

Abstract
The equilibrium properties of a HIV-1-protease precursor are studied by means of an efficient molecular dynamics scheme, which allows for the simulation of the folding of the protein monomers and their dimerization into an active form, and compare them with those of the mature protein. The model can reproduce several experimental observables, including the NMR conformation of the mature dimer, the calorimetry of the system, the effect of the precursor tail on the dimerization constant, the secondary chemical shifts of the monomer and the paramagnetic relaxation enhancement data associated with the conformations of the precursor. It is also found that, while the mature protein can dimerize in a single way, the precursor can populate several dimeric conformations in which the monomers are always native-like, but their binding can be non-native.
Introduction
A key step in the replication of the HIV-1 virus is the maturation of its aspartyl protease (PR) into an active dimeric form. The protease is expressed by the virus as part of a longer polyprotein, in which it can fold into a monomeric, inactive conformation1, 2. The occasional formation of transient active dimers is able to cleave the protease itself and eventually the other viral proteins into their mature form. The blocking of such a transient binding has been suggested as a strategy to inhibit viral replication3.
The minimal system which mimics the properties of the HIV-1 protease precursor was obtained building a construct (SFNFPR) which includes the protease and four residues of its N-terminal flanking sequence in the precursor4. Biochemical studies5 of the inactive D25N variant of SFNFPR indicate that the monomeric structure is identical to that found in the mature dimer, except for the active site (residues 25-27) and residues 3-6 at the N terminus. However, the dimerization constant increases from ∼10 nM of the mature dimer to ∼500 μM of SFNFPRD25N, making it largely monomeric under conditions which mimic cellular environment.
The goal of the present study is to understand the role of the precursor flanking sequence at atomic detail, making use of a computational model of the system and advanced computational algorithms. This problem is particularly hard because of the size of the system, which involves two chains of 103 residues each. For this reason it is not possible to use an explicit solvent description of the system and standard molecular dynamics (MD) tools. Up to date, the largest system studied by MD in explicit solvent to obtain equilibrium properties has been a monomeric 36-residue protein6.
To overcome this problem we made use of an implicit-solvent model of the protein, Medusa force field, and of discrete MD simulations. Medusa force field has proven efficient in folding small proteins to their crystallographic native state ab initio7, to reproduce equilibrium and kinetic properties of proteins up to 100 residues8 and to predict the thermodynamic effect of mutations on a large set of proteins 9. Discrete MD simulations 10 is an event-driven algorithm that allows to use time steps larger than traditional MD, and its speed scales as N log N, making it particularly attractive to study large molecular systems. The parallel version of discrete MD coupled with a replica-exchange algorithm allows studying efficiently the equilibrium properties of large molecular systems11.
The present system displays an additional degree of complexity with respect to the proteins studied in the past with the same model, namely its dimeric character. In fact, here the model has to reproduce two related but distinct phenomena: the folding of the two polypeptidic chain and the recognition between the two structured (or partially structured) molecules. The case of HIV PR is then a challenging test to evaluate the performance of the model.
Consequently, as a first step in the comprehension of the maturation of the HIV PR, we will validate the model to reproduce available experimental data, such as the structure of the mature dimer, the thermodynamics of the system and the NMR data describing the transient dimeric precursor. In particular, paramagnetic relaxation enhancement (PRE) data obtained for SFNFPR12 provide a sensitive atomic-scale test for the model. Moreover, we shall look for equilibrium structural features which hardly can be obtained with available experimental techniques.
Methods
Discrete MD method is an event driven simulation method using a discrete potential energy, relying on the calculation of a sequence of atomic collisions7, 11. The all atom protein model interacts through an implicit solvent force field Medusa, which takes into account van der Waals interaction, hydrogen bonds, electrostatics, dihedral and angular interactions and, in an effective fashion, the interactions mediated by the solvent. We make use of the extended version of the force field, improved from the point of view of long-range charge-charge interactions (electrostatic interactions) and sequence-dependent local backbone interactions 8.
