

Abstract
Arachidonic acid (ARA) undergoes enzyme-mediated oxidative metabolism, resulting in the formation of a number of biologically active metabolites. For over a century, these biochemical transformations have been the target of numerous pharmacological drugs for inflammation and pain. In particular, non-steroidal anti-inflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) selective inhibitors (coxibs) are widely used in the treatment of inflammation and pain. However, gastrointestinal (GI) and cardiovascular adverse effects of NSAIDs and coxibs, and recent findings demonstrating that there are significant risks from the disruption of oxylipin levels when pharmacologically inhibiting a single ARA cascade metabolic pathway, have led to studies involving the simultaneous inhibition of multiple pathways in ARA cascade. These studies suggest that multitarget inhibition represents a new and valuable option to enhance efficacy or reduce side-effects in the treatment of inflammation and pain. This review focuses on the crosstalk within the three pathways of the ARA cascade (cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450)), and summarizes the current and future approaches of multitarget inhibitors for the treatment of eicosanoid driven inflammation and pain.
Keywords: Cyclooxygenase (COX), lipoxygenase (LOX), cytochrome P450 (CYP), soluble epoxide hydrolase (sEH), multitarget inhibitors, dual inhibitors
1. INTRODUCTION
There are three major pathways associated with the arachidonic acid (ARA) cascade, cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways [1, 2]. Since the synthesis of acetylsalicylic acid (Aspirin®) as the first COX inhibitor [3], non-steroidal antiinflammatory drugs (NSAIDs) have become the most widely used pharmaceuticals to treat inflammation and pain world-wide [4]. NSAIDs act by blocking the action of the COX enzymes, COX-1 and COX-2, resulting in the reduction of pro-inflammatory ARA metabolites, and in particular, prostaglandin E2 (PGE2) [5]. COX-1 is reportedly involved in maintaining the homeostatic balance of COX metabolites and is responsible, in part, for normal gastrointestinal (GI) function [6]. Prostacyclin (PGI2) and PGE2 produced in the GI tract play important roles in modulating GI mucosal defense and repair [7]. Therefore, the use of NSAIDs is often limited by side effects emanating from disrupting the levels of these protective COX metabolites. Following the discovery of a second form of COX enzyme (COX-2) which is inducible by cytokines and growth factors [8–10], COX-2 selective inhibitors (coxibs) such as celecoxib (Celebrex®) and rofecoxib (Vioxx®) were subsequently developed in an effort to circumvent these problems [8, 11]. Coxibs are effective against inflammation and pain, with less risk of severe GI toxicity associated with conventional NSAIDs [12]. However, there are safety concerns with coxib use due to an increase in the risk of cardiovascular events associated with the imbalance of the PGI2 and thromboxane (TXA2) metabolite levels [13]. Inhibition of COX-1 reduces platelet-derived TXA2, an eicosanoid which functions as a vasoconstrictor and facilitates platelet aggregation [14]. PGI2 is a major metabolite of ARA in the heart and is associated with vasodilation and the prevention of platelet aggregation [15]. Selective inhibition of COX-2 affects the PGI2/TXA2 ratio to favor TXA2, increasing the risk of mortality from ischemic heart disease [16–18]. These findings were supported by several clinical trials involving coxibs including APPROVE, VIGOR, CLASS, and TARGET [17]. Recently, a metabolomic approach to understand the relationship between adverse cardiovascular events and the use of rofecoxib suggested that this drug acts, in part, through accumulation of 20-hydroxyeicosatetraenoic acid (20-HETE) which is a potent vasoconstrictor among ARA metabolites [19]. In addition, it is reported that chronic use of coxibs also increases GI side effects, albeit the risk is lower than NSAIDs [20, 21]. Treatment of inflammation and pain constitutes significant medical needs because more people are afflicted with these conditions than any other disease state. Thus, there is a growing demand for safer but efficacious NSAIDs or coxibs [4]. Emerging concepts and approaches for the treatment of inflammation and pain have moved towards simultaneously targeting multiple enzymes in the ARA cascade through combination therapy and multitarget inhibitors such as dual inhibitors with the aim of overcoming the risks involved in single enzyme or pathway inhibition.
2. THE ARA CASCADE
ARA, a twenty-carbon fatty acid, is one of the most abundant polyunsaturated fatty acids found in the phospholipid bilayer of cells involved in inflammatory responses [22]. Activation of phospholipase A2 (PLA2, generally cytoplasmic PLA2) [23] and other lipases in response to various stimuli, leads to release of ARA into the cellular milieu. ARA is then metabolized by three major enzymatic pathways, the COX, LOX, and CYP pathways [24]. The ARA metabolites of the COX enzymes, or prostanoids, are primarily pro-inflammatory mediators [25]. The eicosanoid metabolites of the LOX pathway include both pro-inflammatory leukotrienes (LTs) [26] and hydroxyeicosatetraenoic acids (HETEs) [27], as well as mediators of inflammation resolution, the lipoxins (LXs) [28]. The CYP hydroxylases generate the vasoconstrictor 20-HETE [29], and the CYP epoxygenases generate anti-inflammatory epoxyeicosatrienoic acids (EETs) [30]. The EETs are further metabolized by soluble epoxide hydrolase (sEH) to their corresponding diols, dihydroxyeicosatrienoic acids (DHETs), which are reported to have less biological activity [31] (Fig. 1).
Fig. 1.

ARA cascade.
Detailed discussions on each of these metabolic pathways are beyond the scope of this review. Therefore, the focus is on the major enzymes that are targeted in combination therapy by dual inhibitors or multitarget agents, and crosstalk among the ARA metabolizing pathways.
2.1. The COX Pathway
The COX pathway is well studied and has recently been summarized in several reviews [32–34]. It is well established that there are two isoforms of COX, COX-1 and COX-2 in mammals. While COX-1 is constitutively expressed in most mammalian cells under physiological conditions [35], the expression of COX-2 can be induced by pro-inflammatory stimuli such as cytokines, bacterial lipopolysaccharide (LPS), growth factors, and tumor-promoting agents [36, 37]. Recent evidence has shown that COX-2 is constitutively expressed in several tissues including spinal cord [38], brain [39] and kidney [40], and although the roles of constitutively expressed COX-2 are not fully understood, this expression pattern creates challenges in selectively targeting COX-2 for therapeutic purposes [41, 42]. The COX enzymes are dual function enzymes that initially transform ARA into unstable prostaglandin G2 (PGG2) via the COX function, and then into the more stable prostaglandin H2 (PGH2) via the peroxidase (POX) function. PGH2 is then converted to various bioactive prostanoids such as prostaglandins (PGD2, PGE2, PGF2α, and PGI2) and TXA2 by cell- and tissue-selective prostanoid synthases [41–43] (Fig. 2). Among these prostanoid synthases, PGE2 synthases (microsomal PGE synthase-1 (mPGES-1), membrane PGES-2 (mPGES-2), and cytosolic PGES (cPGES)) [44], have recently attracted a great deal of attention as potential targets in the treatment of inflammation and pain [45]. These enzymes are downstream of COX and therefore may not produce the same side-effects as the COX mediated disruption of PGI2 formation. Both mPGES-2 and cPGES are constitutively expressed enzymes, while mPGES-1 is inducible by inflammatory stimuli and primarily coupled with COX-2 [46, 47]. These findings have made mPGES-1 attractive as a potential target for inflammation and pain [45].
Fig. 2.

Prostaglandins, PGI2 and TXA2 from the COX pathway.
Biological Effects of Prostanoids
Prostanoids are autocrine and paracrine lipid mediators, which are widely distributed and involved in a broad range of physiological responses (reviewed in [6]). Typically these metabolites are high-affinity agonists of their respective receptors all of which are G protein-coupled receptors that modulate second messenger levels (e.g., Ca2+, cAMP and inositol phosphates) [48]. Since the initial studies of von Euler describing prostanoids in late 1936 [49], nine human prostanoid receptors have been isolated and cloned including two isoforms activated by PGD2 (DP1, DP2), four by PGE2 (EP1, EP2, EP3, EP4) and three by PGF2α, PGI2, and TXA2 (FP, IP and TP respectively). These receptors and their diverse functions explain much of the multiplicity of the biological effects of prostanoids [50]. The roles and functions of prostanoids vary in a context dependent manner in the body [25]. They have important roles in GI function, with PGE2, PGF2α and PGI2 protecting the gastric mucosa blood flow and stimulating mucus formation as well as bicarbonate secretion [51]. In the cardiovascular system, PGD2, PGE2 and PGI2 mediate vascular tone by eliciting vasodilation, whereas TXA2 is a potent vasoconstrictor [52]. In the airways, PGF2α acts as a bronchoconstrictor and PGE2 acts as a bronchodilator [53]. The role of PGs in inflammatory pain is well established [54]. Specifically PGE2 is one of the most painful endogenous substances known to man and mediates a number of inflammatory processes including redness, swelling, and pain [55]. Binding of PGs to prostanoid receptors sensitizes pain specific neurons in several distinct ways to stimulate pain. For example PGE2 acts on EP1 and EP2 receptors in the peripheral and central nociceptive systems respectively, and PGI2 acts on IP receptors in the peripheral nociceptive system [56]. PGE2 also acts on neurons and contributes to the systemic responses to inflammation such as fever, fatigue, and pain hypersensitivity [57]. These diverse and differential roles of prostanoids obviously require an intricate balance to maintain homeostasis and to avoid overacting inflammatory responses [58].
2.2. The LOX Pathway
There are three LOX isozymes, 5-, 12-, and 15-LOX, that have been identified in human which convert ARA into their respective pro-inflammatory hydroperoxyeicosatetraenoic acids (HPETEs), 5-, 12-, and 15-HPETE respectively [59]. In contrast to other LOX enzymes, 15-LOX also initiates the synthesis of lipoxins (LXs) which are involved in the resolution phase of inflammation [60]. Although 12-LOX and 15-LOX have been implicated in inflammatory diseases such as psoriasis [61] and arthrosclerosis [62], this review will focus on the role of 5-LOX and synthesis of LTs as targets for treating inflammation. 5-LOX is a unique LOX isozyme which utilizes two co-factors, Ca2+ and adenosine triphosphate (ATP) [63], and protein-protein interactions with five-LOX activating protein (FLAP) for catalytic activity [64]. The metabolism of ARA by 5-LOX first generates 5(S)-HPETE [65], and then this intermediate is either reduced to the corresponding alcohol, 5-HETE, or to the very short-lived epoxy-leukotriene, LTA4 [66]. Further systematic metabolism of LTA4 results in a series of LTs starting with the hydrolysis of its epoxide by LTA4 hydrolase (LTA4H) [67] to the corresponding diol, LTB4, or its metabolism to the cysteine-adduct LTC4 [68]. Other cysteinyl-LTs (Cys-LTs), LTD4 and LTE4, are sequentially formed from LTC3 (Fig. 3). Recently a detailed description of the synthesis and biological action of the LOX metabolites has been reviewed [69].
Fig. 3.

