

Abstract
The commensal microbiota of fish skin is suspected to provide a protection against opportunist infections. The skin of fish harbors a complex and diverse microbiota that closely interacts with the surrounding water microbial communities. Up to now there is no clear evidence as to whether the host regulates the recruitment of environmental bacteria to build a specific skin microbiota. To address this question, we detected Quantitative Trait Loci (QTL) associated with the abundance of specific skin microbiota bacterial strains in brook charr (Salvelinus fontinalis), combining 16S RNA tagged-amplicon 454 pyrosequencing with genetic linkage analysis. Skin microbiota analysis revealed high inter-individual variation among 86 F2 fish progeny based upon the relative abundance of bacterial operational taxonomic units (OTUs). Out of those OTUs, the pathogenic strain Flavobacterium psychrophilum and the non-pathogenic strain Methylobacterium rhodesianum explained the majority of inter-individual distances. Furthermore, a strong negative correlation was found between Flavobacterium and Methylobacterium, suggesting a mutually competitive relationship. Finally, after considering a total of 266 markers, genetic linkage analysis highlighted three major QTL associated with the abundance of Lysobacter, Rheinheimera and Methylobacterium. All these three genera are known for their beneficial antibacterial activity. Overall, our results provide evidence that host genotype may regulate the abundance of specific genera among their surface microbiota. They also indicate that Lysobacter, Rheinheimera and Methylobacterium are potentially important genera in providing protection against pathogens.
Introduction
The study of the beneficial effects of bacteria and their influence on host health is a growing field. Namely, research that has explored host microbiota variability in space and time suggests the presence of a host genetic component into the development of mutualism communities [1], [2]. Similar conclusions were found in several studies on twins and on their core gut microbiota [3], [4], [5], [6]. Furthermore, microbiome homeostasis seems to be the key to resistance against some diseases previously considered exclusively influenced by genetic factors [7]. To determine precisely which genes are involved in the recruitment of specific bacterial strains, some studies looked at gene expression in presence of symbiotic bacteria. Recent studies specifically targeted genes already known as being involved in innate or adaptive immunity (for example IgA) [8], [9], [10].
In fish, the influence of host genetic background on commensal bacterial community structure is poorly known. However, advances in the field of probiotic development indicate that endogenous bacteria are able to outcompete pathogens [11], [12]. Results obtained in aquaculture settings are consistent with previous work showing that non-pathogenic bacteria associated with animal mucosa can contribute to the host health by providing protection against pathogen infections [13], [14], [15], [16]. The genetic basis of the host immune response, especially at the major histocompatibility complex (MHC), toll-like receptor (TLR) and immunoglobulin loci, has been well studied [17], [18], [19], [20]. However, aside from the immune response, the influence of the host on the development of bacterial symbiosis remains poorly understood.
Here, the main objective of the present work was to investigate the potential link between host genotype and skin microbiota composition. As a host model, we focused on brook charr (Salvelinus fontinalis), a salmonid that harbors a complex and dynamic skin microbiota [21], [22]. The first specific objective sought to document the genetic variation present in the host population that might underpin the inter-individual variations in the skin microbiota. The second specific objective was to characterize the relationship (cooperation/competition) existing among the different bacterial Operational taxonomic units (OTU) within the skin mucus, which influences the overall structure of the skin microbial community. The third objective was to identify host genetic regions associated to the abundance of specific skin bacterial OTUs (i. e. quantitative trait loci (QTL) associated with the abundance of each constituent bacterial genus). Accordingly, we analyzed the taxonomic structure of skin bacterial community using 16S tagged-amplicon pyrosequencing in 86 F2 fish progenies obtained in the previous work of Sauvage et al. (2012). Inter-individual variation in abundance of each bacterial strain detected in host microbiota was afterwards projected on the results of the genetic linkage map in order to identify quantitative trait loci (QTL) of specific skin bacterial OTUs.
Materials and Methods
Ethics statement
All fish were reared and the experiment conducted strictly following guidelines required by the “Comité de Protection des Animaux de l'Université Laval (CPAUL, http://www.vrr.ulaval.ca/deontologie/cpa/index.html?accueil). The CPAUL reviewed and approved all experimental procedures used in this study.
Fish sampling
The study population was generated using two different populations of brook charr. The first one (D) is a domestic population used in aquaculture for more than 100 years (Pisciculture de la Jacques Cartier - Cap-Santé, Québec, Canada). The other population (L), is an anadromous population from the Laval River near Forestville (North of the St. Lawrence River, Québec, Canada). Breeders from the L population were kept in captivity at the Station aquicole de l'ISMER (Québec, Canada, 48°31′N, 68°28′W) under natural photoperiod and temperature. Crosses between 10 sires of each population (L and D; F0 generation) with 10 dams (L and D) were performed to generate 10 full-sib outbred hybrid families (L×D - F1 generation). Subsequently, six F1 individuals were bi-parentally crossed to obtain three F2 families. The F2 family selected for the present study demonstrated the lowest post-hatch mortality rate (<2%) and the greatest abundance. Fish were raised in the same tank and had fasted for 12 hours before sampling. Fish were measured (mean = 28.8±1.77 cm) and weighed (mean = 276.7±59.48 g) to calculate Fulton index [23].
Mucus was sampled using a sterile swab on the same area for all the fish [24]. We choose to sample the skin between the adipose fin and the caudal fin because this area was undisturbed by fish handling. Samples were stored in a sterile micro-centrifuge tube containing lysis buffer (Tris 50 mM, EDTA 40 mM, sucrose 0.75 g) and immediately stored in −80°C until DNA extraction.
DNA extraction
DNA was extracted using a modified protocol of salt-extraction from Aljanabi & Martinez (1997). During the first lysis step, 22.6 µL of lysozyme (40 mg/mL) was added to the sample and incubated 45 minutes at 37°C. After this step, 22.6 µL of proteinase K (20 mg/µL) and 90 µL of SDS (10%) were added to the lysate and incubated at 55°C over night with agitation. The aqueous phase was transferred into a clean Eppendorf tube containing 600 µL of NaCl 6M, mixed and centrifuged 20 min at 14,800 rpm. The supernatant was transferred again into a clean Eppendorf tube containing 1 volume of cold isopropanol, mixed and stored 30 minutes at −20°C. The mixture was centrifuged 20 minutes at 14,800 rpm and the supernatant was thrown away. The pellet was washed with cold ethanol 70%, air-dried and finally resuspended in 25 µL of sterile MilliQ H2O. Subsequently, DNA integrity and quantity were controlled using a Nanodrop instrument (ND-1000, Nanodrop).
Microbial 16S pyrosequencing
Each DNA sample, the 16S gene was PCR amplified using Takara Ex taq premix (Fisher). All PCR reactions were performed in a reaction volume of 50 µL containing 25 µL of premix Taq, 1 µM of each primer and sterile MilliQ H2O to up to 50 µL. A general reverse primer (R519) combined with B primer (Roche) was used for amplifications in combination with one of 86 uniquely tagged forward primers (F63-targeted) combined with A primer (Roche) (for primer sequences see [25], [26]. The mean length of the amplified fragment was 450 bp. This procedure facilitates the parallel sequencing of thousands of samples on the same run and to reassign each reads to their respective samples. PCR conditions were applied as follows: after a denaturing step of 30 s at 98°C, samples were processed through 30 cycles consisting of 10 sec at 98°C, 30 sec at 55°C, and 30 sec at 72°C. The final extension step was done at 72°C for 4 min 30 sec. Following the amplification step, samples were purified using AMPure Beads (Beckman Coulter Genomics). Samples were adjusted to 100 µL with EB (Qiagen), 63 µL of beads were added. Samples were mixed and incubated for 5 min at RT. Using a Magnetic Particle Concentrator (MPC), the beads were pelleted against the wall of the tube and supernatant was removed. The beads were washed twice with 500 µL of 70% ethanol and incubated for 30 sec each time. Supernatant was removed and beads were air dried for 5 min. Tubes were removed from the MPC and 24 µL of EB were added. Samples were vortexed in order to suspend the beads. Finally, using the MPC, the beads were pelleted against the wall once more and supernatant was transferred to a new clean tube. Samples were quantified with Nanodrop before the amplification step. Amplicons were then quantified with Quant-iT PicoGreen dsDNA Assay Kit and mixed equally before being sent to the Plateforme d'Analyses Biomoléculaires (Institut de Biologie Intégrative et des Systèmes, Université Laval) for pyrosequencing on a 454 GS-FLX DNA Sequencer with the Titanium Chemistry (Roche), according to manufacturer's procedure.
