Manasi Malik, University of Kentucky, Lexington, KY, USA.
Janna H. Neltner, University of Kentucky, Lexington, KY, USA
Eseosa Ighodaro, University of Kentucky, Lexington, KY, USA.
Wang-Xia Wang, University of Kentucky, Lexington, KY, USA.
Bernard R. Wilfred, University of Kentucky, Lexington, KY, USA
Li-San Wang, University of Pennsylvania, Philadelphia, PA, USA.
Walter A. Kukull, University of Washington, Seattle, WA, USA
Kannabiran Nandakumar, Mayo Clinic, Rochester, MN, USA.
Mark L. Farman, University of Kentucky, Lexington, KY, USA
Wayne W. Poon, University of California, Irvine, Irvine, CA, USA
Maria M. Corrada, University of California, Irvine, Irvine, CA, USA
Claudia H. Kawas, University of California, Irvine, Irvine, CA, USA
David H. Cribbs, University of California, Irvine, Irvine, CA, USA
David A. Bennett, Rush University, Chicago, IL, USA
Julie A. Schneider, Rush University, Chicago, IL, USA
Eric B. Larson, Group Health Research Institute, Seattle, WA, USA
Paul K. Crane, University of Washington, Seattle, WA, USA
Otto Valladares, University of Pennsylvania, Philadelphia, PA, USA.
Frederick A. Schmitt, University of Kentucky, Lexington, KY, USA
Richard J. Kryscio, University of Kentucky, Lexington, KY, USA
Gregory A. Jicha, University of Kentucky, Lexington, KY, USA
Charles D. Smith, University of Kentucky, Lexington, KY, USA
Stephen W. Scheff, University of Kentucky, Lexington, KY, USA
Joshua A. Sonnen, University of Utah, Salt Lake City, UT, USA
Jonathan L. Haines, Case Western Reserve University, Cleveland, OH, USA
Margaret A. Pericak-Vance, University of Miami, Miami, FL, USA
Richard Mayeux, Columbia University, New York, NY, USA.
Lindsay A. Farrer, Boston University, Boston, MA, USA
Linda J. Van Eldik, University of Kentucky, Lexington, KY, USA
Craig Horbinski, University of Kentucky, Lexington, KY, USA.
Robert C. Green, Harvard University, Boston, MA, USA
Marla Gearing, Emory University, Atlanta, GA, USA.
Leonard W. Poon, Institute of Gerontology, University of Georgia, Athens, GA, USA
Patricia L. Kramer, Oregon Health and Sciences University, Portland, OR, USA
Randall L. Woltjer, Oregon Health and Sciences University, Portland, OR, USA
Thomas J. Montine, University of Washington, Seattle, WA, USA
Amanda B. Partch, University of Pennsylvania, Philadelphia, PA, USA
Alexander J. Rajic, University of California, Irvine, Irvine, CA, USA
KatieRose Richmire, Group Health Research Institute, Seattle, WA, USA.
Sarah E. Monsell, University of Washington, Seattle, WA, USA
Gerard D. Schellenberg, University of Pennsylvania, Philadelphia, PA, USA
David W. Fardo, Email: david.fardo@uky.edu, Department of Biostatistics, University of Kentucky, 205E Multidisciplinary Science Building, 725 Rose Street, Lexington, KY 40536-0082, USA.