LTs derived from the LOX pathway.
Biological Effects of LTs
LTs are paracrine lipid mediators that play various roles in inflammation and immunological function through specific rhodopsin-like G protein-coupled receptors (reviewed in [70]). Four LT receptors including two cysteinyl LTs receptors (CysLT1 and CysLT2) and two LTs B4 receptors (BLT1 and BLT2) have been isolated and cloned [71–73]. LTB4 is a potent chemotactic agent for inflammatory cells including neutrophils, macrophages and eosinophils [74, 75]. These LTs increase leukocyte tissue infiltration and play an important role in immune reactions by enhancing the release of pro-inflammatory cytokines by macrophages and lymphocytes [76]. Cys-LTs such as LTC4, LTD4, and LTE4, are involved in immediate hypersensitivity reactions and are potent constrictors of airway smooth muscles leading to bronchoconstriction [26, 77]. Cys-LTs also increase vascular permeability leading to edema by contracting endothelial cells (EC) in the microvasculature. LTs also play an important role in GI mucus protection [78, 79]. In addition, they can interact with sensory nerve fibers, leading to changes in their excitability and enhanced release of tachykinins. Therefore LTs are important mediators of various inflammatory diseases and allergic disorders including asthma, rheumatoid arthritis, inflammatory bowel disease, ulcerative colitis, psoriasis and allergic rhinitis [80].
2.3. The Cytochrome P450/sEH Pathway
The role of mammalian CYPs in the oxidative metabolism of ARA was not uncovered until 1981 [81–85]. The CYP enzymes relevant to ARA metabolism include two distinct pathways: the ω-hydroxylases (CYP4A) which produce HETEs such as 20-HETE, and the epoxygenases (CYP2C and CYP2J) which produce the EETs [86]. Research into the biological activity of 20-HETE has shown this metabolite possesses potent vasoconstrictor activity [87]. The EETs have been suggested to be endothelium-derived hyperpolarizing factors (EDHFs) with vasodilatory properties [88], and have various biological activities in several inflammatory diseases [89, 90] and pain [91]. EETs are primarily synthesized in endothelial cells [92–96], and are further metabolized by sEH, to their corresponding and less active diols, DHETs (Fig. 4) [97, 98]. EETs not only decrease inflammation, but also decrease platelet aggregation in order to maintain vascular homeostasis [99]. Importantly, other fatty acids such as the ω-6 linoleic acid (LA) and the ω-3 α-linolenic acid (ALA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are oxidized by CYPs through mechanisms similar to those for ARA [100]. Recent work by Morisseau et al. showed these various epoxy-fatty acids (EpFAs) are efficiently metabolized by sEH to the corresponding diols. The epoxides of DHA have demonstrated to be potently anti-hyperalgesic, although their other biological actions remain unclear [101]. Potential roles of EpFAs in pain are well-described in recent review [102].
Fig. 4.

Epoxyeicosatrienoic acids and 20-HETE from the CYP450 pathways.
Biological Effects of Epoxyeicosatrienoic Acids
The CYP epoxygenases convert ARA to four EET regioisomers, 5,6-, 8,9-, 11,12-, and 14,15-EETs, that function as autocrine and paracrine mediators [103]. The presence of a receptor(s) for EETs has been suggested [48], but membrane receptor(s) or binding protein(s) have yet to be reported. EETs have been shown to exhibit anti-inflammatory effects on the endothelium by inhibiting activity of IKK and TNF-α, thus attenuating cytokine-induced NF-κB activation [93]. This, in turn, suppresses the expression of adhesion molecules such as vascular cell-adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), and E-selectin [93, 104–106]. Inhibition of sEH by small molecules has been shown to reduce inflammatory responses by stabilizing the blood levels of EpFAs including EETs, and indirectly reduce the expression level of COX-2, 5-LOX, iNOS, and VCAM-1 in LPS-induced mouse models of systemic inflammation [107, 108]. These findings are consistent with the opinion that EETs prohibit amplification of an inflammatory event by preventing nuclear translocation of NF-κB, and thus its subsequent transcriptional activity [93, 106]. Animal models of inflammatory pain have shown that exogenous administration of EETs or treatment with sEH inhibitors, results in antihyperalgesic responses [91, 109]. While some types of pain respond well to treatment with opioids, NSAIDs and coxibs, treatment of neuropathic pain including diabetic neuropathy remains a challenge because of side effects and low efficacy of current treatments [110, 111]. Therefore the evidence of sEH inhibitor mediated antihyperalgesia in a diabetic neuropathy model offers an important new approach in meeting this challenge. The biological roles of EETs in inflammatory disease and pain is extensive [112] and in the following articles further information about EETs and specific disease can be found: vascular inflammation [113], pulmonary hypertension [114, 115], metabolic syndrome [116, 117], renal inflammation [118, 119], cardiovascular diseases [120–122], cancer [123], pain [112, 124]. In addition, mono-epoxides of EPA and DHA also possess antihyperalgesic properties [101] and their roles in pain relief have been reviewed [112].
3. CROSSTALK BETWEEN PATHWAYS IN THE ARA CASCADE
It is clear that bioactive lipid mediators from ARA metabolism evoke potent inflammatory and anti-inflammatory responses. Therefore, an intricate communication between the COX, LOX, and CYP pathways would be required to regulate inflammation and inflammation resolution. This communication, or crosstalk, between pathways is far from being well understood, but recent technologies such as lipid profile analysis has enabled the ability to examine changes in the ARA metabolome when inhibiting one or more of the ARA pathways. Coupling this type of technology with a better understanding of the role of certain metabolites in human disease will greatly increase the ability to predict potential risks from inhibiting enzymes in the ARA cascade. The next section will discuss recent findings in the interactions between the multiple pathways.
3.1. Crosstalk Between COX and LOX Pathways
It has been theorized that inhibition of a single pathway in the ARA cascade may lead to the shunting of ARA into the other untargeted pathways within the cascade. For example, inhibiting COX pathway can shunt the metabolism of ARA towards LOX pathway, resulting in an increase synthesis of LTs, and thus potentially diminishing the beneficial effects of reducing prostanoid synthesis [125, 126]. While this theory is currently viewed as an overly simplistic contributor to biological effects, such an event was observed in the case of aspirin-induced asthma, where the inhibition of the COX pathway resulted in a significant increase in the 5-LOX product, LTE4 [127]. Recently, using an LPS-challenged murine model, Liu et al. demonstrated that inhibition of 5-LOX not only reduced both 5-HETE and 15-HETE, but also significantly decreased both PGE2 and TXB2 in the COX metabolites [128]. Cumulatively, these data demonstrate that communication exists not only within the LOX pathway, but between LOX and COX pathways, and this crosstalk is bidirectional. It is unclear if these responses are due to effects on the transcriptional regulation of the associated enzymes, or if this is a result from direct effects on enzyme activity. However, it has been shown that LOX metabolites of ARA and LA can differentially affect the substrate selectivity [129] and reaction mechanism [130] of the LOX enzymes through allosteric regulation. This suggests a direct protein/metabolite interaction may regulate crosstalk within the ARA cascade, and that metabolite interactions may extend into other pathways such LA and ω-3 fatty acids metabolizing cascades.
3.2. Crosstalk Between COX and CYP/sEH Pathways
In addition to the COX/LOX pathway crosstalk, Kozak et al. showed that 11,12-EET, a CYP monooxygenase-derived ARA metabolite, suppresses production of PGE2 in monocytes through modulating COX-2 activity [131], and Schmelzer et al. demonstrated in vivo that co-inhibition of both the COX pathway and CYP pathway (via sEH inhibition) elicited an additive response in alleviating pain [132]. In the latter experiment, there was an observed reduction in the COX-2 protein expression levels and a dramatic shift in the oxylipin metabolomic profile [107]. This work demonstrated that crosstalk within the ARA cascade can, in part, result from effects on transcriptional regulation of proteins involved in ARA metabolism. It was proposed in this work, that increasing the anti-inflammatory EETs level by sEH inhibition, while simultaneously decreasing the pro-inflammatory PGE2 level by the reduction in COX-2 expression, synergistically affected the inflammatory signaling and the associated pain response. It should be noted that this synergistic effect was obtained by a combination of sEH inhibitor and a low-dose of NSAIDs or coxibs that are not effective doses when used alone. These findings are significant in that incidences for the cardiovascular adverse effects of NSAIDs or coxibs are typically higher with chronic high-dose treatment [133]. In addition, the inhibition of sEH alone or in combination with NSAIDs or coxibs did not disrupt the relative ratio of PGI2 to TXA2 in the plasma of LPS-challenged mice. These data indicated that a combination therapy involving sEH and COX-2 inhibition may result in the desired decrease in inflammatory eicosanoids such as PGD2 and PGE2 without the potential cardiovascular risks from the imbalance of the TXA2-to-PGI2 ratio.
3.3. Crosstalk Between LOX and CYP/sEH Pathways
Recent evidence now links the LOX and CYP pathway [134], closing the ARA cascade-feedback loop. In this work, Liu et al. demonstrated that inhibition of sEH in a LPS-challenged murine model significantly decreased the plasma DHETs and TXB2, while unexpectedly reducing two LOX metabolites, 5-HETE and 15-HETE, in plasma [128]. These results suggest that the CYP-generated EETs and their metabolizing enzyme, sEH, are intricately connected to both the LOX and COX pathways. Interestingly, oral administration of acetylsalicylic acid significantly lowered plasma levels of metabolites from all three pathways of the ARA cascade. It is clear from these results that there is a complex mechanism of interaction between the distinct pathways of the ARA cascade, and therefore, disturbances in any one of these pathways may lead to undesirable consequences from the over-production or under-production of critical lipid signaling molecules [115].
4. RATIONALLY DESIGNED MULTITARGET AGENTS
Because selectively blocking one of the pathways in the ARA cascade in most cases also causes the production of undesirable or unexpected metabolites, new approaches have been developed to combine therapies to modulate multiple pathway targets simultaneously [135–137]. However, administering two drugs in combination therapy raises safety concerns and presents unique challenges for optimizing dosage. For example, it is not safe to assume that two drugs with relatively high individual safety profiles will be safe when co-administered [138]. Co-administration treatment requires extensive investigations into optimal dose regiments including safety studies, a complex dosage ranging investigation, and drug-drug interaction analysis, all of which may significantly raise the practical cost and complexity of developing combination therapies [139]. During the drug development process, predicting pharmacodynamic and pharmacokinetic relationships is substantially less complex if multitarget inhibition is obtained from a single agent rather than from combination therapies (co-administration). In addition, improvement of patient compliance is expected with these multitarget inhibitors as fixed-dose drug combinations [140]. For these reasons, interest has grown in designed multiple ligands (also known as DMLs) [135, 141] with the following aims: 1) to enhance drug efficacy, 2) to improve drug safety by acting specifically on multiple targets, 3) to amend patient compliance, and 4) to eliminate the challenges of formulation problems and drug-drug interactions in coadministrative therapies [142]. This review article will focus on current research on DMLs as dual inhibitors which target two or more enzymes in the pathways in ARA cascade.
4.1. COX/5-LOX Dual Inhibitors
Dual inhibition of COX and LOX pathways by a single entity has been attracting attention in academia and industry. Many of the COX/LOX dual inhibitors developed have focused on inhibition of COX and specifically 5-LOX. They can be classified into three classes based on the type of 5-LOX inhibition: redox inhibitors, iron chelators, and nonredox/ non-chelators.
4.1.1. Redox Inhibitors
Benoxaprofen which is a radical scavenger is the first commercially available COX-2/5-LOX inhibitor. However, it has been discontinued due to reports of causing photosensitivity, effects on nail and hair growth, and liver and kidney failure. These side-effects were not proposed to be associated with the inhibition of their target enzymes but remain significant obstacles to their clinical use [143, 144].
Di-tert-Butylphenols
This class of di-tert-butylphenol derivatives is the most extensively studied group of dual inhibitors of COX and 5-LOX enzymes. It was found that naturally occurring diarylheptanoids such as curcumin or yakuchinones exhibited COX and LOX inhibition in vitro [145], but these compounds failed to demonstrate inhibition of LTB4 biosynthesis or anti-inflammatory activity in vivo [146]. Therefore, in an effort to improve both the in vitro and in vivo activities of these diarylheptanoids, various analogues containing similar structural 2,6-di-tert-butylphenols have been developed as dual inhibitors (Fig. 5). Structure-activity relationship (SAR) studies have indicated that 2,6-di-tert-butyl-1-hydroxybenzene substituted in the fourth position is optimum for dual activity. The substituents are either five- or six-membered heterocycles or chains. The phenol moiety possesses the antioxidant and radical scavenging properties which is considered to be responsible for anti-inflammatory potency and low ulcerogenic potential. The therapeutic index of these compounds (ratio of anti-inflammatory efficacy to GI safety profile) has uniformly been shown to be superior to that of conventional NSAIDs [147]. For these reasons, a variety of COX/5-LOX dual inhibitors containing such a phenol have been developed including darbufelone (CI-1004) [148], S-2474 [149], and tebufelone [150]. Among them, S-2474 also displays cytokine-modulating properties and is currently being evaluated in clinical trials for arthritis [147–149, 151–153].
Fig. 5.
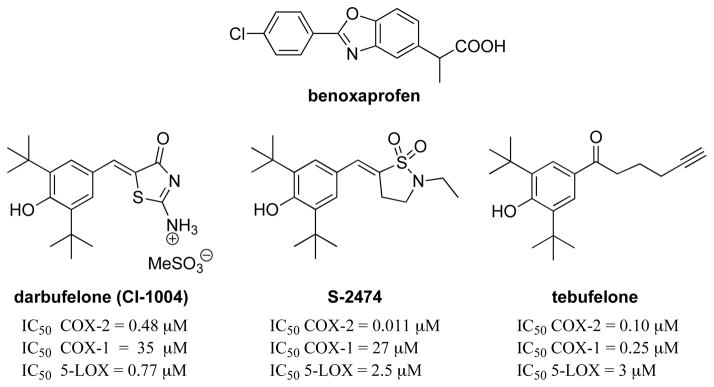
Structures of redox inhibitors.
Pyrazoline Derivatives
BW-755C and phenidone are COX/5-LOX dual inhibitors functioning as one-electron reducing agents (see Fig. 6). These compounds were originally developed as antioxidant 5-LOX inhibitors and later demonstrated to possess inhibitory activity towards COX isozymes [143, 154, 155]. Phenidone significantly decreased eosinophils (ED50 = 15 mg/kg) in bronchoalveolar lavage (BAL) fluid of guinea pigs sensitized to ovalbumin and those challenged with aerosoilized antigen, while indomethacin had no effect in eosinophils accumulation [156].
Fig. 6.