16S sequence analysis
All sequences are available on MG-RAST server (MG-RAST IDs: 4539915.3). The data were analyzed in two steps. First, CLC Genomics Workbench 3.1 (CLC Bio, Aarhus, Denmark CLC workbench BIO) was used to trim sequences for quality, recover and remove the primers' sequences and tags (minimum average quality score: 35 for a window of 50, number of differences to the primer sequence = 0, maximum number of differences to the barcode sequence = 0, number of ambiguous base calls = 0, maximum homopolymer length = 8). In a second step, pre-processing and analysis were performed using the MOTHUR software [27]. All datasets were checked for chimeras with the chimera slayer algorithm implemented in MOTHUR. Standardization of the different samples was done by using the zscore which calculates the normalized abundance as follows: normalized abundance = (abundance - mean) / standard deviation [3], [28], [29]. An alternative method for normalization was also tested, which consisted of subsampling equal number of reads from each sample [30]. This method greatly reduced the numbers of sequences (224 sequences per samples), but gave the same results as zscore (Figure S2, Figure S3, Table S1). Therefore we preferred to keep zscore normalization to base our conclusions on a larger dataset. We used the Operational Taxonomic Unit-based method described by Costello et al. (2009) because it is not biased towards a predefined taxonomy. One index was retained to assess the quality of pyrosequencing: the sequence coverage index (Good's estimator). The sequence coverage index is a metric used to estimate the quality of the depth sequencing. All sequences were clustered into Operational Taxonomic Unit (OTU) using a 97% identity threshold and OTU were classified from phylum to genus using the program MOTHUR with the default settings. For all interesting OTUs (explaining the inter-individual variation or linked to a genetic region), we extracted all the sequences classified in that OTU and used the single best BLAST hit to identify the different species. Statistical differences in the abundance of each OTU were calculated with the software for metagenomic analysis (Metastats).
To visualize potential differences across host progenies in terms of mucus bacterial community structure, distances between those communities were computed using the Yue & Clayton measure of dissimilarity (Thetayc). This index developed by Yue and Clayton (2005) is a measure of dissimilarity between the structures of two communities, meaning that this calculator takes in account the abundances of each OTU. Then, Distances were represented using a dendrograms based on this index and statistical robustness of the dendrogram was determined by a Unifrac weighted test. This test allows determining whether any of the samples have a significantly different structure than the other samples. A random (Monte Carlo) permutation test was performed to test whether or not the distance between two communities was greater than expected by chance alone.
To visualize the distances between communities, we also applied a Principal Coordinate Analysis (PCoA) based on the Yue & Clayton measure of dissimilarity (Thetayc) using an eigenvector-based approach to represent multidimensional data in as few dimensions as possible. This method allows determining which OTUs are responsible for shifting the samples along the PCoA axes by measuring the correlation of the relative abundance of each OTU clustered in genera with the axes of the PCoA dataset. Statistical differences in the abundance of each OTU was calculated to highlight which genera were the causative agents of the differentiation between groups obtained in the dendrogram based upon the TethaYC index [31]. All statistical analyses and graphics were carried out in the R environment (http://www.r-project.org).
QTL detection
QTL analysis was carried out using the [R] package J/qtl [v. 1.3.1, August 2012, http://churchill.jax.org/software/jqtl.shtml/]. QTL were projected on the consensus brook charr linkage map (see Sauvage et al., 2012) according to the following steps. (1) A single QTL analysis was performed using the Haley-Knott (HK) regression method (10,000 permutations) to reveal which LGs were carrying QTL. The most probable position of the QTL was defined at the position giving the largest logarithm of odds (LOD) score indicating the QTL was fixed. (2) A two QTL model based upon the Haley-Knott regression was used to refine the QTL detection across the genome with a resolution of 5 cM and eventually to detect two QTL on a single LG linked to a particular trait. (3) The best model fitting our data was used to compute the percent variance explained (PVE expressed in %) by the QTL. The chromosome-wide and the genome-wide thresholds were calculated for each LG using 10,000 permutations. The 1.5 LOD confidence intervals were determined for all analyses following the Bayesian method implemented in the “bayesint” function in R/qtl. The bayesint function calculates an approximate interval (end points around the maximum LOD) for a given chromosome using the genome scan output.
Results
Mucus samples were obtained from 86 F2 fish progenies (44 males and 42 females) issued from the same family. A total of 87,940 high-quality, classifiable sequences were obtained with an average number of 1022±540 per sample, which were subsequently classified in OTUs [1]. All of these sequences were successfully clustered into 9520 OTUs with 97% identity. A final trimming step was performed to focus on the most abundant OTU (which are represented by at least 10 sequences for the whole project) and result in a dataset containing 71,719 sequences (81% of the initial data set) clustered in 192 OTUs. The depth of sequencing and the coverage were estimated using the Good's estimator index; the coverage found was always higher than 90% except for one sample (#127), which exhibited 76% of coverage (Table 1, Figure S1). The number of OTUs per sample was not equally distributed and the mean number was 156.5±81.7 (ranging from 33 to 368). The alpha-diversity was estimated by calculating the non-parametric index of Shannon (npShannon) because of the non-equally distributed abundance of each OTU in the samples. The npShannon index ranged from 0.29 to 3.45, with a mean index of 1.43±0.83.
Table 1. Descriptive statistics of sequencing to determine the microbiotoa for each individual.
| Sample ID | number of reads | coverage | npshannon index | Number of OTU total | Number of OTU >10 reads |
| 92 | 229 | 0.955975 | 1.690045 | 76 | 19 |
| 93 | 1584 | 0.99131 | 0.436879 | 74 | 28 |
| 94 | 669 | 0.977401 | 1.507996 | 242 | 20 |
| 95 | 738 | 0.986166 | 1.416086 | 226 | 27 |
| 96 | 503 | 0.953079 | 1.395581 | 149 | 29 |