References
- 1.Amador-Ortiz C, Ahmed Z, Zehr C, Dickson DW. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (Berl) 2007;113(3):245–252. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61(5):435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balasubramanian AB, Kawas CH, Peltz CB, Brookmeyer R, Corrada MM. Alzheimer disease pathology and longitudinal cognitive performance in the oldest-old with no dementia. Neurology. 2012;79(9):915–921. doi: 10.1212/WNL.0b013e318266fc77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, Hubbard JL, Koepsell TD, Morris JC, Kukull WA Centers NIAAsD. The National Alzheimer’s Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord. 2007;21(3):249–258. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 5.Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, Kukull WA Centers NI-AsD. The National Alzheimer’s Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18(4):270–277. [PubMed] [Google Scholar]
- 6.Bennett DA, Schneider JA, Aggarwal NT, Arvanitakis Z, Shah RC, Kelly JF, Fox JH, Cochran EJ, Arends D, Treinkman AD, Wilson RS. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology. 2006;27(3):169–176. doi: 10.1159/000096129. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Wilson RS, Arvanitakis Z, Boyle PA, de Toledo-Morrell L, Schneider JA. Selected findings from the Religious Orders Study and Rush memory and aging project. J Alzheimers Dis. 2013;33(Suppl 1):S397–S403. doi: 10.3233/JAD-2012-129007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33(6):403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 9.Brenowitz WD, Monsell SE, Schmitt FA, Kukull WA, Nelson PT. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both Alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimers Dis. 2013 doi: 10.3233/JAD-131880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryan J, Munoz A, Zhang X, Dufer M, Drews G, Krippeit-Drews P, Aguilar-Bryan L. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch: Eur J Physiol. 2007;453(5):703–718. doi: 10.1007/s00424-006-0116-z. [DOI] [PubMed] [Google Scholar]
- 11.Bush WS, Moore JH. Chapter 11: genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi: 10.1371/journal.pcbi.1002822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chutkow WA, Makielski JC, Nelson DJ, Burant CF, Fan Z. Alternative splicing of sur2 Exon 17 regulates nucleotide sensitivity of the ATP-sensitive potassium channel. J Biol Chem. 1999;274(19):13656–13665. doi: 10.1074/jbc.274.19.13656. [DOI] [PubMed] [Google Scholar]
- 13.Chutkow WA, Simon MC, Le Beau MM, Burant CF. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes. 1996;45(10):1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- 14.Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corrada MM, Berlau DJ, Kawas CH. A population-based clinicopathological study in the oldest-old: the 90+ study. Curr Alzheimer Res. 2012;9(6):709–717. doi: 10.2174/156720512801322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corrada MM, Brookmeyer R, Berlau D, Paganini-Hill A, Kawas CH. Prevalence of dementia after age 90: results from the 90+ study. Neurology. 2008;71(5):337–343. doi: 10.1212/01.wnl.0000310773.65918.cd. [DOI] [PubMed] [Google Scholar]
- 17.Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7(1–3):170–174. doi: 10.1159/000289231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, Aronson MK, Crystal HA. Hippocampal sclerosis: a common pathological feature of dementia in very old (≥ 80 years of age) humans. Acta Neuropathol. 1994;88(3):212–221. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 19.Dowling NM, Tomaszewski Farias S, Reed BR, Sonnen JA, Strauss ME, Schneider JA, Bennett DA, Mungas D. Neuropathological associates of multiple cognitive functions in two community-based cohorts of older adults. J Int Neuropsychol Soc. 2010:1–13. doi: 10.1017/S1355617710001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans. Genet Epidemiol. 2008;32(3):227–234. doi: 10.1002/gepi.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engels H, Bosse K, Ehrbrecht A, Zahn S, Hoischen A, Propping P, Bindl L, Reutter H. Further case of Cantu syndrome: exclusion of cryptic subtelomeric chromosome aberrations. Am J Med Genet. 2002;111(2):205–209. doi: 10.1002/ajmg.10560. [DOI] [PubMed] [Google Scholar]
- 22.Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90(3):799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt S, Johnson N, Juettemann T, Kahari AK, Keenan S, Kulesha E, Martin FJ, Maurel T, McLaren WM, Murphy DN, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ruffier M, Sheppard D, Taylor K, Thormann A, Trevanion SJ, Vullo A, Wilder SP, Wilson M, Zadissa A, Aken BL, Birney E, Cunningham F, Harrow J, Herrero J, Hubbard TJ, Kinsella R, Muffato M, Parker A, Spudich G, Yates A, Zerbino DR, Searle SM. Ensembl. Nucleic Acids Res. 2014;201442(Database issue):D749–755. doi: 10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garner C. Upward bias in odds ratio estimates from genome-wide association studies. Genet Epidemiol. 2007;31(4):288–295. doi: 10.1002/gepi.20209. [DOI] [PubMed] [Google Scholar]