The replica-exchange sampling scheme is used to significantly enhance conformational sampling by simulating several replicas of the system at different temperatures. For each system, we first carried out preliminary simulations using 8 replicas, from T=0.5 to T=0.65, and then 20 replicas from T=0.2 to T=0.65 (to 0.70 in the case of the monomer). Attempts to exchange neighbouring replicas are carried out every 1000 steps. Eventually we joined the two sets of simulations of each system, extracting the average energies by means of a weighted histogram algorithm 13.
The simulation box with the periodic boundary condition is taken to be 8 nm and 16 nm each side for the monomer and the WT dimmer/SFNFPR, respectively.
Analysis of the trajectories has been carried out with the help of the Gromacs tools14.
The Cluster analysis was performed with the algorithm described in ref. 15 with a cutoff of 0.7 nm.
Results
One of the main advantages of describing the dynamics of such a large protein as the HIV-1-PR dimer by discrete MD is the possibility of simulating its equilibrium properties. After approximately 3 months of simulation on 20 processors we obtained a good approximation of its equilibrium state (cf. Figs. S1 and S2 in the Supplementary Materials).
As a first control, we have to check that the mature dimer folds into the experimental native conformation. For this purpose, the minimum-energy conformations found in the simulation was compared with the minimized NMR structure 16. The comparison is displayed in Fig. 1 and the resulting RMSD is 0.46 nm, which decreases to 0.41 nm if the termini and the flap regions are not taken into account. No conformations with comparable energies but markedly different from the experimental native conformation were found (see Fig. S3).
Figure 1.
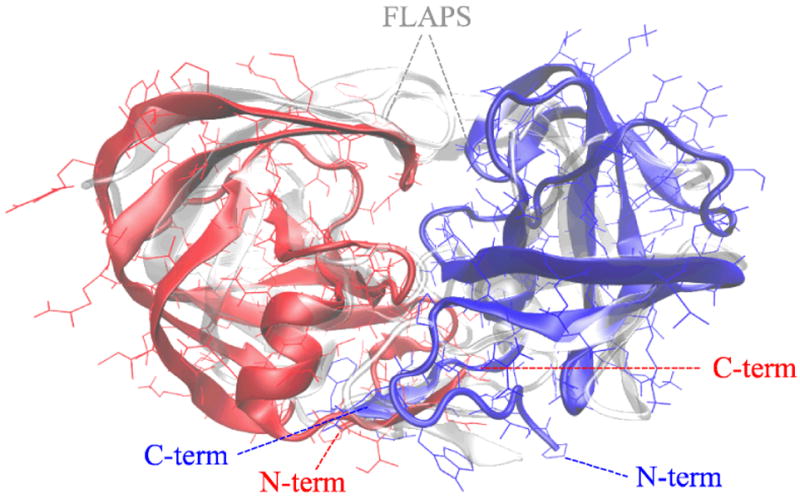
the minimum-energy conformation of the wild-type dimer, whose monomers are coloured in red and blue respectively, superimposed to the average NMR conformation, displayed as a transparent cartoon. The RMSD between simulated and NMR conformations is 0.26 nm, the main differences being at the termini and at the flap region.
The replica-exchange simulations described above can provide also all equilibrium properties of the protease. Figure 2 displays the heat capacities of the monomer, of the dimer and of the precursor protein, calculated with the weighted histogram method 13.
Figure 2.

Heat capacity of monomer (grey curve), mature dimer (red curve) and SFNFPR (blue curve) as a function of temperature, in units normalized to room temperature (see text).
All these systems display a single major peak corresponding to the unfolding of the monomer (cf. Fig. S4). Moreover dimerization enhances the two-state character of the unfolding transition of the monomer, resulting in a more peaked shape of the specific heat. Although the monomeric protein is stable at room temperature, its thermodynamics seems quite far from being two-state. These results are qualitatively in agreement with the results of differential scanning calorimetry described in ref. 17, although for different variants of the protease and under different solution conditions. In Fig. 2 and in the following figures the temperature scale is set in terms of room temperature, determined as that at which the unfolding free energy of the monomer is equal to its experimental value (cf. Fig. S5 and ref. 18).