Structures of pyrazoline derivatives.
4.1.2. Iron Chelators
Hydroxamic Acids
Tepoxalin (Zubrin®) is a COX-2/5-LOX dual inhibitor which exhibited potent anti-inflammatory activity with excellent gastric protection (Fig. 7). It is approved for veterinary use only (in canines) [157]. Oral administration in humans revealed that tepoxalin inhibits whole blood COX, LOX, and platelet function [158] and its further development has been stopped due to its hepatotoxicity [159].
Fig. 7.

Structure of tepoxalin.
Thiophene Derivatives
RWJ 63556 (Fig. 8), was developed as 5-LOX inhibitor [160] that was subsequently found to inhibit COX-2 selectively over COX-1 [161]. This compound displayed significant anti-inflammatory activity in carrageenan-induced inflammation in canines [160] and inhibited the development of neurogenic edema induced by saphenous nerve stimulation [162]. L-652,343 was demonstrated to inhibit COX and 5-LOX in vitro and was efficacious in acute and chronic inflammation models in vivo [163, 164]. However, L-652,343 failed to show inhibition of 5-LOX in vivo in human skin [165] or LTB4 production ex vivo using stimulated human whole blood [166].
Fig. 8.
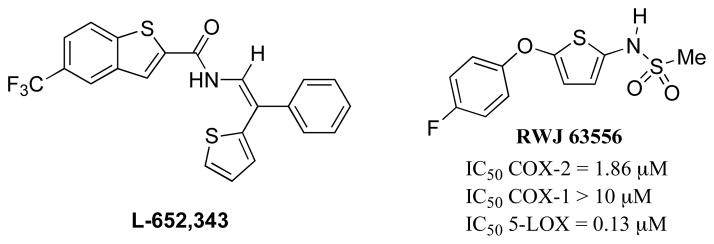
Structures of thiophene based dual inhibitors.
Pyridone Derivatives
Chowdhury et al. developed a COX-2/5-LOX dual inhibitor such as 1 in (Fig. 9) by replacing the tolyl group in celecoxib with the N-difluoromethyl-1,2-dihydropyrid-2-one moiety, which exhibited good anti-inflammatory activity (ED50 = 27.7 mg/kg p.o.) and compared favorably with reference drugs such as celecoxib (ED50 = 10.8 mg/kg p.o.) and ibuprofen (ED50 = 67.4 mg/kg p.o.) [167]. The same strategy has also been applied to NSAIDs [168, 169].
Fig. 9.

Structure of pyridone based dual inhibitor.
4.1.3. Non-Redox, Non-Chelators
Due to the non-selective action of redox and chelating inhibitors which cause undesirable side effects, there was an effort to design a new class of small molecule inhibitors without these actions. Therefore the following compounds are discovered or developed as new class of COX/LOX dual inhibitors.
DHDMBFs
7-tert-Butyl-2,3-dihydro-3,3-dimethylbenzofurans (DHDMBF) was originally found as a metabolite of tebufelone. Interestingly, this compound lacks the anti-oxidant phenolic moiety, but selectively inhibits both 5-LOX and COX-2 enzymes and displays anti-inflammatory activity equivalent to tebufelone in the rat carrageenan paw edema assay [147]. Among DHMBF derivatives, PGV-20229 is of particular interest because it showed excellent gastric safety and was active in a rat arthritis model with an ED50 of 6.6 mg/kg [147]. The acute ulcerogenic dose (UD50) was much higher, with no GI lesions observed at oral doses up to 1000 mg/kg. In addition, in a refed rat antral damage model, the UD50 for PGV-20229 was greater than naproxen and nabumetone and no GI damage was seen in 13-day studies in dogs and rats at oral doses up to 200 mg/kg/day. It has been demonstrated that a large variety of substituents (R) are compatible at the 5 position in DHDMBF (see the general structure in Fig. 10) retaining the anti-inflammatory and analgesic activities [152, 153].
Fig. 10.

Structures of DHDMBFs.
Structural Modification of NSAIDs
In the attempt to find potent COX/LOX dual inhibitors, structural modifications have been performed on conventional NSAIDs (Fig. 11). Tenidap, a COX/5-LOX inhibitor and cytokine modulator, is more effective in the clinical treatment of rheumatic arthritis than diclofenac [170]. However, this compound has been withdrawn from the market due to liver and kidney toxicity attributed to its reactive oxidative metabolites with a thiophene moiety [171–173]. To avoid these toxic side effects, a series of analogues including compound 2 have been developed replacing the thiophene ring in tenidap. These compounds showed comparable or stronger anti-inflammatory and analgesic activities, and better gastric tolerance in vivo than tenidap [174].
Fig. 11.

Structures of NSAID based COX/5-LOX dual inhibitors.
Structural Modification of Coxibs
The combination of a COX-2 pharmacophore and a 5-LOX pharmacophore led to potent selective COX-2/5-LOX inhibitors (Fig. 12). For example, compound 3 is a combination of the pyrazole-based tricyclic moiety found in celecoxib (Pfizer) and the 4-(3-fluoro-5-oxy)phenyl-4-methoxy tetrahydropyran from the 5-LOX inhibitor ZD-2138 (Zeneca) [175]. This dual inhibitor reduced ARA-induced ear edema by i.v. and oral administration. Several others in this class of COX-2/5-LOX dual inhibitors have also developed using similar approach such as compounds 4 (possessing a p-SO2Me COX-2 pharmacophore) [176] and 5 (possessing a 3,4-diaryl-2(5H)furanone COX-2 pharmacophore as in Vioxx®) [177].
Fig. 12.

Structures of coxib based COX-2/5-LOX dual inhibitors.
Pyrrolizine-Based Dual Inhibitors
Licofelone [178, 179] (ML 3000, 2-(6-(4-chlorophenyl)-2,2-dimethyl-7-phenyl-2,3-dihydro-1H-pyrrazolin-5-yl] acetic acid) has shown anti-inflammatory, analgesic and anti-asthmatic effects in several animal models (Fig. 13) [180]. Licofelone is one of the most promising COX/5-LOX inhibitors for human therapy and successfully finished phase III clinical trial for the treatment of osteoarthritis (OA) [181]. Notably, it possesses superior GI protective effect by decreasing gastrotoxic LTs through 5-LOX inhibition [182]. In addition, it exhibits protective anti-thrombotic activity due to its inhibition of COX-1 [183]. This is an excellent example demonstrating that simultaneous inhibition of COX-1, COX-2, and 5-LOX can circumvent the side effects associated with the individual inhibition of these enzymes. Recent study [184] showed that licofelone might suppress PGE2 formation by inhibiting mPGES-1 [185], and not through COX-2 inhibition.
Fig. 13.

Structure of pyrrolizine based COX/5-LOX dual inhibitor.
4.2. COX/sEH DUAL INHIBITORS
Recent work evaluating the co-inhibition of COX-2 and sEH has led to the development of COX-2/sEH dual inhibitors (Fig. 14) [186]. The potencies of these compounds were evaluated using recombinant enzymes assays in vitro and the efficacy of the most promising candidate was evaluated using a LPS induced rat pain model in vivo. The COX-2/sEH dual inhibitor by s.c. injection exhibited anti-allodynic activity that is more efficacious than the same dose of either a celecoxib or a sEH inhibitor (t-AUCB) alone, or the coadministration of sEH inhibitor with celecoxib. This work demonstrated that the use of a dual inhibitor as a DML was more efficacious than combination therapy in a rat pain model. Use of COX-2/sEH dual inhibitors could be an alternative approach for treating inflammation and pain compared to well-studied COX/5-LOX dual inhibitors.
Fig. 14.
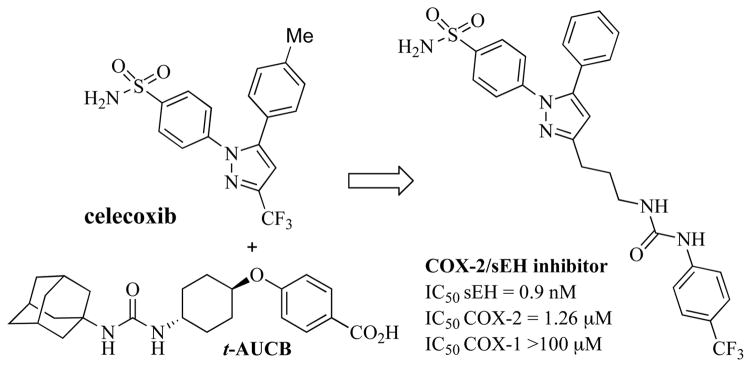
Structure of COX-2/sEH dual inhibitor.
4.3. LOX/sEH Dual Inhibitors
5-LOX/sEH dual inhibitors have been recently discovered by dual-target virtual screening using an in silico approach [187]. Among the eighty compounds selected by the virtual screening, thirty six compounds have been tested with in vitro enzyme assays. One compound has inhibited both 5-LOX and sEH enzymes at a marginal concentration (Fig. 15). Although no biology is associated with 5-LOX/sEH dual inhibitors, further development is warranted based on elevation of LOX products 5-HETE and 15-HETE with long-term sEH inhibition [134].
Fig. 15.
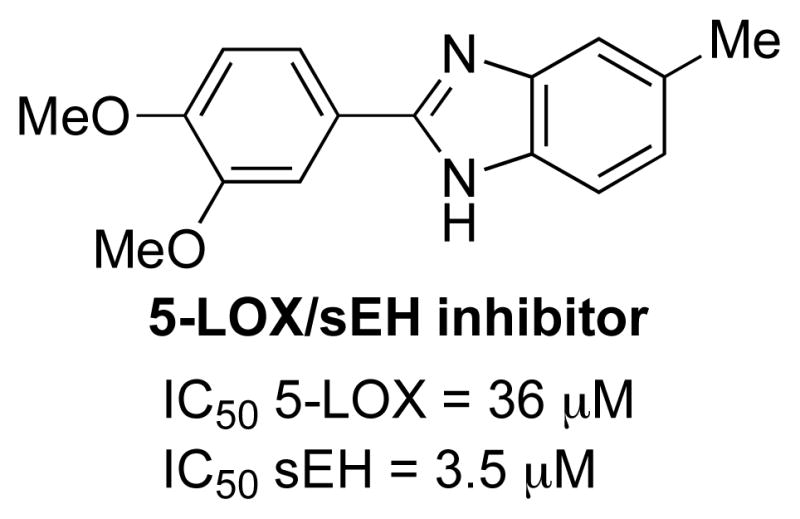
Structure of 5-LOX/sEH dual inhibitor.
4.4. Dual Inhibitors Targeting Enzymes Downstream in the COX or LOX Pathways
In addition to targeting 5-LOX and COX enzymes, a number of alternative multitarget approaches which focus on enzymes downstream of LOX and COX pathways have also been reported. The limitations of NSAIDs or coxibs use are derived from on-target side-effects such as GI injury, renal irritation, cardiovascular events, and off-target side effects such as an increase of LT formation. Therefore a new approach has been developed that targets enzymes downstream in the COX or LOX pathway to minimize potential on-target side-effects.
5-LOX and mPGES-1 Dual Inhibitors
mPGES-1 is one of the most attractive targets for this new approach. As described in section 2.1, unlike other PGE2 synthases, mPGES-1 is inducible by various inflammatory stimuli and is primarily coupled to COX-2 expression [46, 47]. This synthase is a PGE2-forming enzyme downstream of COX in the pathway which has the potential to selectively affect PGE2 and not other prostaglandins or TXA2 synthesis. Recent evidence demonstrated that mPGES-1 knockout mice did not display impaired cardiovascular function [188]. In addition, licofelone (section 4.1.3.), the most promising COX/5-LOX dual inhibitor, is likely suppressing the formation of PGE2 by inhibiting mPGES-1 and not through COX-2 inhibition [184]. Based on these findings several arylpyrrolizines derivatives have been developed as 5-LOX and mPGES-1 dual inhibitors such as 6 (Fig. 16) [189]. In addition, pirinixic acid 7 [190] and 2-mercaptohexanoic acid 8 [191] derivatives have been reported as 5-LOX/mPGES-1 dual inhibitors. In comparison with indomethacin (5 mg/kg), YS121 (1.5 mg/kg) was as efficient in reducing exudates formation and leukocyte infiltration with reduced pleural levels of PGE2 and LTB4 in the carrageenan-induced rat pleurisy model [192]. Recently virtual screening using a comparative model of the human 5-LOX led to the discovery of new 5-LOX/mPGES-1 dual inhibitors 9 and 10 possessing novel scaffolds [193].
Fig. 16.