| 97 | 385 | 0.946203 | 1.774329 | 86 | 33 |
| 98 | 955 | 0.98389 | 0.663968 | 79 | 30 |
| 99 | 770 | 0.978852 | 1.306709 | 114 | 29 |
| 100 | 1018 | 0.985946 | 0.658787 | 107 | 30 |
| 101 | 1482 | 0.994792 | 0.842304 | 188 | 21 |
| 102 | 1667 | 0.992283 | 0.691189 | 96 | 31 |
| 103 | 711 | 0.99179 | 0.946539 | 97 | 23 |
| 104 | 1072 | 0.987599 | 0.719994 | 159 | 25 |
| 105 | 1662 | 0.991645 | 0.371924 | 73 | 26 |
| 106 | 1304 | 0.995734 | 0.658557 | 93 | 18 |
| 107 | 1617 | 0.99416 | 0.290016 | 49 | 21 |
| 108 | 1237 | 0.990451 | 0.634742 | 73 | 30 |
| 109 | 1063 | 0.987778 | 0.679124 | 137 | 26 |
| 110 | 752 | 0.97281 | 0.656197 | 105 | 29 |
| 111 | 731 | 0.986217 | 0.63276 | 79 | 22 |
| 112 | 628 | 0.975779 | 0.630137 | 63 | 23 |
| 113 | 892 | 0.983549 | 0.446389 | 49 | 24 |
| 114 | 1333 | 0.993127 | 0.713271 | 127 | 22 |
| 115 | 1109 | 0.983313 | 1.020009 | 266 | 35 |
| 116 | 532 | 0.967611 | 2.709733 | 269 | 32 |
| 117 | 649 | 0.963145 | 2.108945 | 237 | 41 |
| 118 | 404 | 0.923977 | 2.645631 | 222 | 29 |
| 119 | 969 | 0.97625 | 1.019139 | 177 | 40 |
| 120 | 581 | 0.967647 | 1.975631 | 205 | 26 |
| 121 | 468 | 0.948413 | 2.692236 | 202 | 34 |
| 122 | 744 | 0.980551 | 0.876389 | 117 | 27 |
| 123 | 953 | 0.983204 | 0.832745 | 133 | 32 |
| 124 | 530 | 0.967846 | 2.29117 | 186 | 30 |
| 125 | 797 | 0.977901 | 0.703269 | 79 | 30 |
| 126 | 411 | 0.97561 | 0.904985 | 116 | 18 |
| 127 | 242 | 0.76 | 3.452235 | 182 | 32 |
| 128 | 1230 | 0.989637 | 0.504536 | 69 | 26 |
| 129 | 831 | 0.978723 | 1.537117 | 115 | 41 |
| 130 | 1449 | 0.983421 | 0.548428 | 123 | 38 |
| 131 | 499 | 0.969697 | 2.118408 | 144 | 29 |
| 132 | 431 | 0.945813 | 2.133064 | 181 | 25 |
| 133 | 494 | 0.96114 | 1.408929 | 117 | 35 |
| 134 | 746 | 0.980254 | 0.508333 | 38 | 22 |
| 135 | 662 | 0.977887 | 2.41624 | 180 | 36 |
| 136 | 754 | 0.987755 | 0.956343 | 118 | 18 |
| 137 | 2008 | 0.993154 | 0.557917 | 74 | 30 |
| 138 | 714 | 0.976619 | 1.632525 | 101 | 36 |
| 139 | 798 | 0.967632 | 1.534968 | 129 | 35 |
| 140 | 469 | 0.917197 | 3.267019 | 261 | 35 |
| 141 | 224 | 0.980892 | 0.662835 | 33 | 6 |
| 142 | 517 | 0.957377 | 2.388226 | 180 | 36 |
| 143 | 483 | 0.947195 | 2.446711 | 157 | 36 |
| 144 | 414 | 0.958848 | 3.124298 | 188 | 38 |
| 145 | 930 | 0.975443 | 1.391363 | 168 | 43 |
| 146 | 769 | 0.979661 | 2.024708 | 140 | 28 |
| 147 | 1219 | 0.995122 | 1.641489 | 118 | 17 |
| 148 | 1254 | 0.98244 | 1.127563 | 148 | 40 |
| 149 | 1074 | 0.972678 | 2.309049 | 318 | 55 |
| 150 | 1108 | 0.980583 | 2.793443 | 321 | 47 |
| 151 | 3082 | 0.998643 | 0.332436 | 58 | 16 |
| 152 | 1429 | 0.996561 | 1.820656 | 203 | 25 |
| 153 | 1955 | 0.995286 | 0.815557 | 167 | 27 |
| 154 | 1449 | 0.983366 | 1.74373 | 354 | 54 |
| 155 | 1074 | 0.973611 | 1.716763 | 321 | 41 |
| 156 | 1016 | 0.989316 | 0.588719 | 62 | 21 |
| 157 | 1483 | 0.984836 | 1.374063 | 216 | 46 |
| 158 | 2004 | 0.998944 | 0.791157 | 44 | 10 |
| 159 | 2043 | 0.997359 | 0.812388 | 92 | 18 |
| 160 | 2468 | 0.995741 | 0.305525 | 70 | 23 |
| 161 | 1215 | 0.979566 | 1.928985 | 346 | 48 |
| 162 | 1904 | 0.995286 | 0.821609 | 164 | 37 |
| 163 | 1046 | 0.985799 | 1.569251 | 188 | 40 |
| 164 | 1145 | 0.993814 | 0.744587 | 98 | 19 |
| 165 | 995 | 0.981481 | 1.421104 | 243 | 37 |
| 166 | 725 | 0.97921 | 2.459141 | 211 | 36 |
| 167 | 727 | 0.988743 | 2.142562 | 131 | 24 |
| 168 | 841 | 0.98908 | 2.297402 | 151 | 31 |
| 169 | 552 | 0.968116 | 2.76388 | 178 | 35 |
| 170 | 1204 | 0.99177 | 1.912832 | 162 | 32 |
| 172 | 2010 | 0.993946 | 0.610072 | 155 | 36 |
| 173 | 1454 | 0.987296 | 1.893132 | 316 | 51 |
| 174 | 1056 | 0.979651 | 2.949199 | 335 | 53 |
| 175 | 824 | 0.95098 | 2.845177 | 368 | 46 |
| 176 | 851 | 0.96729 | 2.039242 | 202 | 47 |
| 177 | 778 | 0.985507 | 2.23077 | 221 | 25 |
| 178 | 2142 | 0.998093 | 0.359619 | 52 | 13 |
The coverage was estimated by a Good's estimator index. npshannon: non parametric index of Shannon. OTU classification was done with a treshold of 97% identity.
Phylum, class, and OTU composition of all samples were represented in the Figure 1. Most of the OTUs belong to the Proteobacteria phylum (88.7%) and the Bacteroidetes phyla (9.7%). At the class level, most of the OTUs were classified as Alpha-proteobacteria (78.9%), Gamma-proteobacteria (6.5%) and Flavobacteria (13%). At the OTU level, the most abundant sequence was identified via BLAST as M. rhodesianum (69.8%) followed by F. psychrophilum (8%) (Figure 1 and Table S1).
Figure 1. Taxonomic structure of the bacterial community of fish skin mucus at three different taxonomic levels: Phylum, Classl and OTU.
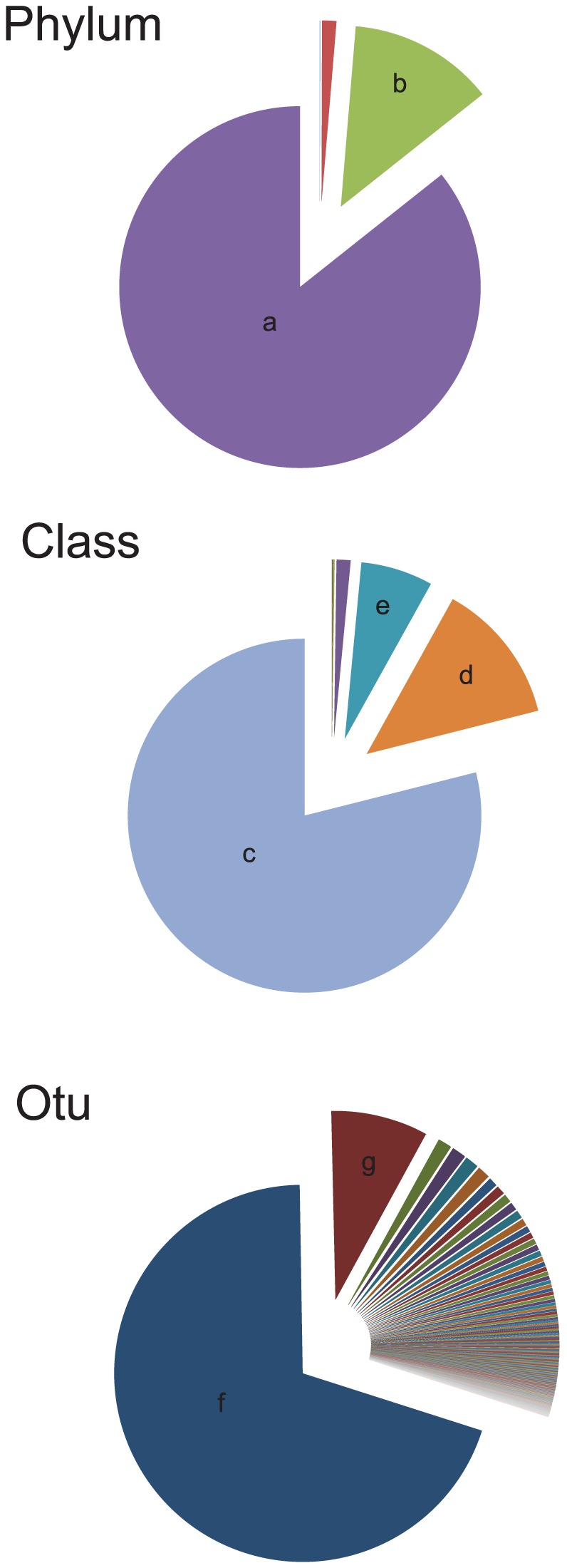
a) Proteobacteria; b) Bacteroidetes, c) Alphaproteobacteria, d) Flavobacteria, e) Gammaproteobacteria, f) OTU 50 (Methylobacterium rhodesianum), g) OTU 36 (Flavobacterium psychrophilum).
Male and female brook charr harbored slightly different microbial communities. The Unifrac weighted distance between samples from males and females was statistically significant indicating that structures of bacterial communities were differentiated according to the sex of the host (Unifrac Score: 0.241695; p-value <0.001). However, there was not a sex difference in the two most abundant species, as this difference was based on 22 OTUs classified on 20 genera and 21 species, all of them being less abundant than 0.1% (Table 2).
Table 2. Taxonomic variation observed between male and female.
| Condition | N° OTU | Best Hit on Blast | Identity | Relative abundance |
| More abundant in female | 82 | Amaricoccus kaplicensis | 98% | 0.00037647 |
| 115 | Loktanella salsilacus | 99% | 0.00015338 | |
| 6 | Methylovirgula ligni | 91% | 0.00026492 | |
| 163 | Micrococcus antarcticus | 99% | 0.00061351 | |
| 88 | Paracoccus yeei | 94% | 0.00019521 | |
| 75 | Pseudochrobactrum kiredjianiae | 99% | 0.00050196 | |
| 128 | Psychrobacter faecalis | 99% | 0.00100392 | |
| 131 | Rheinheimera pacifica | 99% | 0.00057168 | |
| 7, 11 | Singularimonas variicoloris | 89% | 0.00037647 | |
| 104 | Sphingopyxis witflariensis | 99% | 0.00015338 | |
| 113 | Stella humosa | 100% | 0.00018126 | |
| 142 | Stenotrophomonas nitritireducens | 99% | 0.00022309 | |
| More abundant in male | 186 | Flavobacterium aquatile | 97% | 0.00029281 |
| 190 | Hyphomicrobium sulfonivorans | 95% | 0.00039041 | |
| 4 | Methylosinus sporium | 92% | 0.00027887 | |
| 176 | Polynucleobacter necessarius subsp. Asymbioticus | 99% | 0.00015338 | |
| 129 | Pseudomonas hibiscicola | 99% | 0.00075294 | |
| 12 | Pseudorhodoferax soli | 85% | 0.00018126 | |
| 140 | Psychrobacter alimentarius | 99% | 0.00015338 | |
| 116 | Rhodobacter blasticus | 95% | 0.00016732 | |
| 175 | Simplicispira limi | 99% | 0.00034858 |
This table summarize the OTUs differentially abundant between skin mucus communities between male and female (Metastat using a FDR correction, p-value<0.01).