The simulations were carried out in a cubic box of side 16 nm, resulting in a protein concentration [P]=400 μM. The binding probability pbound between the two monomers of the dimer and of the precursor are displayed in Fig. 3 as a function of temperature. The dimerization constant
Figure 3.
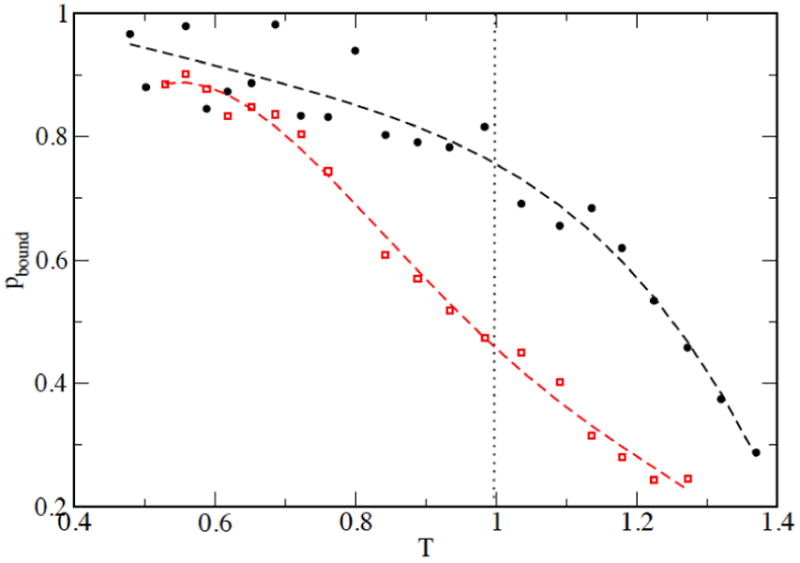
the probability that the two monomers in the dimer (black circles) and in the precursor (red squares) are bound, defined as the fractional number of contacts between CA atoms of the two monomers. A contact between CA atoms is defined if they are closer than 0.5 nm.
at room temperature results to be 93 μM for the dimer and 488 μM for the precursor. Consequently, the precursor dimer results to be considerably less stable than the mature dimer. This is in agreement with data available in the literature, which describe an increase of kD from 5 nM of the wild-type mature dimer to 680 nM of the TFP-p6pol precursor at pH 5 19, from 0.5 μM of the D25N mature dimer to >500 μM of SFNFPRD25N precursor at pH 5 5 and from 10 nM of the D25N mature dimer to 3-6 mM of the SFNFPRD25N precursor at pH 5.8 12.
The average structures at biological temperature of the monomer alone, of the monomer when complexed into the dimer, of the precursor in monomeric state and of the monomer of the precursor in dimeric state are displayed in the left panel of Fig. 4. Their RMSD to the minimized NMR conformation of the monomer are 0.50, 0.38, 0.41 and 0.6 nm, respectively (0.31, 0.32, 0.30 and 0.48 nm not counting the flaps and the termini). This agrees with the NMR data suggesting that both the monomeric and the precursor forms of the protease display an overall native structure 4, 5.
Figure 4.
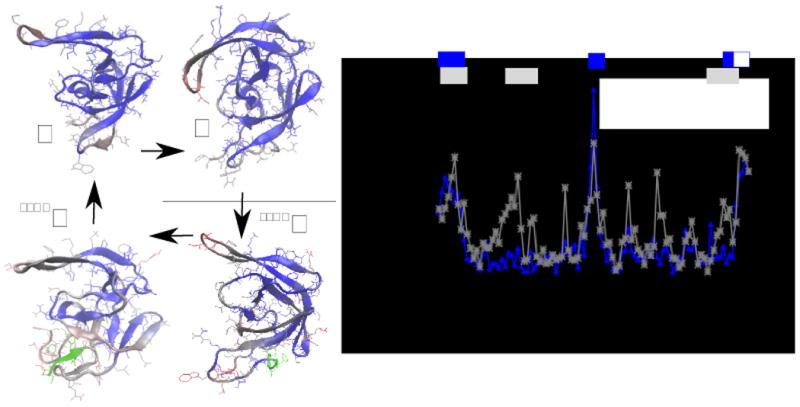
on the left panel, the conformational difference between the average structure of the monomer (M), of the dimer (D), of the monomeric precursor (SFNFM) and of the dimeric precursor (SFNFD). The colors indicate the deviation of the position of each monomer of a structure from that of the structure indicated by the arrow (blue is small, red is large). The green part is the N-terminal tail which characterizes the precursor. On the right panel, the average deviation of the position of residues in conformations M and D (blue line) and in conformations SFNFM and M (gray line). The gray and blue bars on the top of the plot indicate the difference in chemical shifts for the same molecules observed in refs. 5 and 4, respectively. The white square indicates that the monomeric protease was deprived of residues 96-99.
