

Abstract
Retinitis pigmentosa (RP) is a disease characterized by its vast heterogeneity. Many genes are associated with RP, and the disease causing mutations identified in these genes are even more numerous. To date there are 15 genes that cause autosomal dominant RP (adRP) alone. The role of some of these genes, while complex and not completely understood, is somewhat intuitive in that they are involved in pathways such as phototransduction. However, the role of other genes in retinal disease is not as predictable due to their ubiquitous function and/or expression. One such gene is inosine monophosphate dehydrogenase 1 (IMPDH1) IMPDH1 is a gene involved in de novo purine synthesis and is ubiquitously expressed. IMPDH1 mutations account for 2% of all adRP cases and are a rare cause of Leiber Congenital Amaurosis. Despite its ubiquitous expression missense mutations in this gene cause only retinal degeneration. This paradox of tissue specific disease in the presence of ubiquitous expression has only recently begun to be explained. We have shown in a recent study that novel retinal isoforms of IMPDH1 exist and may account for the tissue specificity of disease. We have gone on to characterize these retinal isoforms both in our laboratory and in collaboration with Dr. Lizbeth Hedstrom’s laboratory at Brandeis University (Waltham, MA) in order to understand more about them. We believe that through clarifying the mechanism of disease in RP10 we will be equipped to consider treatment options for this disease.
62.1 Introduction
Mutations in inosine monophosphate dehydrogenase 1 (IMPDH1) cause autosomal dominant retinitis pigmentosa (adRP) (Bowne et al. 2002; Kennan et al. 2002) and are also a rare cause of Leber congenital amaurosis (LCA) (Bowne et al. 2006b). Although it was recently discovered that IMPDH1 is involved in adRP, the functional properties of IMPDH1 have been known since at least the 1950s (e.g. Magasanik et al. 1957). Despite the fact that the IMPDH1 enzyme has been, historically, well studied, the question remains – why do mutations in IMPDH1 cause retinal disease? Our research, and data reported by other groups, suggests several hypotheses.
62.2 Retinitis Pigmentosa
Retinitis pigmentosa (RP) is the most common heritable retinopathy affecting approximately 1 in 3,700 individuals worldwide (Haim 2002). RP is a disease characterized by progressive constriction of visual fields via apoptosis of the rod photoreceptor cells. Patients affected with RP are often left legally or completely blind due to this process (Heckenlively and Daiger 2002).
RP shows phenotypic, genetic, and allelic heterogeneity. The phenotype of RP is heterogeneous in that individuals in the same family and/or even the same sibship may have different symptoms despite carrying an identical genetic mutation. Phenotypic heterogeneity in RP may be attributed to modifying factors and/or environmental factors that have yet to be identified (Daiger, Shankar et al. 2006). Mutations causing autosomal dominant (adRP), autosomal recessive (arRP) or X-linked RP (xlRP) have been identified in a total of 31 genes, with another 9 loci implicated by linkage (http://www.sph.uth.tmc.edu/RetNet) Within these disease-associated genes there may be one or many disease-causing alleles. Approximately 60% of dominant mutations can be identified at this time (Sullivan et al. 2006a, b; Daiger et al. 2007; Gire et al. 2007; Bowne et al. 2008), therefore additional disease-causing genes and/or mutations remain to be found.
62.3 RP10 – Disease Caused by Mutations in IMPDH1
In 2002 our laboratory discovered that missense mutations in the IMPDH1 gene cause adRP (Bowne et al. 2002; Kennan et al. 2002). The IMPDH1 gene is located on chromosome 7q32.1 and accounts for approximately 2.5% of all adRP cases (Bowne et al. 2006b; Sullivan et al. 2006a). In addition to adRP, rare dominant-acting mutations in IMPDH1 cause a congenital form of blindness known as Leber congenital amaurosis or LCA (Bowne et al. 2006). Disease-causing variants in IMPDH1 are mainly missense mutations, the most common of which is the Asp226Asn mutation accounting for about 2% of adRP cases in the United States alone (Wada et al. 2005). The phenotype resulting from IMPDH1 mutations tends to be severe with early onset and rapid progression, sometimes also presenting with cystoid macular edema (CME) (Kozma et al. 2005; Schatz et al. 2005; Wada et al. 2005).