- 25.Gauderman WJ. Sample size requirements for matched case–control studies of gene–environment interaction. Stat Med. 2002;21(1):35–50. doi: 10.1002/sim.973. [DOI] [PubMed] [Google Scholar]
- 26.Harakalova M, van Harssel JJ, Terhal PA, van Lieshout S, Duran K, Renkens I, Amor DJ, Wilson LC, Kirk EP, Turner CL, Shears D, Garcia-Minaur S, Lees MM, Ross A, Venselaar H, Vriend G, Takanari H, Rook MB, van der Heyden MA, Asselbergs FW, Breur HM, Swinkels ME, Scurr IJ, Smithson SF, Knoers NV, van der Smagt JJ, Nijman IJ, Kloosterman WP, van Haelst MM, van Haaften G, Cuppen E. Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat Genet. 2012;44(7):793–796. doi: 10.1038/ng.2324. [DOI] [PubMed] [Google Scholar]
- 27.Hiraki Y, Miyatake S, Hayashidani M, Nishimura Y, Matsuura H, Kamada M, Kawagoe T, Yunoki K, Okamoto N, Yofune H, Nakashima M, Tsurusaki Y, Satisu H, Murakami A, Miyake N, Nishimura G, Matsumoto N. Aortic aneurysm and cranio-synostosis in a family with Cantu syndrome. Am J Med Genet Part A. 2014;164A(1):231–236. doi: 10.1002/ajmg.a.36228. [DOI] [PubMed] [Google Scholar]
- 28.Hoggart CJ, Clark TG, De Iorio M, Whittaker JC, Balding DJ. Genome-wide significance for dense SNP and resequencing data. Genet Epidemiol. 2008;32(2):179–185. doi: 10.1002/gepi.20292. [DOI] [PubMed] [Google Scholar]
- 29.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Ruther E, Schurmann B, Heun R, Kolsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Gallacher J, Hull M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alperovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Bjornsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossu P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm K, Melum E, Franke A, Karlsen TH. SNPexp––a web tool for calculating and visualizing correlation between HapMap genotypes and gene expression levels. BMC Bioinform. 2010;11:600. doi: 10.1186/1471-2105-11-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmans P, Hamshere M, Hollingworth P, Rice F, Tunstall N, Jones S, Moore P, Wavrant DeVrieze F, Myers A, Crook R, Compton D, Marshall H, Meyer D, Shears S, Booth J, Ramic D, Williams N, Norton N, Abraham R, Kehoe P, Williams H, Rudrasingham V, O’Donovan M, Jones L, Hardy J, Goate A, Lovestone S, Owen M, Williams J. Genome screen for loci influencing age at onset and rate of decline in late onset Alzheimer’s disease. Am J Med Genet Part B, Neuropsychiatr Genet: Off Publ Int Soc Psychiatr Genet. 2005;135B(1):24–32. doi: 10.1002/ajmg.b.30114. [DOI] [PubMed] [Google Scholar]
- 32.Hu D, Barajas-Martinez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C, Schimpf R, Cai JJ, Nam GB, Deshmukh P, Scheinman M, Preminger M, Steinberg J, Lopez-Izquierdo A, Ponce-Balbuena D, Wolpert C, Haissaguerre M, Sanchez-Chapula JA, Antzelevitch C. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171(3):431–442. doi: 10.1016/j.ijcard.2013.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16(5):1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 34.Iwasa H, Kurabayashi M, Nagai R, Nakamura Y, Tanaka T. Multiple single-nucleotide polymorphisms (SNPs) in the Japanese population in six candidate genes for long QT syndrome. J Hum Genet. 2001;46(3):158–162. doi: 10.1007/s100380170106. [DOI] [PubMed] [Google Scholar]
- 35.Jicha GA, Abner E, Schmitt FA, Cooper GE, Stiles N, Hamon R, Carr S, Smith CD, Markesbery WR. Clinical features of mild cognitive impairment differ in the research and tertiary clinic settings. Dement Geriatr Cogn Disord. 2008;26(2):187–192. doi: 10.1159/000151635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, Rademakers R, Boeve BF, Parisi JE, Smith GE, Ivnik RJ, Petersen RC, Jack CR, Jr, Dickson DW. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70(19 Pt 2):1850–1857. doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, Petrucelli L, Senjem ML, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Jack CR, Jr, Parisi JE, Petersen RC, Dickson DW. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014 doi: 10.1007/s00401-014-1269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38(6):937–943. doi: 10.1016/j.yjmcc.2005.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, Destefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Hollingworth P, Ramirez A, Hanon O, Fitzpatrick AL, Buxbaum JD, Campion D, Crane PK, Baldwin C, Becker T, Gudnason V, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MJ, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuinness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannfelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley THJr, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltunen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P European Alzheimer’s Disease I, Genetic, Environmental Risk in Alzheimer’s D, Alzheimer’s Disease Genetic C, Cohorts for H, Aging Research in Genomic E. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013 doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemos RR, Oliveira DF, Zatz M, Oliveira JR. Population and computational analysis of the MGEA6 P521A variation as a risk factor for familial idiopathic basal ganglia calcification (Fahr’s disease) J Mol Neurosci: MN. 2011;43(3):333–336. doi: 10.1007/s12031-010-9445-7. [DOI] [PubMed] [Google Scholar]