The structural difference between monomer and dimer, and between monomeric precursor and mature monomer are displayed in the right panel of Fig. 4, where they are compared with the secondary chemical shifts of CA and N atoms reported in 4, 5. The model predicts correctly large deviations between the monomer and the dimer in the termini and in the region 22-32, and between the monomeric precursor and the mature monomer in the termini and in the flap region. The comparison among the four forms of the protease (see also Fig. S6) suggests that all of them are different in the termini (residues 1-15, 95-99) and in the flap region (residues 44-56), while the monomeric precursor displays larger differences involving also the region 18-30 participating to the hydrophobic core.
The width of the thermal fluctuations of the residues of the protease is displayed in Fig. 5 (see also Fig. S7). As concluded in 4 from the absence of NOE signals in the Δ96-99 monomeric variant, the termini and the flap region of the monomer are highly mobile. The simulations suggest a large amount of conformational freedom in these regions also in the dimer and in the dimeric precursor. Concerning the flaps, the fluctuations observed in the simulations of the mature dimer are in agreement with the high temperature factors observed in the crystallographic structure of the protease 20.
Figure 5.
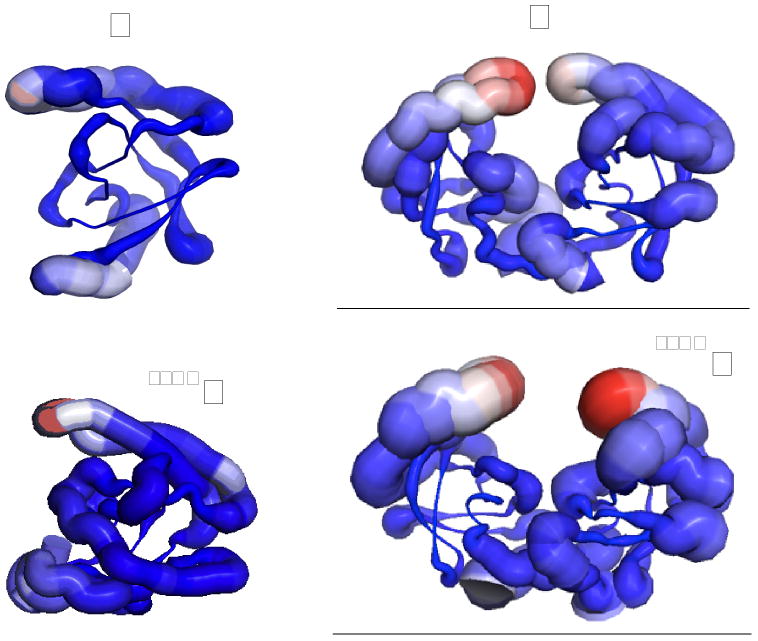
a cartoon representation of the thermal fluctuations of monomer (M), dimer (D) and monomeric precursor (SFNFM) and dimeric precursor (SFNFD) at room temperature. The thicker is the cartoon (also marked in red), the larger the fluctuations around the average structure.
An interesting experiment to characterize the precursor is reported in ref. 12, where PRE data between T12, E34 and V82 of a monomer of the precursor and the core residues (10 to 95) of the other monomer are measured. These data are in good agreement with the average value of 1/r6, normalized accordingly, displayed in Fig. 6. This agreement is definitely not trivial to obtain, since the same calculation carried out in the simulation of the mature dimer displays a completely different pattern (see Fig. S8).