In terms of community structure, clustering based on ThetaYC index clearly defined two groups of samples, whilst a third group was less well defined and composed of highly variable communities (Figure 2). There was little variation among samples in the first and second group (group 1 and group 2). The third group (group 3) was an assemblage of dissimilar communities and is considered as an external group. The variation of the alpha-diversity visualized by the npShannon index (Figure 3) indicated that alpha-diversity increases from group 1 to group 3. Furthermore, the bacterial communities within the third group were more diverse than groups 1 and 2. The same clustering was found with the normalization by subsampling (Figure S2, Figure S3, table S1). The difference between each group was greater than the difference between males and females (Figure 2). The most differentiated groups were groups 1 and 3 (Unifrac Score: 0.710811, p-value <0.001) followed by the distance between groups 2 and 3 (Unifrac Score: 0.685361, p-value <0.001), and the distance between groups 1 and 2 (Unifrac Score: 0.401674, p-value <0.001). This was correlated with the numbers of OTU that were differentially abundant between those 3 groups (Figure 4). The divergence between groups 1 and 2 was based upon 54 OTU classified in 26 genera and 41 species (Table 3). The microbiota of the first group was dominated by M. rhodesianum, and host individuals from this group were tightly clustered. The second group was more diverse, dominated by Flavobacterium and had a lower abundance of M. rhodesianum. For groups 1 and 3, the difference was based upon 109 OTU classified in 45 genera and 70 species (Table 4). Finally, 26 OTU were differently abundant between group 2 and 3 and classified in 17 genera and 22 species (Table 5).
Figure 2. Dendrogram analysis based upon ThetaYC index of bacteria found on the skin of the 86 brook charr individuals.
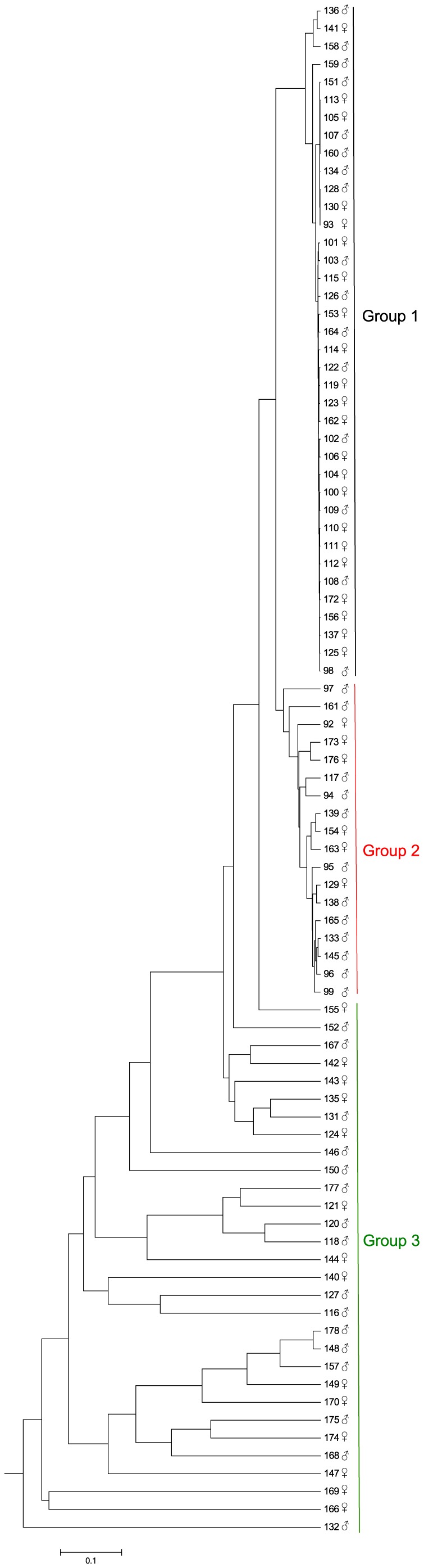
Groups are defined with the Weighted Unifrac distance. The first and second groups are composed of closely related bacterial communities. The third group is an assemblage of dissimilar communities and is considered as an outgroup. The most differentiated groups are groups 1 and 3 (Unifrac Score: 0.710811, p<0.0010) followed by the distance between groups 2 and 3 (Unifrac Score: 0.685361, p<0.0010), and the distance between groups 1 and 2 (Unifrac Score: 0.401674, p<0.0010).
Figure 3. Alpha diversity of each group.

Each group was defined by the dendrogram analysis based upon ThetaYC index (see figure 2). Alpha-diversity was calculated by the non-parametric index of Shannon.
Figure 4. Descriptive analysis of the Metastats.

Numbers indicates the numbers of OTUs differentially present in the different groups of individuals. For details on the identities of the OTUs, please refer to the table 3; 4 and 5.
Table 3. Detailed information related to the OTU differentially abundant between individuals belonging to group 1 and group 2.
| N° OTU | mean abundance in group 1 | mean abundance in group 2 | Best hit on Blast |
| 36 | 0.00484 | 0.037926 | Flavobacterium psychrophilum |
| 122 | 0.000778 | 0.010066 | Pseudomonas cedrina subsp. Fulgida |
| 37 | 0.000471 | 0.006593 | Flavobacterium terrigena |
| 68 | 0.0014 | 0.006629 | Methylocella palustris |
| 128 | 0 | 0.004734 | Psychrobacter faecalis |
| 69 | 0.000531 | 0.004942 | Pseudorhodobacter ferrugineus |
| 190 | 0 | 0.004394 | Hyphomicrobium sulfonivorans |
| 139 | 0.000153 | 0.003833 | Lysobacter capsici |
| 70 | 0.000831 | 0.0043 | Methylobacterium salsuginis |
| 29 | 0.000028 | 0.003344 | Methylobacterium adhaesivum |
| 67 | 0.000518 | 0.003772 | Rhodobacter blasticus |
| 127 | 0.000218 | 0.003382 | Acinetobacter haemolyticus |
| 21 | 0.000117 | 0.002977 | Flavobacterium aquatile |
| 74 | 0 | 0.002743 | Paracoccus yeei |
| 52 | 0.000331 | 0.002911 | Sphingopyxis alaskensis |
| 126 | 0.000969 | 0.003524 | Pseudomonas xanthomarina |
| 89 | 0 | 0.002528 | Sphingomonas dokdonensis |
| 27 | 0.000113 | 0.001814 | Sphingopyxis chilensis |
| 63 | 0.00071 | 0.002374 | Paracoccus haeundaensis |
| 141 | 0.000156 | 0.001642 | Acinetobacter johnsonii |
| 84 | 0.000079 | 0.001484 | Pseudorhodobacter ferrugineus |
| 38 | 0.000063 | 0.001334 | Sejongia jeonii |
| 138 | 0.000014 | 0.001205 | Acinetobacter junii |
| 192 | 0 | 0.001099 | Methylovirgula ligni |
| 81 | 0.000248 | 0.00132 | Ruegeria atlantica |
| 8 | 0 | 0.001062 | Methylosinus sporium |
| 6 | 0 | 0.001033 | Methylovirgula ligni |
| 12 | 0 | 0.001029 | Pseudorhodoferax soli |
| 86 | 0.000282 | 0.00131 | Methylobacterium rhodesianum |
| 32 | 0.000125 | 0.001139 | Methylobacterium zatmanii |
| 22 | 0.000011 | 0.000871 | Flavobacterium psychrophilum |
| 107 | 0.000082 | 0.000932 | Rhodobacter capsulatus |
| 43 | 0.000258 | 0.001098 | Flavobacterium aquatile |
| 94 | 0.000073 | 0.000886 | Rhodobacter sphaeroides |
| 189 | 0.000043 | 0.000809 | Methylomicrobium album |
| 78 | 0.000047 | 0.000761 | Rhodobacter capsulatus |
| 91 | 0.000226 | 0.000929 | Caulobacter leidyia |
| 46 | 0.00002 | 0.000712 | Flavobacterium psychrolimnae |
| 101 | 0.00042 | 0.001111 | Brevundimonas variabilis |
| 154 | 0.00005 | 0.000728 | Methylobacterium adhaesivum |
| 82 | 0.000156 | 0.000815 | Amaricoccus kaplicensis |
| 54 | 0.000176 | 0.000782 | Methylopila capsulata |
| 19 | 0.000154 | 0.000756 | Brevundimonas variabilis |
| 71 | 0 | 0.00054 | Sphingomonas faeni |
| 182 | 0.000046 | 0.000579 | Flavobacterium aquatile |
| 90 | 0.000282 | 0.000812 | Methylobacterium adhaesivum |
| 44 | 0.000094 | 0.000609 | Flavobacterium psychrophilum |
| 143 | 0 | 0.000513 | Dokdonella koreensis |
| 112 | 0 | 0.000511 | Sphingomonas sanguinis |
| 97 | 0 | 0.00049 | Hyphomicrobium facile subsp. Tolerans |
| 105 | 0 | 0.000359 | Paracoccus haeundaensis |
| 137 | 0 | 0.000339 | Acinetobacter johnsonii |
| 75 | 0.000558 | 0 | Pseudochrobactrum kiredjianiae |
| 50 | 0.889343 | 0.677999 | Methylobacterium rhodesianum |
Differentially abundancy were calculated by using Metastats (using a FDR correction, p<0.01).