62.4 IMPDH Structure and Function
IMPDH is enzymatically active in the homotetramer state. Each IMPDH monomer is composed of an eight-stranded alpha/beta barrel structure that performs the enzymatic function, and a flanking subdomain that is composed of two cystathionine β-synthase-like regions, called CBS domains (Carr et al. 1993).
IMPDH is part of a highly conserved class of enzymes found in both prokaryotes and eukaryotes (Senda and Natsumeda 1994). It functions in the de novo purine synthesis pathway. Specifically, IMPDH catalyzes the rate-limiting step of guanine nucleotide synthesis by converting inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) with the concomitant reduction of nicotinamide adenine dinucleotide (NAD+) (Hedstrom 1999). XMP can go on to be converted to guanine monophosphate (GMP) by GMP synthetase (Fig. 62.1).
Fig. 62.1.
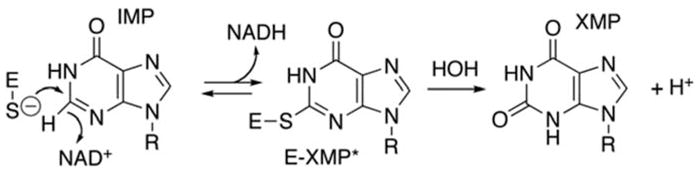
Chemical reaction catalyzed by the inosine monophosphate dehydrogenase (IMPDH) enzyme. IMPDH functions in de novo guanine nucleotide synthesis by converting inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) with the concomitant reduction of nicotinamide adenine dinucleotide (NAD+) (Hedstrom 1999). Figure produced by Dr. Lizbeth Hedstrom, Brandeis University (Waltham, MA)
Mammals have two homologues of the IMPDH gene, IMPDH1 and IMPDH2, which upon translation are 84% identical in humans (Natsumeda et al. 1990). Human IMPDH1 is located on chromosome 7q32.1 and human IMPDH2 is located on chromosome 3p21.2 (Glesne et al. 1993; Gu et al. 1994). The IMPDH isozymes are indistinguishable in enzymatic activity (Carr et al. 1993) but have different inhibitory properties (Hager et al. 1995). Research shows that IMPDH1 and IMPDH2 are differentially expressed between tissues (Natsumeda et al. 1990; Jain et al. 2004). Our analysis using SAGE methodology shows that IMPDH1 mRNA is exceptionally abundant in the retina; the abundance of IMPDH1 in the retina is approximately ten times the average levels found elsewhere in the body, perhaps indicating that retinal tissue has a unique requirement for IMPDH1. IMPDH2 mRNA is scarce in retinal tissue but more abundant elsewhere in the body (Bowne et al. 2006a). Indeed, research shows that the majority of the GTP in mouse retina is produced by IMPDH1, not IMPDH2 or the salvage pathway (Aherne et al. 2004).
62.5 IMPDH Binds Single Stranded Nucleic Acids
In 2004 Dr. Lizbeth Hedstrom’s lab at Brandeis University, Waltham MA, showed that IMPDH proteins bind single-stranded nucleic acids with nanomolar (physiological) affinity (McLean et al. 2004). The binding occurs via the CBS domains and IMPDH appears to bind about 100 bp of single-stranded polynucleotides. The nucleic acid binding activity does not inhibit function of the enzyme, and if the CBS domains are removed from the protein, nucleic acid binding function is ablated.
The majority of the adRP and LCA-associated mutations reside in or near the IMPDH1 CBS domains in the protein structure (Mortimer and Hedstrom 2005; Bowne et al. 2006). To test the effect of the adRP/LCA-associated mutations on the nucleic acid binding activity of IMPDH1 several recombinant proteins were produced. Recombinant wild type IMPDH1 was produced in a typical bacterial system, and IMPDH1 proteins with the following missense mutations were also produced: Arg224Pro, Val268Ile, Asp226Asn, Arg105Trp, Thr116Met, Asn198Lys, and His372Pro (Bowne et al. 2002; Kennan et al. 2002; Bowne et al. 2006b). Two additional genetic variants were tested that are known to be benign, Ala285Thr and His296Arg (Bowne et al. 2006b). When adRP mutations are ‘knocked in’ to the recombinant IMPDH1 protein, the kinetic parameters of the enzyme are unchanged, however the nucleic acid binding property of the protein is altered in most cases (Mortimer and Hedstrom 2005; Bowne et al. 2006b). Binding affinity for ssDNA was decreased in all the known pathogenic mutations by a factor ranging from 7 to 32. In addition, some of the pathogenic variants bound an increased portion of the random pool of ssDNA. As expected, the non-pathogenic variants did not differ significantly from the wild type enzyme in either measure (Mortimer and Hedstrom 2005; Bowne et al. 2006b). These data suggest that IMPDH1 has a unique function that involves binding single stranded nucleic acids.