Figure 6.

the experimental values obtained by PRE in ref. 12 between T12, E34, V82 of a subunit and the core residues (10 to 94) of the other subunit in SFNFPR (black circles), compared to the value of <r--6> calculated from the corresponding simulation at room temperature, normalized accordingly (blue symbols).
From the analysis of the simulated system one can get an insight into the mechanism associated with the dimerization of the precursor. We will focus our attention on the terminal segments, which embody most of the dimerization interface. Figure 7 displays the distribution of RMSD
Figure 7.
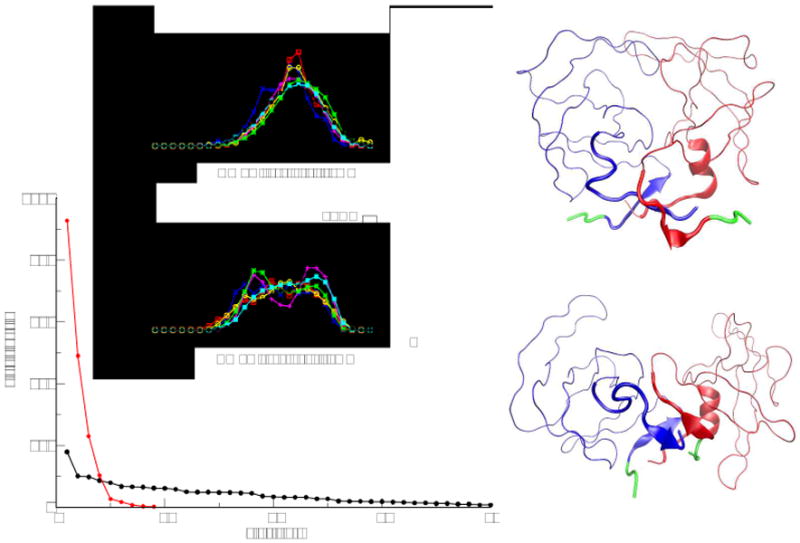
in the lower, left plot the distribution of cluster sizes obtained for the precursor (in black) and for the mature dimer (in red). Above, the distribution of RMSD of the N-terminal segments 1-5 of the dimer and of the precursor at increasing temperature, from 0.8Troom (blue curve) to 1.4Troom (in red). On the right, the average structures of the first two clusters found for the precursor.
The conformations of the precursor and of the mature dimers at room temperature have been further studied by cluster analysis. While the mature dimer displays few, highly populated clusters, essentially accounting for thermal fluctuations around the native conformation, the precursor displays a broad distribution of several clusters (see Fig. 7; cf. also Fig. S9). The most populated of them is native-like, with the precursor tails not interfering with the native binding between the monomers (see upper-right structure in Fig. 7). The other clusters still display native-like monomers, but with various mutual arrangement and displaying the precursor tail interacting with the termini of the protein. The lower-right panel of Fig. 7 shows the second cluster as an example.
To get a further insight in the set of conformations that the precursor can populate, we show in the upper-left panels of Fig. 7 the distribution of RMSD of the N-terminal segment 1-5 to its native conformation for the precursor and, as reference, for the mature dimer. Unlike the mature dimer, the N-terminus of the precursor displays a bimodal distribution, suggesting two possible states. The probability of the more non-native-like state increases with the temperature, suggesting that it is stabilized entropically, and thus is more disordered. Summing up, the precursor can bind the two native monomers in a complicated set of different mutual positions, according to the different ways the precursor tail interacts with the terminals of the protein.
Discussion
The maturation of the HIV-1 protease is particularly cumbersome to study because it involves time and length scales that span many orders of magnitude. Even restricting to a single step of the maturation process, that is the transient dimerization of the precursor protease, one has to face a combination of biophysical phenomena, such as the folding of the monomeric protein, the molecular recognition and binding, the dynamics of the unstructured terminal. Experimental techniques, typically involving NMR, can provide many valuable information on such a process, but are not able to provide a full atomistic picture of the system.