Structures of 5-LOX/mPGES-1 dual inhibitors.
In addition to targeting mPGES-1, downstream enzymes in the LOX or COX pathways such as LTA4H and TXA2 synthase are also being investigated as new approaches for developing dual inhibitors acting in the ARA cascade.
COX-2 and LTA4H Dual Inhibitors
Recently Chen et al. reported dual inhibitors that target COX and the downstream leukotriene metabolizing enzyme, LTA4H, an epoxide hydrolase that catalyzes the conversion of LTA4 into LTB4 (Fig. 17) [194]. These dual inhibitors were developed by combining a COX-2 selective inhibitor nimesulide with an LTA4H inhibitor, 1-(2-(4-phenoxy phenoxy) ethyl)pyrrolidine, however no in vivo efficacy was reported [195].
Fig. 17.

Structure of COX-2/LTA4H and 5-LOX/TXA2 synthase dual inhibitors.
5-LOX and TXA2 Synthase Dual Inhibitors
Hibi et al. reported dual inhibitors such as E3040 targeting both 5-LOX and TXA2 synthase (Fig. 17). E3040 inhibited the production of LTB4 and TXB2, but not PGE2, and was efficacious at a dose of 100 mg/kg p.o. in the induced chronic colitis model compared to sulfasalazine (500 mg/kg) [196]. E6700, having better absorption compared to E3040, was under clinical trial for asthma treatment [197].
4.5. Dual Inhibitors Targeting Enzymes Downstream in the CYP Pathway and Enzymes Outside ARA Cascade
Extensive research on the benefits of therapeutic application of EETs has triggered development of dual inhibitors that include sEH inhibition. This approach is not limited to the enzymes in the arachidonic cascade. Examples here outline the future direction of valuable dual inhibitors for inflammation and pain.
sEH/11β-HSD1 Dual Inhibitors
GlaxoSmithKline (GSK) reported dual inhibitors for sEH and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzymes. 11β-HSD1 converts the inactive glucocorticoids (e.g. cortisone) to the active glucocorticoids (e.g. cortisol) (Fig. 18) [198]. Cortisone injections have been a proven solution for short-term relief of pain associated with swelling from inflammation of a joint, tendon, or bursa. However, their use is restricted due to adverse side effects. Thus, inhibition of sEH in concert with 11β-HSD1 is predicted to yield favorable results for the treatment of diseases mediated by the sEH or 11β-HSD1 [199].
Fig. 18.