Table 4. Detailed information related to the OTU differentially abundant between individuals belonging to group 1 and group 3.
| OTU | mean abundance in group 1 | mean abundance in group 3 | Best hit on Blast |
| 36 | 0.00484 | 0.15627 | Flavobacterium psychrophilum |
| 52 | 0.000331 | 0.052599 | Sphingopyxis alaskensis |
| 51 | 0.002582 | 0.045797 | Sphingomonas paucimobilis |
| 119 | 0.001237 | 0.034255 | Pseudomonas peli |
| 125 | 0.001567 | 0.030681 | Acinetobacter lwoffii |
| 120 | 0.002302 | 0.030821 | Rheinheimera texasensis |
| 63 | 0.00071 | 0.027857 | Paracoccus haeundaensis |
| 122 | 0.000778 | 0.018741 | Pseudomonas cedrina subsp. Fulgida |
| 61 | 0.003372 | 0.02093 | Porphyrobacter dokdonensis |
| 21 | 0.000117 | 0.011826 | Flavobacterium aquatile |
| 127 | 0.000218 | 0.00999 | Acinetobacter haemolyticus |
| 55 | 0 | 0.008105 | Bradyrhizobium pachyrhizi |
| 54 | 0.000176 | 0.007992 | Methylopila capsulata |
| 162 | 0.002112 | 0.009611 | Knoellia sinensis |
| 126 | 0.000969 | 0.008421 | Pseudomonas xanthomarina |
| 67 | 0.000518 | 0.007951 | Rhodobacter blasticus |
| 69 | 0.000531 | 0.007849 | Pseudorhodobacter ferrugineus |
| 81 | 0.000248 | 0.006693 | Ruegeria atlantica |
| 134 | 0.000245 | 0.00635 | Acinetobacter lwoffii |
| 143 | 0 | 0.005914 | Dokdonella koreensis |
| 129 | 0 | 0.005644 | Pseudomonas hibiscicola |
| 124 | 0 | 0.004908 | Nevskia soli |
| 40 | 0.000014 | 0.004576 | Flavobacterium aquatile |
| 38 | 0.000063 | 0.004547 | Sejongia jeonii |
| 68 | 0.0014 | 0.005784 | Methylocella palustris |
| 71 | 0 | 0.004232 | Sphingomonas faeni |
| 79 | 0.000259 | 0.004408 | Rhodobacter capsulatus |
| 78 | 0.000047 | 0.003657 | Rhodobacter capsulatus |
| 37 | 0.000471 | 0.004035 | Flavobacterium terrigena |
| 141 | 0.000156 | 0.003673 | Acinetobacter johnsonii |
| 156 | 0.000033 | 0.003487 | Sphingopyxis alaskensis |
| 39 | 0 | 0.00318 | Flavobacterium psychrophilum |
| 24 | 0.000095 | 0.00295 | Pseudomonas peli |
| 106 | 0.000106 | 0.002945 | Bosea vestrisii |
| 137 | 0 | 0.002615 | Acinetobacter johnsonii |
| 101 | 0.00042 | 0.002997 | Brevundimonas variabilis |
| 97 | 0 | 0.002498 | Hyphomicrobium facile subsp. Tolerans |
| 139 | 0.000153 | 0.002639 | Lysobacter capsici |
| 157 | 0.00006 | 0.002488 | Sphingomonas sanguinis |
| 116 | 0 | 0.002339 | Rhodobacter blasticus |
| 177 | 0.000289 | 0.00255 | Aquabacterium commune |
| 25 | 0.000069 | 0.002236 | Acinetobacter lwoffii |
| 182 | 0.000046 | 0.002172 | Flavobacterium aquatile |
| 47 | 0 | 0.002082 | Flavobacterium psychrophilum |
| 84 | 0.000079 | 0.00214 | Pseudorhodobacter ferrugineus |
| 164 | 0.000219 | 0.002164 | Rhodococcus fascians |
| 105 | 0 | 0.001788 | Paracoccus haeundaensis |
| 43 | 0.000258 | 0.002022 | Flavobacterium aquatile |
| 180 | 0.000017 | 0.001709 | Acidovorax defluvii |
| 92 | 0.000147 | 0.001809 | Sphingopyxis witflariensis |
| 4 | 0 | 0.00161 | Methylosinus sporium |
| 145 | 0 | 0.001417 | Acinetobacter johnsonii |
| 5 | 0 | 0.001415 | Magnetospirillum magnetotacticum |
| 88 | 0.000036 | 0.00143 | Amaricoccus veronensis |
| 3 | 0 | 0.001358 | Rhodobacter changlensis |
| 138 | 0.000014 | 0.001365 | Acinetobacter junii |
| 70 | 0.000831 | 0.002052 | Methylobacterium salsuginis |
| 184 | 0 | 0.001156 | Flavobacterium aquatile |
| 22 | 0.000011 | 0.001144 | Flavobacterium psychrophilum |
| 112 | 0 | 0.001106 | Sphingomonas sanguinis |
| 118 | 0.000062 | 0.001143 | Haematobacter missouriensis |
| 93 | 0.000029 | 0.001093 | Roseomonas mucosa |
| 152 | 0.000028 | 0.001009 | Rhodobacter blasticus |
| 35 | 0.00005 | 0.000936 | Sphingomonas sanguinis |
| 148 | 0.000033 | 0.000915 | Psychrobacter arcticus |
| 172 | 0 | 0.000864 | Sphingomonas sanguinis |
| 146 | 0 | 0.000832 | Pseudomonas mohnii |
| 94 | 0.000073 | 0.000865 | Rhodobacter sphaeroides |
| 140 | 0 | 0.000775 | Psychrobacter alimentarius |
| 160 | 0.000061 | 0.000823 | Sphingomonas faeni |
| 83 | 0.000128 | 0.000877 | Paracoccus aminovorans |
| 42 | 0.000323 | 0.001071 | Flavobacterium psychrophilum |
| 186 | 0 | 0.000711 | Flavobacterium aquatile |
| 44 | 0.000094 | 0.000796 | Flavobacterium psychrophilum |
| 117 | 0.000029 | 0.000731 | Sphingopyxis taejonensis |
| 107 | 0.000082 | 0.000766 | Rhodobacter capsulatus |
| 72 | 0.000432 | 0.001098 | Beijerinckia mobilis |
| 104 | 0.000028 | 0.000693 | Sphingopyxis witflariensis |
| 113 | 0 | 0.000663 | Stella humosa |
| 115 | 0 | 0.000622 | Loktanella salsilacus |
| 108 | 0.000051 | 0.000647 | Novosphingobium panipatense |
| 26 | 0 | 0.000559 | Acinetobacter johnsonii |
| 48 | 0 | 0.00052 | Flavobacterium psychrophilum |
| 166 | 0 | 0.000508 | Rhodococcus fascians |
| 65 | 0.00064 | 0.001148 | Methylobacterium organophilum |
| 103 | 0.000215 | 0.000655 | Sphingomonas ursincola |
| 144 | 0.00007 | 0.000462 | Acinetobacter lwoffii |
| 191 | 0.00006 | 0.000406 | Beijerinckia derxii subsp. Venezuelae |
| 19 | 0.000154 | 0.000498 | Brevundimonas variabilis |
| 178 | 0.000043 | 0.000378 | Pseudorhodoferax soli |
| 23 | 0.000017 | 0.000348 | Flavobacterium psychrophilum |
| 192 | 0 | 0.000328 | Methylovirgula ligni |
| 49 | 0.000058 | 0.000354 | Flavobacterium aquatile |
| 8 | 0 | 0.00029 | Methylosinus sporium |
| 9 | 0.000062 | 0.000245 | Sphingomonas wittichii |
| 169 | 0.000207 | 0.000287 | Corynebacterium tuberculostearicum |
| 111 | 0.000416 | 0.000202 | Mycoplana bullata |
| 7 | 0.000343 | 0 | Singularimonas variicoloris |
| 175 | 0.000348 | 0 | Simplicispira limi |
| 13 | 0.000409 | 0 | Chelatococcus daeguensis |
| 16 | 0.000441 | 0 | Sphingomonas wittichii |
| 150 | 0.000678 | 0.000214 | Sphingomonas wittichii |
| 75 | 0.000558 | 0 | Pseudochrobactrum kiredjianiae |
| 45 | 0.000606 | 0.000032 | Chryseobacterium indologenes |
| 173 | 0.000661 | 0.000034 | Methylobacterium rhodesianum |
| 30 | 0.002932 | 0.001072 | Methylobacterium populi |
| 62 | 0.004156 | 0.001574 | Methylobacterium salsuginis |
| 149 | 0.007494 | 0.00191 | Methylobacterium rhodesianum |
| 50 | 0.889343 | 0.227483 | Methylobacterium rhodesianum |
Differentially abundancy were calculated by using Metastats (using a FDR correction, p<0.01).