When studying the folding of small monomeric proteins or the binding of small molecules, molecular dynamics simulations carried out with standard explicit-solvent force fields can be of help, complementing the experimental data and giving an atomistic description of the system. But in the case of such a complicated phenomenon as the dimerization of HIV-1-PR precursor, the possibility of obtaining an equilibrium sampling of two separate chains of 103 amino acids each is computationally not feasible. Since now, the largest computational efforts to calculate the equilibrium properties of biomolecules involve monomeric chains of less than one fourth the size of the present system 6, or the binding of molecules of few tens of atoms to a protein 21.
A good trade-off between computational economy and a realistic description of the system comes from implicit-solvent protein models. Beside involving a much smaller number of atoms than the corresponding explicit-solvent representation of the system, these models offer a further advantage, allowing the use of computational algorithms 10 that are more efficient than plain molecular dynamics.
Of course, in order to rely on such simplified models it is necessary to validate them, a process that for explicit-solvent models involved the last two decades. In the case of the Medusa force field, several studies 7, 8 show its ability to identify correctly the native state of proteins up to one hundred residues, and to account for their thermodynamical properties. In the present work we increase our expectations and challenge the model to reproduce a more complicated phenomenon, involving the folding and dimerization of a large protein, perturbed by the interaction with the precursor tail.
The model can reproduce to a significant degree of approximation the NMR conformation of the mature dimer, the two-state thermodynamics of the dimer and the broadening of the calorimetric peak in the monomer, and the effect of the precursor tail on the dimerization constant. To a more atomistic level, the model can predict correctly the structural differences between monomer and dimer, and between monomer and precursor, as reported by secondary chemical shifts in NMR experiments. Also the PRE data describing the interaction of the monomers in the precursor are reproduced correctly.
The cluster analysis of the conformations visited by the precursor at room temperature indicates that multiple quaternary structures are possible, according to the different ways the precursor tail interacts with the termini of the protein, while the tertiary structure of the monomers remain overall native-like. This finding is qualitatively in agreement with the analysis carried out in ref. 12, where the authors back-calculate PRE data from rigid-body sampling of the relative positions of the native monomers. They conclude that a single conformation is not able to reproduce the experimental PRE, the average of 4 conformations is the minimum to obtain good results, while a larger number of conformations would be an over-fitting of the data. Our simulations indicate that indeed the quaternary structure of the precursor involves multiple states, but the number of these states (although depending on the precise definition) is larger than 4. Since many of these states correspond to an arrangement of the monomers quite different from the native one, we expect them to be enzymatically inactive, and consequently we do not expect a simple relation between the dimerization constant of the protein and its Michaelis constant.
Conclusions
Computational modelling of biological systems can be a very useful tool to complement and interpret experimental data, provided that a suitable model is chosen. Not all models are suitable to describe all phenomena. In the case of studying the equilibrium properties associated with the maturation of the HIV-1-PR precursor, explicit-solvent models, although very reliable for many purposes, are not useful. We have shown that a simpler model can reproduce a large amount of experimental data and help to understand at atomistic level the equilibrium properties of the protease precursor.