62.6 Retinal Isoforms of IMPDH1
Although alternate starts of transcription, and thus alternate transcripts of IMPDH1 were previously identified, only one protein was known to be produced from these transcripts (Gu and Mitchell 1997). This traditionally-studied form of IMPDH1 is referred to as ‘canonical IMPDH1’ or IMPDH1 (514) IMPDH1 (514) results from the transcription and translation of exons 1–14, excluding exon 13b (see genomic structure, Fig. 62.2a) Upon translation, the IMPDH1 (514) protein is 514 amino acids in length and predicted to be 55.6 kD in molecular weight.
Fig. 62.2.
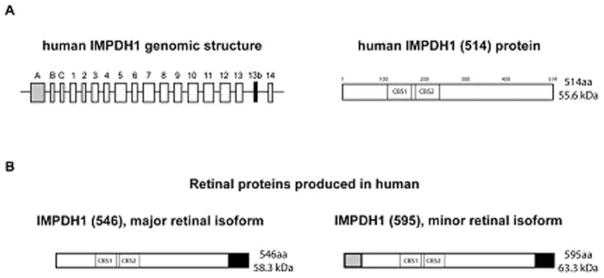
(a) Genomic structure of IMPDH1 in humans and the ‘canonical’ IMPDH1 (514) protein. The ‘canonical’ IMPDH1 (514) protein results from translation of exons 1–14. (b) Retinal-specific isoforms of IMPDH1 in human. The black portion of the proteins represents the amino acid sequence resulting from the addition of exon 13b in the spliced transcript. The grey portion of the protein represents the sequence resulting from the addition of exon A in the spliced transcript. IMPDH1 (546) is the most abundant isoform found in human retina and results from the translation of exons 1–14 and exon 13b. The less abundant isoform in human retina is IMPDH1 (595) which is translated from exons 1–14 and exons A and 13b. CBS1 and CBS2 illustrate the approximate location of the cystathionine β-synthase-like (CBS) domains in the proteins
In 2006 our laboratory discovered that retinal-specific isoforms of IMPDH1 exist (Bowne et al. 2006). A series of experiments using human and mouse retinal cDNA were performed to identify the predominant IMPDH1 transcript in the retinal tissue in these species and results showed several novel mRNA transcripts. These novel transcripts result from both alternate splicing events and alternate starts of transcription and translation. Results showed that exon A is transcribed in retina and is spliced, in frame, directly to exon 1, leading to a novel protein sequence at the N terminus upon translation. In addition, the data showed that a novel 17 bp exon exists between exons 13 and 14, exon 13b. Exon 13b causes a frame shift in transcription that abolishes the stop codon, and leads to a novel protein sequence at the C terminus, as well. Exon 13b was present in almost all the retinal transcripts analyzed (Bowne et al. 2006a).
There are two predominant transcripts present in human and mouse retina. One transcript includes exons 1 through 14, but also contains exons A and 13b. The other most abundant transcript in human and mouse retina also includes exons 1 through 14 and exon 13b, but not exon A (Fig. 62.2b) (Bowne et al. 2006a). Upon translation these two transcripts result in proteins that are referred to as ‘1+13b’ or IMPDH1 (546), and ‘A+13b’ or IMPDH1 (595), respectively. The names IMPDH1 (546) and IMPDH1 (595), based on the amino acid lengths of the retinal isoforms, will be the nomenclature used here.