Structures of 11β-HSD1/sEH dual inhibitors and sorafenib.
Sorafenib
In a recent study which examined the structural similarities of the FDA-approved anti-cancer drug sorafenib (Nexavar ®) and sEH inhibitors, Liu et al. observed that sorafenib possesses potent in vitro and in vivo sEH inhibitory activity (IC50 = 12 nM) in addition to multi-kinase inhibition (Fig. 18) [200]. Therefore, it has been suggested that sEH inhibition might ameliorate the toxicity of sorafenib during treatment through demonstrated anti-inflammatory and antihypertensive effects. However, the contribution of sEH inhibition on the efficacy of sorafenib has yet to be fully described.
5. CONCLUSION
Inflammation and pain are extremely complex biological processes that involve a great number of lipid mediators providing a theoretically wide range of possible targets for pharmacological treatment. Of the current available drugs that inhibit a single branch of the ARA cascade, inhibition of the COX pathway with NSAIDs is by far the most commonly used approach in the treatment of inflammation and inflammatory pain. This drug class, as well as the second generation coxibs, is not without serious GI and cardiovascular risks which stem from an imbalance in homeostatic eicosanoid metabolite levels. In addition, it is clear that inhibiting a single pathway can affect the metabolite output of the other pathways and crosstalk. This has led to a significant improvement in our understanding of the on-target adverse effect risks of COX inhibition [201]. However these serious side-effects from inhibiting either of the COX enzymes remain and the risks are not dogmatically separated as were previously thought [202]. While attempts to mitigate side-effects based on this historical dogma drove the exploration of dual COX/LOX inhibition, little success has come from this approach in the past decades. Several of these compounds, especially COX/5-LOX inhibitors, have been extensively studied and exhibited significant increases in efficacy both in vitro and in vivo without the undesired on-target side-effects associated with single pathway inhibition. However the off-target toxicity and adverse events limited many of these early dual inhibitors. Many of these initial dual inhibitors or multitarget drugs were not rationally designed; rather were subsequently found to have activity towards unintended targets. For example, compounds possessing anti-oxidant or iron-chelating properties originally developed as 5-LOX inhibitors, also possessed COX inhibitory activity due to their non-selective inhibition which eventually resulted in off-target adverse effects. Therefore the need persists for safer and effective multitarget agents by rational design. Recent advances such as the recent X-ray crystal structure of human 5-LOX [203] and computer-aided drug design (CADD), will allow medicinal chemists to rationally design new therapies through virtual screening using molecular docking, ligand-based and structure-based approaches. In addition, emerging roles of CYP metabolites in inflammation, pain, and other diseases will expand the scope of multitarget agents in ARA cascade. Dual inhibitors that target sEH and COX enzymes are an example of such a novel approach that holds great promise for treating pain and inflammation. This approach was fundamentally different from the previously developed COX/LOX dual inhibitors since the object was not to reduce undesirable metabolites, but to increase the lifetime of the CYP pathway metabolites (e.g., EETs) through sEH inhibition. The beneficial effects of the stabilized EETs allow dose-sparing of COX inhibitors, thereby potentially attenuating GI or pro-thrombic side-effects. Although sEH inhibitors share some of the same indications as coxibs, the synergy observed in lowering prostaglandins with combination therapy [132] and in antihyperalgesia with the dual inhibitor [186] merits inhibiting both target enzymes. Another promising approach is the development of dual inhibitors targeting enzymes down-stream of the COX or LOX enzymes to prevent unavoidable on-target side-effects. While these new approaches are novel, recent results from use of these multitarget agents or dual inhibitors indicate they are a viable alternative pharmacological strategy for treatment of inflammation and pain.
Acknowledgments
This work was supported in part by NIEHS grant ES02710, NIEHS Superfund grant P42 ES04699, NIHLB grant HL059699 and the CounterACT Program, National Institutes of Health Office of the Director, and the National Institute of Neurological Disorders and Stroke, Grant Number U54 NS079202. Aaron T. Wecksler was supported by Award Number T32CA108459 from the National Institutes of Health. Karen M. Wagner was supported by NIEHS T32ES007059 and NIH 5T32DC008072-05. Bruce D. Hammock is a George and Judy Senior Fellow of the American Asthma Society.
ABBREVIATIONS
- 11β-HSD1
11β-Hydroxysteroid Dehydroge nase Type 1
- 20-HETE
20-Hydroxyeicosatetraenoic Acid
- 5-LOX
5-Lipoxygenase
- ALA
α-Linolenic Acid
- ARA
Arachidonic Acid
- ATP
Adenosine Triphosphate
- BAL
Bronchoalveolar Lavage
- BLT1 & BLT2
Two Leukotriene B4 Receptors
- CADD
Computer-Aided Drug Design
- COX
Cyclooxygenase
- COX-2
Cyclooxygenase-2
- cPGES
Cytosolic Prostaglandin E2 Synthase
- CYP450
Cytochrome P450
- CysLT1 / CysLT2
Cysteinyl Leukotrienes Receptors
- Cys-LTs
Cysteinyl-Leukotrienes
- DHA
Docosahexaenoic Acid
- DHDMBF
7-tert-Butyl-2,3-dihydro-3,3-dimethylbenzofurans
- DHETs
Dihydroxyeicosatrienoic Acids
- DMLs
Designed Multiple Ligands
- EC
Endothelial Cells
- EDHFs
Endothelium-Derived Hyperpolarizing Factors
- EETs
Epoxyeicosatrienoic Acids
- EPA
Eicosapentaenoic Acid
- EpFAs
Epoxy-Fatty Acids
- GI
Gastrointestinal
- HETEs
Hydroxyeicosatetraenoic Acids
- HPETEs
Hydroperoxyeicosatetraenoic Acids
- ICAM-1
Intracellular Adhesion Molecule-1
- LA
Linoleic Acid
- LOX
Lipoxygenase
- LPS
Lipopolysaccharide
- LTA4H
Leukotriene A4 Hydrolase
- LTs
Leukotrienes
- mPGES-1
Microsomal Prostaglandin E2 Synthase-1
- mPGES-2
Membrane Prostaglandin E2 Synthase-2
- NSAIDs
Non-Steroidal Anti-Inflammatory Drugs
- OA
Osteoarthritis
- PGE2
Prostaglandin E2
- PGG2
Prostaglandin G2
- PGH2
Prostaglandin H2
- PGI2
Prostaglandin I2 (Prostacyclin)
- PLA2
Phospholipase A2
- POX
Peroxidase
- SAR
Structure-Activity Relationship
- sEH
Soluble Epoxide Hydrolase
- t-AUCB
trans-4-[4-(3-Adamantan-1-ylureido)-cyclohexyloxy]-benzoic acid
- TXA2 synthase
Thromboxane A2 Synthase
- VCAM-1
Vascular Cell-Adhesion Molecule-1
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Piomelli D. Arachidonic acid in cell signaling. Curr Opin Cell Biol. 1993;5:274–280. doi: 10.1016/0955-0674(93)90116-8. [DOI] [PubMed] [Google Scholar]
- 2.Capdevila JH, Falck JR, Estabrook RW. Cytochrome P450 and the arachidonate cascade. FASEB J. 1992;6:731–736. doi: 10.1096/fasebj.6.2.1537463. [DOI] [PubMed] [Google Scholar]
- 3.Vane JR, Flower RJ, Botting RM. History of aspirin and its mechanism of action. Stroke. 1990;21(12 Suppl):IV12–23. [PubMed] [Google Scholar]
- 4.Melnikova I. Pain market. Nat Rev Drug Discov. 2010;9:589–590. doi: 10.1038/nrd3226. [DOI] [PubMed] [Google Scholar]
- 5.Green GA. Understanding NSAIDs: from aspirin to COX-2. Clin Cornerstone. 2001;3:50–60. doi: 10.1016/s1098-3597(01)90069-9. [DOI] [PubMed] [Google Scholar]
- 6.Griswold DE, Adams JL. Constitutive cyclooxygenase (COX-1) and inducible cyclooxygenase (COX-2): rationale for selective inhibition and progress to date. Med Res Rev. 1996;16:181–206. doi: 10.1002/(SICI)1098-1128(199603)16:2<181::AID-MED3>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 7.Wallace JL, Ma L. Inflammatory mediators in gastrointestinal defense and injury. Exp Biol Med. 2001;226:1003–1015. doi: 10.1177/153537020122601107. [DOI] [PubMed] [Google Scholar]
- 8.Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DA, Carlton DP, McIntyre TM, Zimmerman GA, Prescott SM. Molecular cloning of human prostaglandin endoperoxide synthase type II and demonstration of expression in response to cytokines. JBiol Chem. 1993;268:9049–9054. [PubMed] [Google Scholar]
- 10.Fletcher BS, Lim RW, Varnum BC, Kujubu DA, Koski RA, Herschman HR. Structure and expression of TIS21, a primary response gene induced by growth factors and tumor promoters. J Biol Chem. 1991;266:14511–14518. [PubMed] [Google Scholar]
- 11.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. New Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 12.Scheiman JM. Gastroduodenal safety of cyclooxygenase-2 inhibitors. Curr Pharm Des. 2003;9:2197–2206. doi: 10.2174/1381612033454018. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 14.Patscheke H. Thromboxane A2/prostaglandin H2 receptor antagonists. A new therapeutic principle. Stroke. 1990;21:IV139–142. [PubMed] [Google Scholar]
- 15.Moncada S, Vane JR. The role of prostacyclin in vascular tissue. Fed Proc. 1979;38:66–71. [PubMed] [Google Scholar]
- 16.Bing RJ, Lomnicka M. Why do cyclooxygenase-2 inhibitors cause cardiovascular events? J Am Coll Cardiol. 2002;39:521–522. doi: 10.1016/s0735-1097(01)01749-1. [DOI] [PubMed] [Google Scholar]
- 17.Fitzgerald GA. Coxibs and cardiovascular disease. New Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- 19.Liu JY, Li N, Yang J, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc Natl Acad Sci USA. 2010;107:17017–17022. doi: 10.1073/pnas.1011278107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia Rodriguez LA, Barreales Tolosa L. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology. 2007;132:498–506. doi: 10.1053/j.gastro.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Hippisley-Cox J, Coupland C, Logan R. Risk of adverse gastrointestinal outcomes in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ. 2005;331:1310–1316. doi: 10.1136/bmj.331.7528.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calder PC. Long-chain fatty acids and inflammation. Proc Nutr Soc. 2012:1–6. doi: 10.1017/S0029665112000067. [DOI] [PubMed] [Google Scholar]
- 23.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith W. Eicosanoid nomenclature. Prostaglandins. 1989;38:125–133. doi: 10.1016/0090-6980(89)90021-x. [DOI] [PubMed] [Google Scholar]
- 25.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuelsson B. Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science. 1983;220:568–575. doi: 10.1126/science.6301011. [DOI] [PubMed] [Google Scholar]
- 27.Spokas EG, Rokach J, Wong PY. Leukotrienes, lipoxins, and hydroxyeicosatetraenoic acids. Methods Mol Biol. 1999;120:213–247. doi: 10.1385/1-59259-263-5:213. [DOI] [PubMed] [Google Scholar]
- 28.McMahon B, Mitchell S, Brady HR, Godson C. Lipoxins: revelations on resolution. Trends Pharmacol Sci. 2001;22:391–395. doi: 10.1016/s0165-6147(00)01771-5. [DOI] [PubMed] [Google Scholar]
- 29.Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J Smooth Muscle Res. 2005;41:175–193. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- 30.Sarkis A, Lopez B, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids in hypertension. Curr Opin Nephrol Hypertens. 2004;13:205–214. doi: 10.1097/00041552-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Smith WL, Urade Y, Jakobsson PJ. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev. 2011;111:5821–5865. doi: 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santovito D, Mezzetti A, Cipollone F. Cyclooxygenase and prostaglandin synthases: roles in plaque stability and instability in humans. Curr Opin Lipidol. 2009;20:402–408. doi: 10.1097/MOL.0b013e32832fa22c. [DOI] [PubMed] [Google Scholar]
- 34.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Isakson P. Distribution of COX-1 and COX-2 in normal and inflamed tissues. Adv Exp Med Biol. 1997;400A:167–170. doi: 10.1007/978-1-4615-5325-0_24. [DOI] [PubMed] [Google Scholar]
- 36.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 37.Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 38.Beiche F, Scheuerer S, Brune K, Geisslinger G, Goppelt-Struebe M. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Lett. 1996;390:165–169. doi: 10.1016/0014-5793(96)00604-7. [DOI] [PubMed] [Google Scholar]