Table 5. Detailed information related to the OTU differentially abundant between individuals belonging to group 2 and group 3.
| N° OTU | mean abundance in group 2 | mean abundance in group 3 | Best hit on Blast |
| 63 | 0.002374 | 0.027857 | Paracoccus haeundaensis |
| 55 | 0 | 0.008105 | Bradyrhizobium pachyrhizi |
| 54 | 0.000782 | 0.007992 | Methylopila capsulata |
| 134 | 0.000155 | 0.00635 | Acinetobacter lwoffii |
| 129 | 0 | 0.005644 | Pseudomonas hibiscicola |
| 143 | 0.000513 | 0.005914 | Dokdonella koreensis |
| 124 | 0.000109 | 0.004908 | Nevskia soli |
| 40 | 0.000306 | 0.004576 | Flavobacterium aquatile |
| 123 | 0.000266 | 0.004124 | Acinetobacter haemolyticus |
| 71 | 0.00054 | 0.004232 | Sphingomonas faeni |
| 38 | 0.001334 | 0.004547 | Sejongia jeonii |
| 156 | 0.000306 | 0.003487 | Sphingopyxis alaskensis |
| 39 | 0.000144 | 0.00318 | Flavobacterium psychrophilum |
| 78 | 0.000761 | 0.003657 | Rhodobacter capsulatus |
| 137 | 0.000339 | 0.002615 | Acinetobacter johnsonii |
| 47 | 0.000087 | 0.002082 | Flavobacterium psychrophilum |
| 164 | 0.000236 | 0.002164 | Rhodococcus fascians |
| 4 | 0 | 0.00161 | Methylosinus sporium |
| 182 | 0.000579 | 0.002172 | Flavobacterium aquatile |
| 3 | 0 | 0.001358 | Rhodobacter changlensis |
| 184 | 0 | 0.001156 | Flavobacterium aquatile |
| 42 | 0.000252 | 0.001071 | Flavobacterium psychrophilum |
| 12 | 0.001029 | 0 | Pseudorhodoferax soli |
| 6 | 0.001033 | 0 | Methylovirgula ligni |
| 27 | 0.001814 | 0.000082 | Sphingopyxis chilensis |
| 50 | 0.677999 | 0.227483 | Methylobacterium rhodesianum |
Differentially abundancy were calculated by using Metastats (using a FDR correction, p<0.01).
PCoA analysis displayed the same patterns of divergence between the three groups (Figure 5). Each group found in the Unifrac analysis was highlighted in the PCoA. To understand which OTUs were responsible for the differentiation of the cluster on the PCoA axes 1 and 2, individual correlation coefficients were calculated. Three OTUs were highly correlated with the two axes (σ>0.6); OTU 36, 50 and 149. Those OTU belonged to two species: Flavobacterium psychrophilum (OTU 36) and M. Rhodesianum (OTU 50 and 149). The negative correlation between Methylobacterium and Flavobacterium was further confirmed by the co-occurrence analysis, which identified potential taxonomic interactions between genera. Two significant correlations between genera were detected: a positive correlation between Maritimibacter and Micrococcus (σ = 0.69, p-value <0.01) and a negative correlation between Methylobacterium and Flavobacterium (σ = −0.63, p-value <0.01). Furthermore, variance analysis of OTU 36′ abundance showed that it was influenced by the Fulton index (Shapiro test for Normality: p-value = 0.07, Linear model: p-value = 0.02741, F = 5.0388, Rsq = −0.1496). A negative relationship between Fulton index and the OTU 36 was observed (Figure 6).
Figure 5. PCoA analysis of the microbiome for all 86 F2 individuals based on the Yue & Clayton measure of dissimilarity (Thetayc).

Black circles represent individuals belonging to the group 1, red circles represent individuals belonging to the group 2 and green circles represent individuals belonging to the group 3. Arrows represent genus, which are significantly correlated with the axis. The first and second axes represented 24.5% and 7.7% of the variation respectively. The R-squared between the original distance matrix and the distance between the points in 2D PCoA space was 0.87.
Figure 6. Relationship between the Fulton Index and and the abundance of the OTU 36.

A linear regression is observed based on a linear model (Shapiro test for Normality: p-value = 0.07, Linear model: p-value = 0.02741, F = 5.0388, Rsq = 0.04536).
In the F2 fish progeny, three significant QTLs (at the genome-wide level) were identified on two linkage groups (LG 11 and LG 16). One major QTL per strain was detected respectively for Lysobacter, Rheinheimera and Methylobacterium counts (LOD score = 9.89, 7.46 and 3.48 respectively). For each of these traits, the total PVE (percent variance explained) of the QTL were estimated to 17.01%, 31.05% and 41.1% for Methylobacterium, Rheinheimera and Lysobacter respectively. The most probable positions of these QTL, their respective 95% CIs, the closest linked molecular markers (one per QTL) as well as additive and dominance effects are presented in Table 6.
Table 6. Descriptive statistics, including the LOD score, the position, 95% CI, PVE (%), the associated P-value of each QTL linked to every phenotype related to bacteria counts trait (LOD, Log10 of the odd ratio; 95% CI, 95% confidence interval; PVE, percent variance explained) [23].
| Phenotype | Linkage Group | Pos (cM) | 95% CI (cM) | LOD | p value (F) | PVE (in %) | Nearest marker |
| Lysobacter | 11 | 62 | 56.2–68.2 | 9.89 | 0.000 | 41.1 | sf003455 |
| Rheinheimera | 16 | 42.8 | 39.3–45.5 | 7.46 | 0.000 | 31.05 | SFO-D91 |
| Methylobacterium | 16 | 39 | 34.5–43.5 | 3.48 | 0.001 | 17.02 | SFO-D91 |
Discussion
Inter-individual variations in host microbiota has been well documented (e.g. [1]. Such variations occur even in hosts with identical genetic background, as observed in monozygotic twins [3] indicating that both genetic and environmental conditions play a role in the modulation of host microbiota. The gold standard forward genetics technique to identify areas of the genome that relate to certain phenotypes is to make a cross between genetically divergent individuals [32]. In this study, we focused on the structural variation of the skin microbiota (i.e abundance of each bacterial genus) of an F2 intercross originally generated from two genetically contrasted strains of brook charr. To our knowledge, this is the first study that identified host genomic regions associated with the abundance of specific microbiota strains in a non-model vertebrate.
The deep taxonomic analysis of the fish skin microbiota indicates that two phyla dominate: Proteobacteria (88.7%), followed by Bacteroidetes (9.7%). Those two phyla are mainly represented by a single OTU each; 50 and 36 respectively. OTU 50 is classified as M. rhodesianum. OTU 36 is classified as F. psychrophilum, the causative agent of the cold water disease, a pervasive infectious disease in farmed fish [33], [34]. This disease especially affects salmonids at early life-stages, and it is well documented that both stressful conditions, and injuries facilitate infection triggering [33], [35]. According to the dendrogram (Figure 2), each individual's bacterial communities clustered into three groups. Similar clustering was also found with the second normalization method (subsampling of the same number of sequences for each samples). The same result was obtained with PCoA analysis and furthermore the Pearson correlation indicates that the genus Flavobacterium and Methylobacterium are negatively correlated. In the PCoA, two species (F. psychrophilum and M. rhodesianum) are correlated in opposite ways with the axis 1 which discriminate the three groups. We assume that two or more species which seem to be mutually exclusive are involved in a negative relationship [36]. All together those results indicate that the antagonistic relationship between those two dominant species is very likely playing a key role in shaping the structure of the individual's microbiota.