We also performed experiments to characterize the IMPDH1 (546) and (595) isoforms in other species. Retinal tissues from several mammalian species, including sheep, dog, mouse, cow, pig, rat, and human, were analyzed by western blot for the various IMPDH1 protein isoforms. Antibodies specific to each IMPDH1 isoform were used to visualize the proteins in each species. These data show that the retinal isoforms of IMPDH1 are not only transcribed but also translated in the retina of human and in all other species tested. Secondly, the relative abundances of the two retinal proteins are different among the species observed, with humans being different from all the rest. Specifically, data showed that in most species IMPDH1 (595) is the most abundant isoform. In pig and sheep the isoforms appear to be equal in abundance, and human is unique in that IMPDH1 (546) is the abundant isoform. Lastly, the sizes of the retinal isoforms differ slightly between species (Spellicy et al. 2007). This indicates there may be slight sequence differences and/or post-translational modifications between species.
Despite the fact that IMPDH1 (514) is ubiquitously expressed in the body, disease resulting from IMPDH1 mutations is limited strictly to the retina. The discovery of the retinal isoforms of IMPDH1 provides a possible explanation for this long-standing mystery – that the mechanism of disease is specific to the retinal isoforms and therefore does not manifest in other tissues.
62.7 Kinetic and Nucleic Acid Binding Properties of Retinal IMPDH1
In collaboration with Dr. Hedstrom’s laboratory we repeated the kinetic and nucleic acid binding experiments using the retinal isoforms of IMPDH1. Measuring NAD+ production as an indicator of enzyme activity we showed that the IMPDH1 (546) and IMPDH1 (595) are able to convert IMP to XMP with kinetic parameters similar to that of IMPDH1 (514) Cellular localization experiments were also performed in vivo for both retinal isoforms and showed a distribution identical to IMPDH1 (514) (Xu et al. 2008).
Nucleic acid binding studies were performed using radiolabeled single-stranded nucleic acids and two different membranes to measure nucleic acid affinity of the proteins (hybond, which binds free nucleic acids, and nitrocellulose, which binds protein and protein-nucleic acid complexes) The ratio of free and bound single-stranded nucleotides was used to calculate the affinity of IMPDH (546) and IMPDH1 (595) for polynucleotides (McLean et al. 2004; Mortimer and Hedstrom 2005; Bowne et al. 2006b; Xu et al. 2008). Interestingly, the retinal isoforms of IMPDH1 failed to bind polynucleotides almost completely (Xu et al. 2008). However, recent research from Dr. Hedstrom’s laboratory shows that IMPDH1 (595) and (546) both associate with polyribosomes translating RNA in bovine retina. In addition, inclusion of the Asp226Asn mutation in either of these protein isoforms inhibits the interaction, sometimes completely (Mortimer et al. 2008). It is possible that the additional protein sequence at the C terminus of the protein blocks this activity, or that another protein component is needed in vivo to potentiate this interaction.
62.8 Conclusion
Elucidating the mechanism of retinal disease in RP10 presents a formidable task. Many questions remain about the function of IMPDH1 as it relates specifically to the retina. Strides have been made towards starting to address these questions; discovery of the retinal-specific isoforms may help explain the tissue specificity of disease, and data suggest that retinal IMPDH1 may have a novel function in RNA transcription, translation, or stabilization. We are optimistic that these novel findings are a first step in understanding retinal disease caused by IMPDH1 mutations.