- 39.Maslinska D, Kaliszek A, Opertowska J, Toborowicz J, Deregowski K, Szukiewicz D. Constitutive expression of cyclooxygenase-2 (COX-2) in developing brain. A. Choroid plexus in human fetuses. Folia Neuropathol. 1999;37:287–291. [PubMed] [Google Scholar]
- 40.Komhoff M, Wang JL, Cheng HF, Langenbach R, McKanna JA, Harris RC, Breyer MD. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000;57:414–422. doi: 10.1046/j.1523-1755.2000.00861.x. [DOI] [PubMed] [Google Scholar]
- 41.Ohki S, Ogino N, Yamamoto S, Hayaishi O. Prostaglandin hydroperoxidase, an integral part of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem. 1979;254:829–836. [PubMed] [Google Scholar]
- 42.Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostag Oth Lipid M. 2002;68–69:115–128. doi: 10.1016/s0090-6980(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 43.Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 44.Thoren S, Weinander R, Saha S, Jegerschold C, Pettersson PL, Samuelsson B, Hebert H, Hamberg M, Morgenstern R, Jakobsson PJ. Human microsomal prostaglandin E synthase-1: purification, functional characterization, and projection structure determination. J Biol Chem. 2003;278:22199–22209. doi: 10.1074/jbc.M303227200. [DOI] [PubMed] [Google Scholar]
- 45.Friesen RW, Mancini JA. Microsomal prostaglandin E2 synthase-1 (mPGES-1): a novel anti-inflammatory therapeutic target. J Med Chem. 2008;51:4059–4067. doi: 10.1021/jm800197b. [DOI] [PubMed] [Google Scholar]
- 46.Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, Ohmiya Y, Watanabe K, Kudo I. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 47.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Ikeda T, Fueki M, Ueno A, Oh S, Kudo I. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 48.Wong PY, Lai PS, Falck JR. Mechanism and signal transduction of 14 (R), 15 (S)-epoxyeicosatrienoic acid (14,15-EET) binding in guinea pig monocytes. Prostag Oth Lipid M. 2000;62(4):321–333. doi: 10.1016/s0090-6980(00)00079-4. [DOI] [PubMed] [Google Scholar]
- 49.von Euler US. On the specific vaso-dilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin) J Physiol-London. 1936;88:213–234. doi: 10.1113/jphysiol.1936.sp003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Woodward DF, Jones RL, Narumiya S International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 51.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. New Engl J Med. 1999;340:1888–1899. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 52.Moncada S, Vane JR. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol Rev. 1978;30:293–331. [PubMed] [Google Scholar]
- 53.Orehek J, Douglas JS, Lewis AJ, Bouhuys A. Prostaglandin regulation of airway smooth muscle tone. Nat New Biol. 1973;245:84–85. doi: 10.1038/newbio245084a0. [DOI] [PubMed] [Google Scholar]
- 54.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 55.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 56.Zeilhofer HU. Prostanoids in nociception and pain. Biochem Pharmacol. 2007;73:165–174. doi: 10.1016/j.bcp.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 57.Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002;8:390–396. doi: 10.1016/s1471-4914(02)02383-3. [DOI] [PubMed] [Google Scholar]
- 58.Khanapure SP, Garvey DS, Janero DR, Letts LG. Eicosanoids in inflammation: biosynthesis, pharmacology, and therapeutic frontiers. Curr Top Med Chem. 2007;7:311–340. doi: 10.2174/156802607779941314. [DOI] [PubMed] [Google Scholar]
- 59.Kuhn H. Structural basis for the positional specificity of lipoxygenases. Prostag Oth Lipid M. 2000;62:255–270. doi: 10.1016/s0090-6980(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 60.Serhan CN, Hamberg M, Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc Natl Acad Sci USA. 1984;81:5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hussain H, Shornick LP, Shannon VR, Wilson JD, Funk CD, Pentland AP, Holtzman MJ. Epidermis contains platelet-type 12-lipoxygenase that is overexpressed in germinal layer keratinocytes in psoriasis. Am J physiol. 1994;266:C243–253. doi: 10.1152/ajpcell.1994.266.1.C243. [DOI] [PubMed] [Google Scholar]
- 62.Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 63.Rouzer CA, Kargman S. Translocation of 5-lipoxygenase to the membrane in human leukocytes challenged with ionophore A23187. J Biol Chem. 1988;263:10980–10988. [PubMed] [Google Scholar]
- 64.Ford-Hutchinson AW. FLAP: a novel drug target for inhibiting the synthesis of leukotrienes. Trends Pharmacol Sci. 1991;12:68–70. doi: 10.1016/0165-6147(91)90500-r. [DOI] [PubMed] [Google Scholar]
- 65.Borgeat P, Hamberg M, Samuelsson B. Transformation of arachidonic acid, homo-gamma-linolenic acid by rabbit polymorphonuclear leukocytes. Monohydroxy acids from novel lipoxygenases. J Biol Chem. 1976;251:7816–7820. [PubMed] [Google Scholar]
- 66.Rouzer CA, Matsumoto T, Samuelsson B. Single protein from human leukocytes possesses 5-lipoxygenase and leukotriene A4 synthase activities. Proc Natl Acad Sci USA. 1986;83:857–861. doi: 10.1073/pnas.83.4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Radmark O, Shimizu T, Jornvall H, Samuelsson B. Leukotriene A4 hydrolase in human leukocytes. Purification and properties. J Biol Chem. 1984;259:12339–12345. [PubMed] [Google Scholar]
- 68.Lam BK, Frank Austen K. Leukotriene C4 synthase. A pivotal enzyme in the biosynthesis of the cysteinyl leukotrienes. Am J Respir Crit Care Med. 2000;161:S16–19. doi: 10.1164/ajrccm.161.supplement_1.ltta-4. [DOI] [PubMed] [Google Scholar]
- 69.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 70.Peters-Golden M, Henderson WR., Jr Leukotrienes. New Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 71.Tager AM, Luster AD. BLT1 and BLT2: the leukotriene B(4) receptors. Prostag Leukotr Ess. 2003;69:123–134. doi: 10.1016/s0952-3278(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 72.Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, Williams DL, JrZeng Z, Liu Q, Ma L, Clements MK, Coulombe N, Liu Y, Austin CP, George SR, O’Neill GP, Metters KM, Lynch KR, Evans JF. Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem. 2000;275:30531–30536. doi: 10.1074/jbc.M003490200. [DOI] [PubMed] [Google Scholar]
- 73.Lynch KR, O’Neill GP, Liu Q, Im DS, Sawyer N, Metters KM, Coulombe N, Abramovitz M, Figueroa DJ, Zeng Z, Connolly BM, Bai C, Austin CP, Chateauneuf A, Stocco R, Greig GM, Kargman S, Hooks SB, Hosfield E, Williams DL, JrFord-Hutchinson AW, Caskey CT, Evans JF. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 74.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 1980;286:264–265. doi: 10.1038/286264a0. [DOI] [PubMed] [Google Scholar]
- 75.Bjork J, Hedqvist P, Arfors KE. Increase in vascular permeability induced by leukotriene B4 and the role of polymorphonuclear leukocytes. Inflammation. 1982;6:189–200. doi: 10.1007/BF00916243. [DOI] [PubMed] [Google Scholar]
- 76.Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- 77.Hay DW, Torphy TJ, Undem BJ. Cysteinyl leukotrienes in asthma: old mediators up to new tricks. Trends Pharmacol Sci. 1995;16:304–309. doi: 10.1016/s0165-6147(00)89059-8. [DOI] [PubMed] [Google Scholar]
- 78.Rioux KP, Wallace JL. Mast cell activation augments gastric mucosal injury through a leukotriene-dependent mechanism. Am J Physiol. 1994;266:G863–869. doi: 10.1152/ajpgi.1994.266.5.G863. [DOI] [PubMed] [Google Scholar]
- 79.Vaananen PM, Keenan CM, Grisham MB, Wallace JL. Pharmacological investigation of the role of leukotrienes in the pathogenesis of experimental NSAID gastropathy. Inflammation. 1992;16:227–240. doi: 10.1007/BF00918812. [DOI] [PubMed] [Google Scholar]
- 80.Lewis RA, Austen KF, Soberman RJ. Leukotrienes, other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. New Engl J Med. 1990;323:645–655. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 81.Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci USA. 1981;78:5362–5366. doi: 10.1073/pnas.78.9.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Capdevila J, Marnett LJ, Chacos N, Prough RA, Estabrook RW. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proc Natl Acad Sci USA. 1982;79:767–770. doi: 10.1073/pnas.79.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morrison AR, Pascoe N. Metabolism of arachidonate through NADPH-dependent oxygenase of renal cortex. Proc Natl Acad Sci USA. 1981;78:7375–7378. doi: 10.1073/pnas.78.12.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oliw EH, Oates JA. Oxygenation of arachidonic acid by hepatic microsomes of the rabbit. Mechanism of biosynthesis of two vicinal dihydroxyeicosatrienoic acids. Biochim Biophys Acta. 1981;666:327–340. doi: 10.1016/0005-2760(81)90291-5. [DOI] [PubMed] [Google Scholar]
- 85.Oliw EH, Guengerich FP, Oates JA. Oxygenation of arachidonic acid by hepatic monooxygenases. Isolation and metabolism of four epoxide intermediates. J Biol Chem. 1982;257:3771–3781. [PubMed] [Google Scholar]
- 86.Elbekai RH, El-Kadi AO. Cytochrome P450 enzymes: central players in cardiovascular health and disease. Pharmacol Therapeut. 2006;112:564–587. doi: 10.1016/j.pharmthera.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 87.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 88.Carroll MA, McGiff JC. A new class of lipid mediators: cytochrome P450 arachidonate metabolites. Thorax. 2000;55(Suppl 2):S13–16. doi: 10.1136/thorax.55.suppl_2.S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 90.Kaspera R, Totah RA. Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathophysiology. Expert Opin Drug Metab Toxicol. 2009;5:757–771. doi: 10.1517/17425250902932923. [DOI] [PubMed] [Google Scholar]
- 91.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostag Oth Lipid M. 2007;82:42–49. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, Falck JR, Capdevila JH. Characterization of 5,6-and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–27146. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 93.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- 95.Rosolowsky M, Campbell WB. Synthesis of hydroxyeicosatetraenoic (HETEs) and epoxyeicosatrienoic acids (EETs) by cultured bovine coronary artery endothelial cells. Biochim Biophys Acta. 1996;1299:267–277. doi: 10.1016/0005-2760(95)00216-2. [DOI] [PubMed] [Google Scholar]
- 96.Fleming I. DiscrEET regulators of homeostasis: epoxyeico satrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol Sci. 2007;28:448–452. doi: 10.1016/j.tips.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 97.Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevila JH. Regio- and enantiofacial selectivity of epoxyeicosatrienoic acid hydration by cytosolic epoxide hydrolase. J Biol Chem. 1993;268:6402–6407. [PubMed] [Google Scholar]
- 98.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 99.Imig JD. Epoxides and soluble epoxide hydrolase in cardio vascular physiology. Physiol Rev. 2012;92:101–130. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arnold C, Konkel A, Fischer R, Schunck WH. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol Rep. 2010;62:536–547. doi: 10.1016/s1734-1140(10)70311-x. [DOI] [PubMed] [Google Scholar]
- 101.Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD. Naturally occurring mono epoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51:3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wagner K, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostag Oth Lipid M. 2011;96:76–83. doi: 10.1016/j.prostaglandins.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Falck JR, Reddy LM, Reddy YK, Bondlela M, Krishna UM, Ji Y, Sun J, Liao JK. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett. 2003;13:4011–4014. doi: 10.1016/j.bmcl.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 106.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeico satrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 107.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norwood S, Liao J, Hammock BD, Yang GY. Epoxyeico satrienoic acids and soluble epoxide hydrolase: potential therapeutic targets for inflammation and its induced carcinogenesis. Am J Transl Res. 2010;2:447–457. [PMC free article] [PubMed] [Google Scholar]