- 41.Leverenz JB, Agustin CM, Tsuang D, Peskind ER, Edland SD, Nochlin D, DiGiacomo L, Bowen JD, McCormick WC, Teri L, Raskind MA, Kukull WA, Larson EB. Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol. 2002;59(7):1099–1106. doi: 10.1001/archneur.59.7.1099. [DOI] [PubMed] [Google Scholar]
- 42.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 44.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT National Institute on A, Alzheimer’s A. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montine TJ, Sonnen JA, Montine KS, Crane PK, Larson EB. Adult Changes in Thought study: dementia is an individually varying convergent syndrome with prevalent clinically silent diseases that may be modified by some commonly used therapeutics. Curr Alzheimer Res. 2012;9(6):718–723. doi: 10.2174/156720512801322555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mortimer JA. The Nun Study: risk factors for pathology and clinical-pathologic correlations. Curr Alzheimer Res. 2012;9(6):621–627. doi: 10.2174/156720512801322546. [DOI] [PubMed] [Google Scholar]
- 47.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, Decarli C, Dekosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011 doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Davis DG, Poduska JW, Patel E, Mendiondo MS, Markesbery WR. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20(1):66–79. doi: 10.1111/j.1750-3639.2008.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, Abner EL, Smith CD, Van Eldik LJ, Kryscio RJ, Scheff SW. Alzheimer’s disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121(5):571–587. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson PT, Schmitt FA, Lin Y, Abner EL, Jicha GA, Patel E, Thomason PC, Neltner JH, Smith CD, Santacruz KS, Sonnen JA, Poon LW, Gearing M, Green RC, Woodard JL, Van Eldik LJ, Kryscio RJ. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134(Pt 5):1506–1518. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson PT, Smith CD, Abner EL, Wilfred BJ, Wang WX, Neltner JH, Baker M, Fardo DW, Kryscio RJ, Scheff SW, Jicha GA, Jellinger KA, Van Eldik LJ, Schmitt FA. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol. 2013;126(2):161–177. doi: 10.1007/s00401-013-1154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neltner JH, Abner EL, Baker S, Schmitt FA, Kryscio RJ, Jicha GA, Smith CD, Hammack E, Kukull WA, Brenowitz WD, Van Eldik LJ, Nelson PT. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain. 2013 doi: 10.1093/brain/awt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117(2):137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 55.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res. 2013;112(7):1059–1072. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oliveira JR, Sobrido MJ, Spiteri E, Hopfer S, Meroni G, Petek E, Baquero M, Geschwind DH. Analysis of candidate genes at the IBGC1 locus associated with idiopathic basal ganglia calcification (“Fahr’s disease”) J Mol Neurosci: MN. 2007;33(2):151–154. [PubMed] [Google Scholar]
- 57.Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25(4):364–368. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parsons MJ, Lester KJ, Barclay NL, Nolan PM, Eley TC, Gregory AM. Replication of Genome-Wide Association Studies (GWAS) loci for sleep in the British G1219 cohort. Am J Med Genet Part B, Neuropsychiatr Genet: Off Publ Int Soc Psychiatr Genet. 2013;162B(5):431–438. doi: 10.1002/ajmg.b.32106. [DOI] [PubMed] [Google Scholar]
- 59.Poon LW, Clayton GM, Martin P, Johnson MA, Courtenay BC, Sweaney AL, Merriam SB, Pless BS, Thielman SB. The Georgia Centenarian Study. Int J Aging Hum Dev. 1992;34(1):1–17. doi: 10.2190/8M7H-CJL7-6K5T-UMFV. [DOI] [PubMed] [Google Scholar]
- 60.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, Willer CJ. Locus-Zoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, Finch N, Rutherford NJ, Crook RJ, Josephs KA, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Caselli RJ, Wszolek ZK, Uitti RJ, Feldman H, Hutton ML, Mackenzie IR, Graff-Radford NR, Dickson DW. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17(23):3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rauramaa T, Pikkarainen M, Englund E, Ince PG, Jellinger K, Paetau A, Alafuzoff I. Consensus recommendations on pathologic changes in the hippocampus: a postmortem multi-center inter-rater study. J Neuropathol Exp Neurol. 2013;72(6):452–461. doi: 10.1097/NEN.0b013e318292492a. [DOI] [PubMed] [Google Scholar]