Because all fish progenies were reared in the same tank, and under the same environmental conditions since birth, environment had likely little influence on the phenotypic variation observed in this study. Yet, the individual's microbiota either clustered in one of two closely related groups, or formed divergent outliers. Sex-specific variations had little influence on individual clustering since both males and females were present in each defined group and there was no correlation between sex and groups. The difference between male and female is based on 22 OTUs which are less abundant than 0.1% of the community and are then classified as part of the rare biosphere. Previous studies observed similar individual variations of the microbiota in human or mouse [1], [2], [32].
Three OTUs classified into two species were responsible for the inter-individual differentiation; M. rhodesianum and F. psychrophilum. Interestingly, Flavobacterium and Methylobacterium, which are the most abundant genera, negatively co-occurred in the samples, indicating that the relationship between those two genera is based on competition. Furthermore, a strong negative correlation was found between the OTU 36 and 50, thus supporting a strong antagonistic relationship between the two species, F. psychrophilum and M. rhodesianum.
M. rhodesianum produces poly-β-hydroxybutyrate, a polymer of short-chain fatty acid, known to inhibit the growth of pathogens like enterobacteria and Vibrio sp. [37], [38], [39], [40], [41], [42]. Taken together, this suggests that a reduction of M. rhodesianum abundance allows colonization or over-growth by other bacteria, some of which are pathogenic.
Evidently, a change in skin microbiota taxonomic structure favors opportunist bacteria and especially opportunistic pathogens like F. psychrophilum, Acinetobacter haemolyticus, Acinetobacter johnsonii and Acinetobacter junii, which have a higher abundance in group 2 compared with group 1 Such a disturbance in the microbiome homeostasis is called dysbiosis, and its occurrence enlightens the importance of the function of M. rhodesianum in controlling the balance between both other commensal bacteria and opportunistic pathogens. Furthermore, Flavobacterium psychrophilum was negatively correlated to both M. rhodesianum abundance and Fulton index. The Fulton index is a condition factor used as a proxy for fish health status. Therefore, it suggests that skin microbiota from the weakest host individuals were unable to prevent colonization by the opportunistic pathogen F. psychrophilum. Furthermore, skin microbiota taxonomic structure varies significantly across the F2 progeny. This result added to the fact that i the genetics of the host is the only variable in the experiment, and ii the finding of three QTLs associated to the abundance of bacterial genera with high PVE, suggest that host genotype influences the abundance of commensal strains e.g. Methylobacterium, which will regulate the abundance of F. psychrophilum.
We found three QTL associated with the abundance of three genera: Lysobacter, Rheinheimera and Methylobacterium, all of which were observed to provide antimicrobial compounds [39], [40], [41], [42], [43], [44], [45], [46], [47]. These finding strongly suggests that host genotype influences abundance of specific commensal strains, and possibly targets their recruitment. The most compelling evidence concerns the QTL associated with Methylobacterium (PVE = 17.01%): as previously described above, Methylobacterium is influential on the structure and the homeostasis of the microbiota. Its abundance is inversely correlated with those of the pathogen F. psychrophilum. The targeted recruitment of M. rhodesianum mediated by the host genotype is therefore a strategy to prevent pathogen growth via harnessing antagonistic relationship towards resources use [48]. To our knowledge, this is only the second study that identified QTL associated with microbial variation among individuals. A study on murine gut microbiota showed similar results, in which McKnite et al. (2012) found 5 QTL located on four chromosomes influencing the variation of different taxa. The variance explained by their QTL is in the same range of our (20% to 27% of the variance explained for McKnite et al. (2012)). We also found QTLs with a major effect on the variance of genus abundances (PVE ranging for 17.02 to 41.1%). Linkage analysis on genus abundance data strongly evidenced the influence of host genetics on the modulation and/or recruitment for those genera. Therefore, brook charr immunity involves both specific commensal strains recruitment and nuclear gene expression, as previously evidenced in [49], those are under the control of the host genotype.
The study of the genetic architecture underlying the regulation of bacterial abundance further highlights the coevolutionary pattern between host, commensals, and pathogens. However, identifying genes located in the genomic regions (QTL) linked to the abundance of microbial partners will be far more challenging as the genome of brook charr is currently not fully sequenced and poorly annotated compared to the mouse genome. The markers surrounding the QTL regions may define regions to be deeply sequenced in future work to identify potential underlying genes and their associated functions. Evidently, those markers will be invaluable to conduct highly innovative genetic breeding programs targeting disease resistant host strains via the recruitment of highly resilient microbiota.
Supporting Information
Rarefaction curves for each group defined by the dendrogram analysis based upon ThetaYC index (see figure 2 ). Each group curve reaches a plateau thus indicating the depth of sequencing is sufficient.
(EPS)
Alpha diversity of each group normalized by subsampling of the same number of reads per sample. Each group was defined by the dendrogram analysis based upon ThetaYC index performed on the normalized dataset. Alpha-diversity was calculated by the non-parametric index of Shannon. Statistical differences (represented by the asterisk) were calculated with a wilcoxon test with a correction for multiple testing (Holm, p-values <1.10−4).
(EPS)
PCoA analysis of the microbiome for all 86 F2 individuals. This PCoA was performed on a normalized dataset with the second method of normalization (subsampling of the same number of reads per sample). Black circles represent individuals belonging to the group 1, red circles represent individuals belonging to the group 2 and green circles represent individuals belonging to the group 3. Arrows represent genera, which are significantly correlated with the axis. The first and second axes represented 24.5% and 7.8% of the variation respectively. The R-squared between the original distance matrix and the distance between the points in 2D PCoA space was 0.88.
(EPS)
Differentiation between the three groups of OTU calculated with an analysis of similarity (ANOSIM, statistic R ). The q-values represent p-values corrected with Bonferroni and considered as significant when p<0.05. Note: The analysis was performed on a dataset normalized by two methods: zscore and subsampling. Both analyses gave the same results, each group being significantly different from the two others.
(DOC)
Acknowledgments
We thank Martin Llewellyn, Scott A. Pavey, the associate editor of Plos One, and two anonymous reviewers for critical reading of the manuscript.
Funding Statement
This work was financially supported by a Partnership grant (Strategic program) of the “Conseil de recherches en sciences naturelles et en génie” (CRSNG) to Louis Bernatchez, Nicolas Derome, and Céline Audet and the “Collaborative Research And Training Experience” program (CREATE). This is a contribution to the research program of Resources Aquatiques Québec (RAQ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, et al. (2009) Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, et al. (2010) From the cover: Forensic identification using skin bacterial communities. Proceedings of the National Academy of Sciences 107: 6477–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, et al. (2010) Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proceedings of the National Academy of Sciences 107: 7503–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, et al. (2007) The human microbiome project. Nature 449: 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turnbaugh PJ, Henrissat B, Gordon JI (2010) Viewing the human microbiome through three-dimensional glasses: integrating structural and functional studies to better define the properties of myriad carbohydrate-active enzymes. Acta Crystallographica Section F 66: 1261–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turnbaugh PJ, Gordon JI (2009) The core gut microbiome, energy balance and obesity. The Journal of Physiology 587: 4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, et al. (2011) Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 5: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peterson DA, McNulty NP, Guruge JL, Gordon JI (2007) IgA Response to Symbiotic Bacteria as a Mediator of Gut Homeostasis. Cell Host & Microbe 2: 328–339. [DOI] [PubMed] [Google Scholar]
- 9. Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, et al. (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456: 507–510. [DOI] [PubMed] [Google Scholar]
- 10.Macpherson AJ, Geuking MB, McCoy KD (2011) Immunoglobulin A: a bridge between innate and adaptive immunity. Current Opinion in Gastroenterology 27: 529–533 510.1097/MOG.1090b1013e32834bb32805 [DOI] [PubMed]
- 11. Boutin S, Bernatchez L, Audet C, Derôme N (2012) Antagonistic effect of indigenous skin bacteria of brook charr (Salvelinus fontinalis) against Flavobacterium columnare and F.psychrophilum . Veterinary Microbiology 155: 355–361. [DOI] [PubMed] [Google Scholar]