- 109.Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeico satrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci USA. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zeilhofer HU, Brune K. Analgesic strategies beyond the inhibition of cyclooxygenases. Trends Pharmacol Sci. 2006;27:467–474. doi: 10.1016/j.tips.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 112.Wagner K, Inceoglu B, Gill SS, Hammock BD. Epoxy genated fatty acids and soluble epoxide hydrolase inhibition: novel mediators of pain reduction. J Agric Food Chem. 2011;59:2816–2824. doi: 10.1021/jf102559q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stables MJ, Gilroy DW. Old and new generation lipid mediators in acute inflammation and resolution. Prog Lipid Res. 2011;50:35–51. doi: 10.1016/j.plipres.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 114.Townsley MI, King JA, Alvarez DF. Ca2+ channels and pulmonary endothelial permeability: insights from study of intact lung and chronic pulmonary hypertension. Microcirculation. 2006;13:725–739. doi: 10.1080/10739680600930362. [DOI] [PubMed] [Google Scholar]
- 115.Loot AE, Fleming I. Cytochrome P450-derived epoxyeico satrienoic acids and pulmonary hypertension: central role of transient receptor potential C6 channels. J Cardiovasc Pharmacol. 2011;57:140–147. doi: 10.1097/FJC.0b013e3181ed088d. [DOI] [PubMed] [Google Scholar]
- 116.Mustafa S, Sharma V, McNeill JH. Insulin resistance and endothelial dysfunction: Are epoxyeicosatrienoic acids the link? Exp Clin Cardiol. 2009;14:e41–50. [PMC free article] [PubMed] [Google Scholar]
- 117.Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, Tsai HJ, Imig JD, Haj FG, Hammock BD. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA. 2011;108:9038–9043. doi: 10.1073/pnas.1103482108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hao CM, Breyer MD. Roles of lipid mediators in kidney injury. Semin Nephrol. 2007;27:338–351. doi: 10.1016/j.semnephrol.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 119.Imig JD. Targeting epoxides for organ damage in hypertension. J Cardiovasc Pharmacol. 2010;56:329–335. doi: 10.1097/FJC.0b013e3181e96e0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu X, Zhang XA, Wang DW. The roles of CYP450 epoxygenases and metabolites, epoxyeicosatrienoic acids, in cardiovascular and malignant diseases. Adv Drug Deliv Rev. 2011;63:597–609. doi: 10.1016/j.addr.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 121.Sato M, Yokoyama U, Fujita T, Okumura S, Ishikawa Y. The roles of cytochrome p450 in ischemic heart disease. Curr Drug Metab. 2011;12:526–532. doi: 10.2174/138920011795713715. [DOI] [PubMed] [Google Scholar]
- 122.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Panigrahy D, Kaipainen A, Greene ER, Huang S. Cytochrome P450-derived eicosanoids: the neglected pathway in cancer. Cancer Metastasis Rev. 2010;29:723–735. doi: 10.1007/s10555-010-9264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wagner K, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostag Oth Lipid M. 2011;96:76–83. doi: 10.1016/j.prostaglandins.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Asako H, Kubes P, Wallace J, Gaginella T, Wolf RE, Granger DN. Indomethacin-induced leukocyte adhesion in mesenteric venules: role of lipoxygenase products. Am J Physiol. 1992;262:G903–908. doi: 10.1152/ajpgi.1992.262.5.G903. [DOI] [PubMed] [Google Scholar]
- 126.Gilroy DW, Tomlinson A, Willoughby DA. Differential effects of inhibitors of cyclooxygenase (cyclooxygenase 1 and cyclooxygenase 2) in acute inflammation. Eur J Pharmacol. 1998;355:211–217. doi: 10.1016/s0014-2999(98)00508-1. [DOI] [PubMed] [Google Scholar]
- 127.Knapp HR, Sladek K, Fitzgerald GA. Increased excretion of leukotriene E4 during aspirin-induced asthma. J Lab Clin Med. 1992;119:48–51. [PubMed] [Google Scholar]
- 128.Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem pharmacol. 2010;79:880–887. doi: 10.1016/j.bcp.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wecksler AT, Kenyon V, Deschamps JD, Holman TR. Substrate specificity changes for human reticulocyte and epithelial 15-lipoxygenases reveal allosteric product regulation. Bio chemistry. 2008;47:7364–7375. doi: 10.1021/bi800550n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wecksler AT, Kenyon V, Garcia NK, Deschamps JD, van der Donk WA, Holman TR. Kinetic and structural investigations of the allosteric site in human epithelial 15-lipoxygenase-2. Biochemistry. 2009;48:8721–8730. doi: 10.1021/bi9009242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kozak W, Aronoff DM, Boutaud O, Kozak A. 11,12-epoxyeicosatrienoic acid attenuates synthesis of prostaglandin E2 in rat monocytes stimulated with lipopolysaccharide. Exp Biol Med. 2003;228:786–794. doi: 10.1177/15353702-0322807-03. [DOI] [PubMed] [Google Scholar]
- 132.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roumie CL, Arbogast PG, Mitchel EF, JrGriffin MR. Prescriptions for chronic high-dose cyclooxygenase-2 inhibitors are often inappropriate and potentially dangerous. J Gen Intern Med. 2005;20:879–883. doi: 10.1111/j.1525-1497.2005.0173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, Morisseau C, Hwang SH, Tsai C, Hammock BD, Schaefer L, Geisslinger G, Amann K, Brandes RP. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS One. 2010;5:e11979. doi: 10.1371/journal.pone.0011979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discov Today. 2004;9:641–651. doi: 10.1016/S1359-6446(04)03163-0. [DOI] [PubMed] [Google Scholar]
- 136.Millan MJ. Multi-target strategies for the improved treatment of depressive states: Conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Therapeut. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 137.Keith CT, Borisy AA, Stockwell BR. Multicomponent therapeutics for networked systems. Nat Rev Drug Discov. 2005;4:71–78. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- 138.Sreedhar D, Subramanian G, Udupa N. Combination drugs: Are they rational? Curr SciIndia. 2006;91:406. [Google Scholar]
- 139.Frantz S. The trouble with making combination drugs. Nat Rev Drug Discov. 2006;5:881–882. doi: 10.1038/nrd2188. [DOI] [PubMed] [Google Scholar]
- 140.Bergmann JF. Review: fixed-dose drug combinations improve medication compliance compared with free-drug regimens. Evid Based Med. 2008;13:18. doi: 10.1136/ebm.13.1.18. [DOI] [PubMed] [Google Scholar]
- 141.Morphy R, Rankovic Z. Designing multiple ligands - medicinal chemistry strategies and challenges. Curr Pharm Des. 2009;15:587–600. doi: 10.2174/138161209787315594. [DOI] [PubMed] [Google Scholar]
- 142.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 143.Leval X, Julemont F, Delarge J, Pirotte B, Dogne JM. New trends in dual 5-LOX/COX inhibition. Curr Med Chem. 2002;9:941–962. doi: 10.2174/0929867024606713. [DOI] [PubMed] [Google Scholar]
- 144.Brahams D. Late benoxaprofen claims. Lancet. 1991;337:483–484. doi: 10.1016/0140-6736(91)93412-3. [DOI] [PubMed] [Google Scholar]
- 145.Flynn DL, Rafferty MF, Boctor AM. Inhibition of 5-hydroxyeicosatetraenoic acid (5-HETE) formation in intact human neutrophils by naturally-occurring diarylheptanoids: inhibitory activities of curcuminoids and yakuchinones. Prostaglandins Leukot Med. 1986;22:357–360. doi: 10.1016/0262-1746(86)90146-0. [DOI] [PubMed] [Google Scholar]
- 146.Flynn DL, Belliotti TR, Boctor AM, Connor DT, Kostlan CR, Nies DE, Ortwine DF, Schrier DJ, Sircar JC. Styrylpyrazoles styrylisoxazoles, styrylisothiazoles. Novel 5-lipoxygenase and cyclooxygenase inhibitors. J Med Chem. 1991;34:518–525. doi: 10.1021/jm00106a006. [DOI] [PubMed] [Google Scholar]
- 147.Janusz JM, Young PA, Ridgeway JM, Scherz MW, Enzweiler K, Wu LI, Gan L, Darolia R, Matthews RS, Hennes D, Kellstein DE, Green SA, Tulich JL, Rosario-Jansen T, Magrisso IJ, Wehmeyer KR, Kuhlenbeck DL, Eichhold TH, Dobson RL, Sirko SP, Farmer RW. New cyclooxygenase-2/5-lipoxygenase inhibitors1. 7-tert-buty1-2,3-dihydro-3,3-dimethylbenzofuran derivatives as gastrointestinal safe antiinflammatory and analgesic agents: discovery and variation of the 5-keto substituent. J Med Chem. 1998;41:1112–1123. doi: 10.1021/jm970679q. [DOI] [PubMed] [Google Scholar]
- 148.Unangst PC, Connor DT, Cetenko WA, Sorenson RJ, Kostlan CR, Sircar JC, Wright CD, Schrier DJ, Dyer RD. Synthesis and biological evaluation of 5-[[3,5-bis(1,1-dimethylethyl)-4-hydroxyphenyl]methylene]oxazoles, -thiazoles, and -imidazoles: novel dual 5-lipoxygenase and cyclooxygenase inhibitors with antiinflammatory activity. J Med Chem. 1994;37:322–328. doi: 10.1021/jm00028a017. [DOI] [PubMed] [Google Scholar]
- 149.Inagaki M, Tsuri T, Jyoyama H, Ono T, Yamada K, Kobayashi M, Hori Y, Arimura A, Yasui K, Ohno K, Kakudo S, Koizumi K, Suzuki R, Kawai S, Kato M, Matsumoto S. Novel antiarthritic agents with 1,2-isothiazolidine-1,1-dioxide (gamma-sultam) skeleton: cytokine suppressive dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. J Med Chem. 2000;43:2040–2048. doi: 10.1021/jm9906015. [DOI] [PubMed] [Google Scholar]
- 150.Weisman SM, Doyle MJ, Wehmeyer KR, Hynd BA, Eichhold TH, Clear RM, Coggeshall CW, Kuhlenbeck DL. Effects of tebufelone (NE-11740), a new anti-inflammatory drug, on arachidonic acid metabolism. Agents Actions. 1994;41:156–163. doi: 10.1007/BF02001910. [DOI] [PubMed] [Google Scholar]
- 151.Unangst PC, Shrum GP, Connor DT, Dyer RD, Schrier DJ. Novel 1,2,4-oxadiazoles and 1,2,4-thiadiazoles as dual 5-lipoxygenase and cyclooxygenase inhibitors. J Med Chem. 1992;35:3691–3698. doi: 10.1021/jm00098a015. [DOI] [PubMed] [Google Scholar]
- 152.Janusz JM, Young PA, Ridgeway JM, Scherz MW, Enzweiler K, Wu LI, Gan L, Chen J, Kellstein DE, Green SA, Tulich JL, Rosario-Jansen T, Magrisso IJ, Wehmeyer KR, Kuhlenbeck DL, Eichhold TH, Dobson RL. New cyclooxygenase-2/5-lipoxygenase inhibitors. 3. 7-tert-butyl-2, 3-dihydro-3,3-dimethylbenzofuran derivatives as gastrointestinal safe antiinflammatory and analgesic agents: variations at the 5 position. J Med Chem. 1998;41:3515–3529. doi: 10.1021/jm9802416. [DOI] [PubMed] [Google Scholar]
- 153.Janusz JM, Young PA, Scherz MW, Enzweiler K, Wu LI, Gan L, Pikul S, McDow-Dunham KL, Johnson CR, Senanayake CB, Kellstein DE, Green SA, Tulich JL, Rosario-Jansen T, Magrisso IJ, Wehmeyer KR, Kuhlenbeck DL, Eichhold TH, Dobson RL. New cyclooxygenase-2/5-lipoxygenase inhibitors. 2. 7-tert-butyl-2,3-dihydro-3,3-dimethyl benzofuran derivatives as gastrointestinal safe antiinflammatory and analgesic agents: variations of the dihydrobenzofuran ring. J Med Chem. 1998;41:1124–1137. doi: 10.1021/jm970680p. [DOI] [PubMed] [Google Scholar]
- 154.Stanton BJ, Coupar IM. The effect of BW 755C and nordihydroguaiaretic acid in the rat isolated perfused mesenteric vasculature. Prostaglandins Leukot Med. 1986;25:199–207. doi: 10.1016/0262-1746(86)90066-1. [DOI] [PubMed] [Google Scholar]
- 155.Radmark O, Malmsten C, Samuelsson B. The inhibitory effects of BW 755C on arachidonic acid metabolism in human polymorphonuclear leukocytes. FEBS lett. 1980;110:213–215. doi: 10.1016/0014-5793(80)80075-5. [DOI] [PubMed] [Google Scholar]
- 156.Gulbenkian AR, Fernandez X, Kreutner W, Minnicozzi M, Watnick AS, Kung T, Egan RW. Anaphylactic challenge causes eosinophil accumulation in bronchoalveolar lavage fluid of guinea pigs. Modulation by betamethasone, phenidone, indomethacin, WEB 2086; and a novel antiallergy agent, SCH 37224. Am Rev Respir Dis. 1990;142:680–685. doi: 10.1164/ajrccm/142.3.680. [DOI] [PubMed] [Google Scholar]
- 157.Argentieri DC, Ritchie DM, Ferro MP, Kirchner T, Wachter MP, Anderson DW, Rosenthale ME, Capetola RJ. Tepoxalin: a dual cyclooxygenase/5-lipoxygenase inhibitor of arachidonic acid metabolism with potent anti-inflammatory activity and a favorable gastrointestinal profile. J Pharmacol Exp Ther. 1994;271:1399–1408. [PubMed] [Google Scholar]
- 158.Waldman SA, Vitow C, Osborne B, Gillen L, Argentieri DC, Wong FA, Smith IL, Chow AT, Misiti J, Bjornsson TD. Pharmacokinetics and pharmacodynamics of tepoxalin after single oral dose administration to healthy volunteers. J Clin Pharmacol. 1996;36:462–468. doi: 10.1002/j.1552-4604.1996.tb05033.x. [DOI] [PubMed] [Google Scholar]