Supplementary Material
References
- 1.Louis JM, Nashed NT, Parris KD, Kimmel AR, Jerina DM. Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc Natl Acad Sci USA. 1994;91(17):7970–7974. doi: 10.1073/pnas.91.17.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis JM, Wondrak EM, Kimmel AR, Wingfield PT, Nashed NT. Proteolytic processing of HIV-1 protease precursor, kinetics and mechanism. J Biol Chem. 1999;274(33):23437–23442. doi: 10.1074/jbc.274.33.23437. [DOI] [PubMed] [Google Scholar]
- 3.Louis JM, Aniana A, Weber IT, Sayer JM. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc Natl Acad Sci USA. 2011;108(22):9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishima R, Torchia DA, Lynch SM, Gronenborn AM, Louis JM. Solution structure of the mature HIV-1 protease monomer: insight into the tertiary fold and stability of a precursor. J Biol Chem. 2003;278(44):43311–43319. doi: 10.1074/jbc.M307549200. [DOI] [PubMed] [Google Scholar]
- 5.Ishima R, Torchia DA, Louis JM. Mutational and structural studies aimed at characterizing the monomer of HIV-1 protease and its precursor. J Biol Chem. 2007;282(23):17190–17199. doi: 10.1074/jbc.M701304200. [DOI] [PubMed] [Google Scholar]
- 6.Piana S, Lindorff-Larsen K, Shaw DE. Protein folding kinetics and thermodynamics from atomistic simulation. Proc Natl Acad Sci USA. 2012;109(44):17845–17850. doi: 10.1073/pnas.1201811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding F, Tsao D, Nie H, Dokholyan NV. Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure. 2008;16(7):1010–1018. doi: 10.1016/j.str.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shirvanyants D, Ding F, Tsao D, Ramachandran S, Dokholyan NV. Discrete Molecular Dynamics: An Efficient And Versatile Simulation Method For Fine Protein Characterization. J Phys Chem B. 2012;116:8372–8382. doi: 10.1021/jp2114576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin S, Ding F, Dokholyan NV. Eris: an automated estimator of protein stability. Nat Methods. 2007;4(6):466–467. doi: 10.1038/nmeth0607-466. [DOI] [PubMed] [Google Scholar]
- 10.Dokholyan NV, Buldryev SV, Stanley HE, Shakhnovich EI. Discrete molecular dynamics studies of the folding of a protein-like model. Folding and Design. 1998;3:577–587. doi: 10.1016/S1359-0278(98)00072-8. [DOI] [PubMed] [Google Scholar]
- 11.Proctor EA, Ding F, Dokholyan NV. Discrete molecular dynamics. WIREs Comput Mol Sci. 2011;1(1):80–92. [Google Scholar]
- 12.Tang C, Louis JM, Aniana A, Suh JY, Clore GM. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455(7213):693–696. doi: 10.1038/nature07342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrenberg A, Swendsen R. Optimized Monte Carlo data analysis. Phys Rev Lett. 1989;63(12):1195–1198. doi: 10.1103/PhysRevLett.63.1195. [DOI] [PubMed] [Google Scholar]
- 14.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN. SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Research. 2010;38:W657–W661. doi: 10.1093/nar/gkq498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide Folding: When Simulation Meets Experiment. Angew Chem Int Ed. 1999;38(1-2):236–240. [Google Scholar]
- 16.Yamazaki T, Hinck AP, WANG YX, Nicholson LK, Torchia DA, Wingfield P, Stahl SJ, Kaufman JD, Chang CH, Domaille PJ, Lam PY. Three-dimensional solution structure of the HIV-1 protease complexed with DMP323, a novel cyclic urea-type inhibitor, determined by nuclear magnetic resonance spectroscopy. Protein Sci. 1996;5(3):495–506. doi: 10.1002/pro.5560050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayer JM, Liu F, Ishima R, Weber IT, Louis JM. Effect of the active site D25N mutation on the structure, stability, and ligand binding of the mature HIV-1 protease. J Biol Chem. 2008;283(19):13459–13470. doi: 10.1074/jbc.M708506200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noel AF, Bilsel O, Kundu A, Wu Y, Zitzewitz JA, Matthews CR. The folding free-energy surface of HIV-1 protease: insights into the thermodynamic basis for resistance to inhibitors. J Mol Biol. 2009;387(4):1002–1016. doi: 10.1016/j.jmb.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6(9):868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 20.Spinelli S, Liu QZ, Alzari PM, Hirel PH, Poljak RJ. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie. 1991;73(11):1391–1396. doi: 10.1016/0300-9084(91)90169-2. [DOI] [PubMed] [Google Scholar]
- 21.Limongelli V, Bonomi M, Parrinello M. Funnel metadynamics as accurate binding free-energy method. Proc Natl Acad Sci USA. 2013;110(16):6358–6363. doi: 10.1073/pnas.1303186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.