References
- Aherne A, Kennan A, et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum Mol Genet. 2004;13(6):641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- Bowne S, Liu Q, et al. Why do mutations in the ubiquitously expressed housekeeping gene IMPDH1 cause retina-specific photoreceptor degeneration? Invest Ophthalmol Vis Sci. 2006;47(9):3754–3765. doi: 10.1167/iovs.06-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne S, Sullivan L, et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002a;11(5):559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne S, Sullivan L, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47(1):34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne S, Sullivan L, et al. Mutations in the TOPORS gene cause 1% of autosomal dominant retinitis pigmentosa. Mol Vis. 2008b;14:922–927. [PMC free article] [PubMed] [Google Scholar]
- Carr S, Papp E, et al. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268(36):27286–27290. [PubMed] [Google Scholar]
- Daiger S, Bowne S, et al. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125(2):151–158. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger S, Shankar S, et al. Genetic factors modifying clinical expression of autosomal dominant RP. Adv Exp Med Biol. 2006;572:3–8. doi: 10.1007/0-387-32442-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gire A, Sullivan L, et al. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol Vis. 2007;13:1970–1975. [PubMed] [Google Scholar]
- Glesne D, Collart F, et al. Chromosomal localization and structure of the human type II IMP dehydrogenase gene (IMPDH2) Genomics. 1993;16(1):274–277. doi: 10.1006/geno.1993.1177. [DOI] [PubMed] [Google Scholar]
- Gu J, Kaiser-Rogers K, et al. Assignment of the human type I IMP dehydrogenase gene (IMPDH1) to chromosome 7p31.3-p32. Genomics. 1994;24(1):179–181. doi: 10.1006/geno.1994.1597. [DOI] [PubMed] [Google Scholar]
- Gu J, Mitchell B. Regulation of the human inosine monophosphate dehydrogenase type I gene: utilization of alternative promoters. J Biol Chem. 1997;272:4458–4466. doi: 10.1074/jbc.272.7.4458. [DOI] [PubMed] [Google Scholar]
- Hager P, Collart F, et al. Recombinant human inosine monophosphate dehydrogenase type I and type II proteins. Biochem Pharmacol. 1995;49(9):1323–1329. doi: 10.1016/0006-2952(95)00026-v. [DOI] [PubMed] [Google Scholar]
- Haim M. The epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand. 2002;233:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- Heckenlively J, Daiger S. In: Emery and Rimoin’s principals and practices of medical genetics. 4. Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Churchill Livingston; London: 2002. pp. 3555–3593. [Google Scholar]
- Hedstrom L. IMP dehydrogenase: mechanism of action and inhibition. Curr Med Chem. 1999;6:545–560. [PubMed] [Google Scholar]
- Jain J, Almquist S, et al. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem Pharmacol. 2004;67:767–776. doi: 10.1016/j.bcp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- Kennan A, Aherne A, et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho−/− mice. Hum Mol Genet. 2002;11(5):547–558. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- Kozma P, Hughbanks-Wheaton D, et al. Phenotypic characterization of a large family with RP10 autosomal-dominant retinitis pigmentosa: an Asp226Asn mutation in the IMPDH1 gene. Am J Ophthalmol. 2005;140(5):858–867. doi: 10.1016/j.ajo.2005.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magasanik B, Moyed H, et al. Enzymes essential for the biosynthesis of nucleic acid guanine; inosine 5′-phosphate dehydrogenase of aerobacter aerogenes. J Biol Chem. 1957;226(1):339–350. [PubMed] [Google Scholar]
- McLean J, Hamaguchi N, et al. Inosine 5′-monophosphate dehydrogenase binds nucleic acids in vitro and in vivo. Biochem J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer S, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390(Pt 1):41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer S, Xu D, et al. IMP dehydrogenase type 1 associates with polyribosomes translating Rhodopsin RNA. J Biol Chem. 2008;283(52):36354–36360. doi: 10.1074/jbc.M806143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsumeda Y, Ohno S, et al. Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem. 1990;265(9):5292–5295. [PubMed] [Google Scholar]
- Schatz P, Ponjavic V, et al. Clinical phenotype in a Swedish family with a mutation in the IMPDH1 gene. Ophthalmic Genet. 2005;26(3):119–124. doi: 10.1080/13816810500229090. [DOI] [PubMed] [Google Scholar]
- Senda M, Natsumeda Y. Tissue-differential expression of two distinct genes for human IMP dehydrogenase (E.C.1.1.1.205) Life Sci. 1994;54(24):1917–1926. doi: 10.1016/0024-3205(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Spellicy C, Daiger S, et al. Characterization of retinal inosine monophosphate dehydrogenase 1 in several mammalian species. Mol Vis. 2007;13:1866–1872. [PubMed] [Google Scholar]
- Sullivan L, Bowne S, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006a;47(7):3052–3064. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan L, Bowne S, et al. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006b;47(10):4579–4588. doi: 10.1167/iovs.06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Sandberg M, et al. Screen of IMPDH1 gene among patients with dominant retinitis pigmentosa and clinical features associated with the most common mutation, Asp226Asn. Invest Ophthalmol Vis Sci. 2005;46(5):1735–1741. doi: 10.1167/iovs.04-1197. [DOI] [PubMed] [Google Scholar]
- Xu D, Cobb G, et al. Retinal isoforms of inosine 5′-monophosphate dehydrogenase type 1 are poor nucleic acid binding proteins. Arch Biochem Biophys. 2008;472:100–104. doi: 10.1016/j.abb.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]