- 159.Knight EV, Kimball JP, Keenan CM, Smith IL, Wong FA, Barrett DS, Dempster AM, Lieuallen WG, Panigrahi D, Powers WJ, Szot RJ. Preclinical toxicity evaluation of tepoxalin, a dual inhibitor of cyclooxygenase and 5-lipoxygenase, in Sprague-Dawley rats and beagle dogs. Fundam Appl Toxicol. 1996;33:38–48. doi: 10.1006/faat.1996.0141. [DOI] [PubMed] [Google Scholar]
- 160.Beers SA, Malloy EA, Wu W, Wachter M, Ansell J, Singer M, Steber M, Barbone A, Kirchner T, Ritchie D, Argentieri D. N-(5-substituted) thiophene-2-alkylsulfonamides as potent inhibitors of 5-lipoxygenase. Bioorg Med, Chem. 1997;5:779–786. doi: 10.1016/s0968-0896(97)00025-4. [DOI] [PubMed] [Google Scholar]
- 161.Kirchner T, Argentieri DC, Barbone AG, Singer M, Steber M, Ansell J, Beers SA, Wachter MP, Wu W, Malloy E, Stewart A, Ritchie DM. Evaluation of the antiinflammatory activity of a dual cyclooxygenase-2 selective/5-lipoxygenase inhibitor, RWJ 63556; in a canine model of inflammation. J Pharmacol Exp Ther. 1997;282:1094–1101. [PubMed] [Google Scholar]
- 162.Le Filliatre G, Sayah S, Latournerie V, Renaud JF, Finet M, Hanf R. Cyclo-oxygenase and lipoxygenase pathways in mast cell dependent-neurogenic inflammation induced by electrical stimu lation of the rat saphenous nerve. Br J Pharmacol. 2001;132:1581–1589. doi: 10.1038/sj.bjp.0703950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bailey PJ, Dallob AL, Allison DL, Anderson RL, Bach T, Durette PL, Hand KM, Hopple SL, Luell S, Meurer R, et al. Pharmacology of the dual inhibitor of cyclooxygenase and 5-lipoxygenase 3-hydroxy-5-trifluoromethyl-N-(2-(2-thienyl)-2-phe nyl-ethenyl)-benzo (b)thiophene-2-carboxamide. Arzneimittel forschung. 1988;38:372–378. [PubMed] [Google Scholar]
- 164.Tischler A, Bailey P, Dallob A, Witzel B, Durette P, Rupprecht K, Allison D, Dougherty H, Humes J, Ham E, et al. L-652,343: a novel dual 5-lipoxygenase/cyclooxygenase inhibitor. Adv Prostaglandin Thromboxane Leukot Res. 1986;16:63–66. [PubMed] [Google Scholar]
- 165.Barr RM, Black AK, Dowd PM, Koro O, Mistry K, Isaacs JL, Greaves MW. The in vitro 5-lipoxygenase and cyclooxygenase inhibitor L-652,343 does not inhibit 5-lipoxygenase in vivo in human skin. Br J Clin Pharmacol. 1988;25:23–26. doi: 10.1111/j.1365-2125.1988.tb03277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gresele P, Arnout J, Deckmyn H, Vermylen J. L-652,343, a novel dual cyclo/lipoxygenase inhibitor, inhibits LTB4-production by stimulated human polymorphonuclear cells but not by stimulated human whole blood. Biochem Pharmacol. 1987;36:3529–3531. doi: 10.1016/0006-2952(87)90336-4. [DOI] [PubMed] [Google Scholar]
- 167.Chowdhury MA, Abdellatif KR, Dong Y, Das D, Suresh MR, Knaus EE. Synthesis of celecoxib analogues possessing a N-difluoromethyl-1,2-dihydropyrid-2-one 5-lipoxygenase pharma cophore: biological evaluation as dual inhibitors of cyclooxy genases and 5-lipoxygenase with anti-inflammatory activity. J Med Chem. 2009;52:1525–1529. doi: 10.1021/jm8015188. [DOI] [PubMed] [Google Scholar]
- 168.Chowdhury MA, Abdellatif KR, Dong Y, Das D, Yu G, Velazquez CA, Suresh MR, Knaus EE. Synthesis and biological evaluation of salicylic acid and N-acetyl-2-carboxy benzenesulfonamide regioisomers possessing a N-difluoromethyl-1,2-dihydropyrid-2-one pharmacophore: dual inhibitors of cyclo oxygenases and 5-lipoxygenase with anti-inflammatory activity. Bioorg Med Chem Lett. 2009;19:6855–6861. doi: 10.1016/j.bmcl.2009.10.083. [DOI] [PubMed] [Google Scholar]
- 169.Yu G, Praveen Rao PN, Chowdhury MA, Abdellatif KR, Dong Y, Das D, Velazquez CA, Suresh MR, Knaus EE. Synthesis and biological evaluation of N-difluoromethyl-1,2-dihydropyrid-2-one acetic acid regioisomers: dual inhibitors of cyclooxygenases and 5-lipoxygenase. Bioorg Med Chem Lett. 2010;20:2168–2173. doi: 10.1016/j.bmcl.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 170.Wylie G, Appelboom T, Bolten W, Breedveld FC, Feely J, Leeming MR, Le Loet X, Manthorpe R, Marcolongo R, Smolen J. A comparative study of tenidap, a cytokine-modulating anti-rheumatic drug, and diclofenac in rheumatoid arthritis: a 24-week analysis of a 1-year clinical trial. Br J Rheumatol. 1995;34:554–563. doi: 10.1093/rheumatology/34.6.554. [DOI] [PubMed] [Google Scholar]
- 171.Aleo MD, Wang T, Giebisch G, Sanders MJ, Walsh AH, Lopez-Anaya A. Model development and analysis of tenidap-induced proteinuria in the rat. J Pharmacol Exp Ther. 1996;279:1318–1326. [PubMed] [Google Scholar]
- 172.Nelson SD. Structure toxicity relationships--how useful are they in predicting toxicities of new drugs? Adv Exp Med Biol. 2001;500:33–43. doi: 10.1007/978-1-4615-0667-6_4. [DOI] [PubMed] [Google Scholar]
- 173.Fouda HG, Avery MJ, Dalvie D, Falkner FC, Melvin LS, Ronfeld RA. Disposition and metabolism of tenidap in the rat. Drug Metab Dispos. 1997;25:140–148. [PubMed] [Google Scholar]
- 174.Lai Y, Ma L, Huang W, Yu X, Zhang Y, Ji H, Tian J. Synthesis and biological evaluation of 3-[4-(amino/methyl sulfonyl)phenyl]methylene-indolin-2-one derivatives as novel COX-1/2 and 5-LOX inhibitors. Bioorg Med Chem Lett. 2010;20:7349–7353. doi: 10.1016/j.bmcl.2010.10.056. [DOI] [PubMed] [Google Scholar]
- 175.Barbey S, Goossens L, Taverne T, Cornet J, Choesmel V, Rouaud C, Gimeno G, Yannic-Arnoult S, Michaux C, Charlier C, Houssin R, Henichart JP. Synthesis and activity of a new methoxytetrahydropyran derivative as dual cyclooxygenase-2/5-lipoxygenase inhibitor. Bioorg Med Chem Lett. 2002;12:779–782. doi: 10.1016/s0960-894x(02)00013-6. [DOI] [PubMed] [Google Scholar]
- 176.Rao PN, Chen QH, Knaus EE. Synthesis and structure-activity relationship studies of 1,3-diarylprop-2-yn-1-ones: dual inhibitors of cyclooxygenases and lipoxygenases. J Med Chem. 2006;49:1668–1683. doi: 10.1021/jm0510474. [DOI] [PubMed] [Google Scholar]
- 177.Chen QH, Rao PN, Knaus EE. Synthesis and biological evaluation of a novel class of rofecoxib analogues as dual inhibitors of cyclooxygenases (COXs) and lipoxygenases (LOXs) Bioorg Med Chem. 2006;14:7898–7909. doi: 10.1016/j.bmc.2006.07.047. [DOI] [PubMed] [Google Scholar]
- 178.Laufer SA, Augustin J, Dannhardt G, Kiefer W. (6,7-Diaryldihydropyrrolizin-5-yl)acetic acids, a novel class of potent dual inhibitors of both cyclooxygenase and 5-lipoxygenase. J Med Chem. 1994;37:1894–1897. doi: 10.1021/jm00038a021. [DOI] [PubMed] [Google Scholar]
- 179.Cossy J, Belotti D. Synthesis of ML-3000, an Inhibitor of Cyclooxygenase and 5-Lipoxygenase. J Org Chem. 1997;62:7900–7901. doi: 10.1021/jo971480o. [DOI] [PubMed] [Google Scholar]
- 180.Rotondo S, Dell’Elba G, Krauze-Brzosko K, Manarini S, Martelli N, Pecce R, Evangelista V, Cerletti C. Licofelone, a dual lipoxygenase-cyclooxygenase inhibitor, downregulates polymorphonuclear leukocyte and platelet function. Eur J Pharmacol. 2002;453:131–139. doi: 10.1016/s0014-2999(02)02385-3. [DOI] [PubMed] [Google Scholar]
- 181.Kulkarni SK, Singh VP. Licofelone--a novel analgesic and antiinflammatory agent. Curr Top Med Chem. 2007;7:251–263. doi: 10.2174/156802607779941305. [DOI] [PubMed] [Google Scholar]
- 182.Alvaro-Gracia JM. Licofelone--clinical update on a novel LOX/COX inhibitor for the treatment of osteoarthritis. Rheu matology. 2004;43(Suppl 1):i21–25. doi: 10.1093/rheumatology/keh105. [DOI] [PubMed] [Google Scholar]
- 183.Vidal C, Gomez-Hernandez A, Sanchez-Galan E, Gonzalez A, Ortega L, Gomez-Gerique JA, Tunon J, Egido J. Licofelone, a balanced inhibitor of cyclooxygenase and 5-lipoxygenase, reduces inflammation in a rabbit model of atherosclerosis. J Pharmacol Exp Ther. 2007;320:108–116. doi: 10.1124/jpet.106.110361. [DOI] [PubMed] [Google Scholar]
- 184.Koeberle A, Siemoneit U, Buhring U, Northoff H, Laufer S, Albrecht W, Werz O. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- 185.Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- 186.Hwang SH, Wagner KM, Morisseau C, Liu JY, Dong H, Wecksler AT, Hammock BD. Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem. 2011;54:3037–3050. doi: 10.1021/jm2001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Moser D, Wisniewska JM, Hahn S, Achenbach J, Buscato E, Klingler FM, Hofmann B, Steinhilber D, Proschak E. Dual-Target Virtual Screening by Pharmacophore Elucidation and Molecular Shape Filtering. ACS Med Chem Lett. 2012;3:155–158. doi: 10.1021/ml200286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liedtke AJ, Keck PR, Lehmann F, Koeberle A, Werz O, Laufer SA. Arylpyrrolizines as inhibitors of microsomal prostaglandin E2 synthase-1 (mPGES-1) or as dual inhibitors of mPGES-1 and 5-lipoxygenase (5-LOX) J Med Chem. 2009;52:4968–4972. doi: 10.1021/jm900481c. [DOI] [PubMed] [Google Scholar]
- 190.Koeberle A, Zettl H, Greiner C, Wurglics M, Schubert-Zsilavecz M, Werz O. Pirinixic acid derivatives as novel dual inhibitors of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Med Chem. 2008;51:8068–8076. doi: 10.1021/jm801085s. [DOI] [PubMed] [Google Scholar]
- 191.Greiner C, Zettl H, Koeberle A, Pergola C, Northoff H, Schubert-Zsilavecz M, Werz O. Identification of 2-mercaptohexanoic acids as dual inhibitors of 5-lipoxygenase and microsomal prostaglandin E(2) synthase-1. Bioorg Med Chem. 2011;19:3394–3401. doi: 10.1016/j.bmc.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 192.Koeberle A, Rossi A, Zettl H, Pergola C, Dehm F, Bauer J, Greiner C, Reckel S, Hoernig C, Northoff H, Bernhard F, Dotsch V, Sautebin L, Schubert-Zsilavecz M, Werz O. The molecular pharmacology and in vivo activity of 2-(4-chloro-6-(2,3-dimethylphenylamino)pyrimidin-2-ylthio)octanoic acid (YS121), a dual inhibitor of microsomal prostaglandin E2 synthase-1 and 5-lipoxygenase. J Pharmacol Exp Ther. 2010;332:840–848. doi: 10.1124/jpet.109.160663. [DOI] [PubMed] [Google Scholar]
- 193.Wu Y, He C, Gao Y, He S, Liu Y, Lai L. Dynamic modeling of human 5-lipoxygenase-inhibitor interactions helps to discover novel inhibitors. J Med Chem. 2012;55:2597–2605. doi: 10.1021/jm201497k. [DOI] [PubMed] [Google Scholar]
- 194.McGee JE, Fitzpatrick FA. Erythrocyte-neutrophil interactions: formation of leukotriene B4 by transcellular biosynthesis. Proc Natl Acad Sci USA. 1986;83:1349–1353. doi: 10.1073/pnas.83.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen Z, Wu Y, Liu Y, Yang S, Chen Y, Lai L. Discovery of dual target inhibitors against cyclooxygenases and leukotriene A4 hydrolyase. J Med Chem. 2011;54:3650–3660. doi: 10.1021/jm200063s. [DOI] [PubMed] [Google Scholar]
- 196.Hibi S, Okamoto Y, Tagami K, Numata H, Kobayashi N, Shinoda M, Kawahara T, Murakami M, Oketani K, Inoue T, et al. Novel dual inhibitors of 5-lipoxygenase and thromboxane A2 synthetase: synthesis and structure-activity relationships of 3-pyridylmethyl-substituted 2-amino-6-hydroxybenzothiazole deriva tives. J Med Chem. 1994;37:3062–3070. doi: 10.1021/jm00045a011. [DOI] [PubMed] [Google Scholar]
- 197.Hibi S, Okamoto Y, Tagami K, Numata H, Kobayashi N, Shinoda M, Kawahara T, Harada K, Miyamoto K, Yamatsu I. Structure-activity relationships of (E)-3-(1,4-benzoquinonyl)-2-[(3-pyridyl)-alkyl]-2-propenoic acid derivatives that inhibit both 5-lipoxygenase and thromboxane A2 synthetase. J Med Chem. 1996;39:3148–3157. doi: 10.1021/jm950725r. [DOI] [PubMed] [Google Scholar]
- 198.Draper N, Stewart PM. 11beta-hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J Endocrinol. 2005;186:251–271. doi: 10.1677/joe.1.06019. [DOI] [PubMed] [Google Scholar]
- 199.Marino JPBCA, Eidam P, Mcatee JJ. sEH AND 11β-HSD1 DUAL INHIBITORS. 2010/011917 A1. WO. 2008 Jul 25;
- 200.Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol Cancer Ther. 2009;8:2193–2203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal antiinflammatory drugs. Lancet. 2008;371:270–273. doi: 10.1016/S0140-6736(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 202.Marnett LJ. The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol. 2009;49:265–290. doi: 10.1146/annurev.pharmtox.011008.145638. [DOI] [PubMed] [Google Scholar]
- 203.Gilbert NC, Bartlett SG, Waight MT, Neau DB, Boeglin WE, Brash AR, Newcomer ME. The structure of human 5-lipoxygenase. Science. 2011;331:217–219. doi: 10.1126/science.1197203. [DOI] [PMC free article] [PubMed] [Google Scholar]