- 12. Balcázar J, de Blas I, Ruiz-Zarzuela I, Vendrell D, Muzquiz J (2004) Probiotics: a tool for the future of fish and shellfish health management. Journal of Aquaculture in the Tropics 19: 239–242. [Google Scholar]
- 13. Backhed F (2005) Host-bacterial mutualism in the human intestine. Science 307: 1915–1920. [DOI] [PubMed] [Google Scholar]
- 14. Hooper LV, Gordon JI (2001) Commensal host-bacterial relationships in the gut. Science 292: 1115–1118. [DOI] [PubMed] [Google Scholar]
- 15. O'Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Reports 7: 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stecher B, Hardt W-D (2008) The role of microbiota in infectious disease. Trends in Microbiology 16: 107–114. [DOI] [PubMed] [Google Scholar]
- 17. Loiseau C, Richard M, Garnier S, Chastel O, Julliard R, et al. (2009) Diversifying selection on MHC class I in the house sparrow (Passer domesticus). Molecular Ecology 18: 1331–1340. [DOI] [PubMed] [Google Scholar]
- 18. Krawczyk M, Reith W (2006) Regulation of MHC class II expression, a unique regulatory system identified by the study of a primary immunodeficiency disease. Tissue Antigens 67: 183–197. [DOI] [PubMed] [Google Scholar]
- 19. Dionne M, Miller KM, Dodson JJ, Bernatchez L (2009) MHC standing genetic variation and pathogen resistance in wild Atlantic salmon. Philosophical Transactions of the Royal Society B-Biological Sciences 364: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alexander C, Rietschel ET (2001) Invited review: Bacterial lipopolysaccharides and innate immunity. Journal of Endotoxin Research 7: 167–202. [PubMed] [Google Scholar]
- 21.Boutin S, Audet C, Derôme N (2013) Probiotic treatment by indigenous bacteria decreases mortality without disturbing the natural microbiota of Salvelinus fontinalis. Canadian Journal of Microbiology In press. [DOI] [PubMed]
- 22. Boutin S, Bernatchez L, Audet C, Derôme N (2013) Network Analysis Highlights Complex Interactions between Pathogen, Host and Commensal Microbiota. PLoS ONE 8: e84772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sauvage C, Vagner M, Derôme N, Audet C, Bernatchez L (2012) Coding gene single nucleotide polymorphism mapping and quantitative trait loci detection for physiological reproductive traits in brook charr, Salvelinus fontinalis . G3: Genes|Genomes|Genetics 2: 379–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livia L, Antonella P, Hovirag L, Mauro N, Panara F (2006) A nondestructive, rapid, reliable and inexpensive method to sample, store and extract high-quality DNA from fish body mucus and buccal cells. Molecular Ecology Notes 6: 257–260. [Google Scholar]
- 25. Turner S, Pryer KM, Miao VPW, Palmer JD (1999) Investigating deep phylogenetic relationships among Cyanobacteria and plastids by small subunit rRNA sequence analysis. Journal of Eukaryotic Microbiology 46: 327–338. [DOI] [PubMed] [Google Scholar]
- 26. Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, et al. (1998) Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Applied and Environmental Microbiology 64: 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schloss P, Westcott S, Ryabin T, Hall J, Hartmann M, et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Siddharth J, Holway N, Parkinson SJ (2013) A Western Diet Ecological Module Identified from the ‘Humanized’ Mouse Microbiota Predicts Diet in Adults and Formula Feeding in Children. PLoS ONE 8: e83689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeshita T, Suzuki N, Nakano Y, Yasui M, Yoneda M, et al. (2012) Discrimination of the oral microbiota associated with high hydrogen sulfide and methyl mercaptan production. Sci Rep 2. [DOI] [PMC free article] [PubMed]
- 30. Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM (2004) A taxa-area relationship for bacteria. Nature 432: 750–753. [DOI] [PubMed] [Google Scholar]
- 31. White JR, Nagarajan N, Pop M (2009) Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Computational Biology 5: e1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, et al. (2012) Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE 7: e39191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Starliper CE (2011) Bacterial coldwater disease of fishes caused by Flavobacterium psychrophilum . Journal of Advanced Research 2: 97–108. [Google Scholar]
- 34. Wiklund T, Madsen L, Bruun MS, Dalsgaard I (2000) Detection of Flavobacterium psychrophilum from fish tissue and water samples by PCR amplification. Journal of Applied Microbiology 88: 299–307. [DOI] [PubMed] [Google Scholar]
- 35. Madetoja J, Nyman P, Wiklund T (2000) Flavobacterium psychrophilum, invasion into and shedding by rainbow trout Oncorhynchus mykiss . Diseases of Aquatic Organisms 43: 27–38. [DOI] [PubMed] [Google Scholar]
- 36. Faust K, Raes J (2012) Microbial interactions: from networks to models. Nat Rev Micro 10: 538–550. [DOI] [PubMed] [Google Scholar]
- 37. Defoirdt T, Halet D, Vervaeren H, Boon N, Van de Wiele T, et al. (2007) The bacterial storage compound poly-β-hydroxybutyrate protects Artemia franciscana from pathogenic Vibrio campbellii . Environmental Microbiology 9: 445–452. [DOI] [PubMed] [Google Scholar]
- 38. Halet D, Defoirdt T, Van Damme P, Vervaeren H, Forrez I, et al. (2007) Poly-β-hydroxybutyrate-accumulating bacteria protect gnotobiotic Artemia franciscana from pathogenic Vibrio campbellii . FEMS Microbiology Ecology 60: 363–369. [DOI] [PubMed] [Google Scholar]
- 39. Yellore Desai (1998) Production of poly-3-hydroxybutyrate from lactose and whey by Methylobacterium sp. ZP24. Letters in Applied Microbiology 26: 391–394. [DOI] [PubMed] [Google Scholar]
- 40. Mothes G, Ackermann J-U, Babel W (1998) Regulation of poly(β-hydroxybutyrate) synthesis in Methylobacterium rhodesianum MB 126 growing on methanol or fructose. Archives of Microbiology 169: 360–363. [DOI] [PubMed] [Google Scholar]
- 41. Bormann EJ, Roth M (1999) The production of polyhydroxybutyrate by Methylobacterium rhodesianum and Ralstonia eutropha in media containing glycerol and casein hydrolysates. Biotechnology Letters 21: 1059–1063. [Google Scholar]
- 42. Bormann EJ, Roth M, Linβ W, Leiβner M (1999) Selection of capsule deficient strains of Methylobacterium rhodesianum producing polyhydroxybutyrate. Biotechnology Techniques 13: 539–544. [Google Scholar]
- 43. Balachandran C, Duraipandiyan V, Ignacimuthu S (2012) Cytotoxic (A549) and antimicrobial effects of Methylobacterium sp. isolate (ERI-135) from Nilgiris forest soil, India. Asian Pacific Journal of Tropical Biomedicine 2: 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Korotkova N, Chistoserdova L, Lidstrom ME (2002) Poly-β-Hydroxybutyrate Biosynthesis in the Facultative Methylotroph Methylobacterium extorquens AM1: Identification and Mutation of gap11, gap20, and phaR. Journal of Bacteriology 184: 6174–6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen W-M, Lin C-Y, Sheu S-Y (2010) Investigating antimicrobial activity in Rheinheimera sp. due to hydrogen peroxide generated by l-lysine oxidase activity. Enzyme and Microbial Technology 46: 487–493. [DOI] [PubMed] [Google Scholar]
- 46.Chen W-M, Lin C-Y, Young C-C, Sheu S-Y (2010) Rheinheimera aquatica sp. nov., antimicrobial activity-producing bacterium isolated from freshwater culture pond. Seoul, COREE, REPUBLIQUE DE: Korean Society for Applied Microbiology. 7 p. [DOI] [PubMed] [Google Scholar]
- 47.Reichenbach H (2006) The genus Lysobacter. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors.The Prokaryotes: Springer New York. pp. 939–957. [Google Scholar]
- 48. Dillon R, Charnley K (2002) Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Research in Microbiology 153: 503–509. [DOI] [PubMed] [Google Scholar]
- 49. Langevin C, Blanco M, Martin SAM, Jouneau L, Bernardet J-F, et al. (2012) Transcriptional responses of resistant and susceptible fish clones to the bacterial pathogen Flavobacterium psychrophilum . PLoS ONE 7: e39126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rarefaction curves for each group defined by the dendrogram analysis based upon ThetaYC index (see figure 2 ). Each group curve reaches a plateau thus indicating the depth of sequencing is sufficient.
(EPS)
Alpha diversity of each group normalized by subsampling of the same number of reads per sample. Each group was defined by the dendrogram analysis based upon ThetaYC index performed on the normalized dataset. Alpha-diversity was calculated by the non-parametric index of Shannon. Statistical differences (represented by the asterisk) were calculated with a wilcoxon test with a correction for multiple testing (Holm, p-values <1.10−4).
(EPS)
PCoA analysis of the microbiome for all 86 F2 individuals. This PCoA was performed on a normalized dataset with the second method of normalization (subsampling of the same number of reads per sample). Black circles represent individuals belonging to the group 1, red circles represent individuals belonging to the group 2 and green circles represent individuals belonging to the group 3. Arrows represent genera, which are significantly correlated with the axis. The first and second axes represented 24.5% and 7.8% of the variation respectively. The R-squared between the original distance matrix and the distance between the points in 2D PCoA space was 0.88.
(EPS)
Differentiation between the three groups of OTU calculated with an analysis of similarity (ANOSIM, statistic R ). The q-values represent p-values corrected with Bonferroni and considered as significant when p<0.05. Note: The analysis was performed on a dataset normalized by two methods: zscore and subsampling. Both analyses gave the same results, each group being significantly different from the two others.
(DOC)