- 63.Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, Parisi JE, Petersen RC, Graff-Radford NR, Younkin SG, Dickson DW, Rademakers R. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology. 2012;79(7):717–718. doi: 10.1212/WNL.0b013e318264e3ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169(4):1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, Cooper G, Mendiondo M, Danner DD, Van Eldik LJ, Caban-Holt A, Lovell MA, Kryscio RJ. University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res. 2012;9(6):724–733. doi: 10.2174/156720512801322591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scott WK, Grubber JM, Abou-Donia SM, Church TD, Saunders AM, Roses AD, Pericak-Vance MA, Conneally PM, Small GW, Haines JL. Further evidence linking late-onset Alzheimer disease with chromosome 12. JAMA J Am Med Assoc. 1999;281(6):513–514. doi: 10.1001/jama.281.6.513. [DOI] [PubMed] [Google Scholar]
- 67.Scurr I, Wilson L, Lees M, Robertson S, Kirk E, Turner A, Morton J, Kidd A, Shashi V, Stanley C, Berry M, Irvine AD, Goudie D, Turner C, Brewer C, Smithson S. Cantu syndrome: report of nine new cases and expansion of the clinical phenotype. Am J Med Genet Part A. 2011;155A(3):508–518. doi: 10.1002/ajmg.a.33885. [DOI] [PubMed] [Google Scholar]
- 68.Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81(2):133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- 69.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62(4):406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 70.Stouffer SA, Suchman EA, De Vinney LC, Star SA, Williams RM. The American Soldier: adjustment during army life. Princeton University Press: Princeton; 1949. [Google Scholar]
- 71.Team RC. R: a language and environment for statistical computing. 2013 http://wwwR-project.org/
- 72.Troncoso JC, Kawas CH, Chang CK, Folstein MF, Hedreen JC. Lack of association of the apoE4 allele with hippocampal sclerosis dementia. Neurosci Lett. 1996;204(1–2):138–140. doi: 10.1016/0304-3940(96)12331-4. [DOI] [PubMed] [Google Scholar]
- 73.Tschanz JT, Treiber K, Norton MC, Welsh-Bohmer KA, Toone L, Zandi PP, Szekely CA, Lyketsos C, Breitner JC. A population study of Alzheimer’s disease: findings from the Cache County Study on memory, health, and aging. Care Manag J. 2005;6(2):107–114. doi: 10.1891/cmaj.6.2.107. [DOI] [PubMed] [Google Scholar]
- 74.Tyas SL, Snowdon DA, Desrosiers MF, Riley KP, Markesbery WR. Healthy ageing in the Nun Study: definition and neuropathologic correlates. Age Ageing. 2007;36(6):650–655. doi: 10.1093/ageing/afm120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.van Bon BW, Gilissen C, Grange DK, Hennekam RC, Kayserili H, Engels H, Reutter H, Ostergaard JR, Morava E, Tsiakas K, Isidor B, Le Merrer M, Eser M, Wieskamp N, de Vries P, Steehouwer M, Veltman JA, Robertson SP, Brunner HG, de Vries BB, Hoischen A. Cantu syndrome is caused by mutations in ABCC9. Am J Hum Genet. 2012;90(6):1094–1101. doi: 10.1016/j.ajhg.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van Langenhove T, van der Zee J, Van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration–amyotrophic lateral sclerosis spectrum. Ann Med. 2012;44(8):817–828. doi: 10.3109/07853890.2012.665471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu J, Hu J, Chen YP, Takeo T, Suga S, Dechon J, Liu Q, Yang KC, St John PA, Hu G, Wang H, Wakui M. Iptakalim modulates ATP-sensitive K(+) channels in dopamine neurons from rat substantia nigra pars compacta. J Pharmacol Exp Ther. 2006;319(1):155–164. doi: 10.1124/jpet.106.106286. [DOI] [PubMed] [Google Scholar]
- 78.Ye B, Kroboth SL, Pu JL, Sims JJ, Aggarwal NT, McNally EM, Makielski JC, Shi NQ. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ Res. 2009;105(11):1083–1093. doi: 10.1161/CIRCRESAHA.109.195040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep. 2008;8(5):363–370. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 80.Zarow C, Weiner MW, Ellis WG, Chui HC. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav. 2012;2(4):435–442. doi: 10.1002/brb3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhong H, Prentice RL. Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics. 2008;9(4):621–634. doi: 10.1093/biostatistics/kxn001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou F, Yao HH, Wu JY, Ding JH, Sun T, Hu G. Opening of microglial K(ATP) channels inhibits rotenone-induced neuroinflammation. J Cell Mol Med. 2008;12(5A):1559–1570. doi: 10.1111/j.1582-4934.2007.00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.