

Abstract
Protein kinase C (PKC) engenders motility through phosphorylation of α-tubulin at Ser-165 in non-transformed MCF-10A cells. Live cell imaging explored the impact of PKC-mediated phosphorylation on microtubule (MT) dynamics. MTs fluorescently labeled with GFP-α-tubulin were treated with diacylglycerol (DAG)-lactone (a membrane-permeable PKC activator), or co-transfected with a pseudo-phosphorylated S165D-α6-tubulin mutant. Each condition increased the dynamicity of MTs by stimulating the rate and duration of the growth phase and decreasing the frequency of catastrophe. In MDA-MB-231 metastatic breast cells where the intrinsic PKC activity is high, these MT growth parameters were also high but could be suppressed by expression of phosphorylation-resistant S165N-α6-tubulin or by treatment with a pan-PKC inhibitor (bis-indoleylmaleimide). Sub-cellular fractionation and immunofluorescence of MCF-10A cells showed that phosphorylation (via DAG-lactone) or pseudo-phosphorylation of α6-tubulin increased its partitioning into MTs as compared to controls, and produced longer, more stable MTs. Following expression of the plus-end binding protein GFP-EB1, DAG-lactone accelerated the formation and increased the number of nascent MTs. Expression of S165D-α6-tubulin promoted Rac1 activation and Rac1-dependent cell motility. These findings call attention to PKC-mediated phosphorylation of α-tubulin as a novel mechanism for controlling the dynamics of MTs that result in cell movement.
Keywords: live cell imaging, mutant, partitioning, Rac1, motility
INTRODUCTION
Microtubules (MTs) are cytoskeletal polymeric structures that are assembled from heterodimers of α- and β-tubulin proteins [Draber and Draberova, 2012]. They originate at the microtubule organizing center (MTOC) and continue to grow towards the cell periphery during which they undergo frequent alternating growth and shortening episodes termed ‘dynamic instability’ [Desai and Mitchison, 1997; Komarova e al., 2002]. Several parameters used to characterize MT dynamic instability include the rates of growth and shortening that together define dynamicity, and the frequency of transitioning between the two phases termed ‘catastrophe’ and ‘rescue’ [Dhamodharan et al., 1995]. The term ‘dynamicity’ is defined as the sum of the distances for growth and shortening events divided by the total observation time. The complexity entailed in MT instability is essential to the biological functions that MTs perform during cell division, migration, and intracellular trafficking [Piehl and Cassimeris, 2003; Etienne-Manneville, 2013]. Because of their central role in mitosis and cell migration, MTs provide an attractive target for chemotherapy against rapidly growing tumg tumors of the breast, ovary and lung [Honore et al., 2005; Pasquier and Kavallaris, 2008; Risinger and Mooberryor cells such as in lymphoma and leukemia, metastatic cancers, and slow growin, 2012].
The dynamic parameters that define the behavior of MTs have been used to characterize the mechanism of action of several MT-directed anti-mitotic drugs [Kamath et al., 2006; Chan et al., 2011]. MT stabilizing drugs include paclitaxel (Taxol), which increases the net polymer mass of MTs in cells by increasing the ratio of MT-incorporated (insoluble) tubulin to monomeric (soluble) tubulin to 90:10. Destabilizing drugs such as nocodazole or vinblastine inhibit MT formation, or depolymerize existing polymers [Dhamodharan et al., 1995; Yvon et al., 1999]. Certain post-translational modifications, such as acetylation of α-tubulin (Lys-40), also result in altered dynamic behavior of MTs [Chao et al., 2012]. Although β-tubulin phosphorylation by Cdk1 during mitosis was previously reported [Fourest-Lieuvin et al., 2006], no studies have yet addressed whether phosphorylation of α-tubulin subunits plays a role in the functional behavior of MTs.
Protein kinase C (PKC) is a serine/threonine protein kinase that is known to regulate the actin cytoskeleton [Larsson, 2006], and is an important factor in breast cancer progression [Lonne et al., 2010]. PKC exists as a family of structurally-related isoforms that are defined by the mechanism of their activation in the cell [Steinberg, 2008]. The conventional isoforms (α, β, γ) are activated by Ca+2 and diacylglycerol (DAG), the novel isoforms (δ, ε, η, θ) are activated by DAG only, whereas the atypical isoforms (ι/λ, ζ) are unresponsive to both Ca+2 and DAG. In view of their complexity with regard to regulation, sub-cellular location and slight differences in active site structure, individual PKC isoforms may select specific substrates as well as substrates shared with other isoforms. Recent work from this laboratory has begun to shed light on this important area [Abeyweera and Rotenberg, 2007; Abeyweera et al., 2009; Chen et al., 2012].
Non-motile, non-transformed MCF-10A human breast cells contain very low levels of PKC isoforms, especially PKCα, and therefore provide an ideal environment for identifying and characterizing PKC substrates. Our previous studies showed that either over-expression of PKCα in MCF-10A cells or treatment with DAG-lactone resulted in aggressive cell movement [Sun and Rotenberg, 1999; Abeyweera et al., 2009]. A reagent commonly used in our studies is DAG-lactone, a cell-permeable analogue that selectively activates PKCα and, to a lesser degree, other DAG-stimulated PKC isoforms [Garcia-Bermejo et al., 2002]. By applying the traceable kinase method with MCF-10A cells [Abeyweera et al., 2007], we showed that α6-tubulin is a PKC substrate that undergoes phosphorylation at Ser-165 in response to DAG-lactone treatment [Abeyweera et al., 2009]. Mutation of Ser-165 to the pseudo-phosphorylated mutant (S165D-α6-tubulin) followed by its expression in MCF-10A cells promoted cell motility to the same extent as that produced in these cells by PKCα overexpression or DAG-lactone treatment. Furthermore, phosphorylation of α6-tubulin in cells treated with DAG-lactone could be almost entirely blocked by either treatment with a pan-PKC inhibitor (bis-indoleylmaleimide; BIM), or expression of a site-specific mutant of α6-tubulin that was resistant to phosphorylation (S165N-α6-tubulin) [Abeyweera et al., 2009]. The physiological significance of phospho-Ser-165 in α-tubulin was also demonstrated with human breast tumor cells that express intrinsically high levels of PKC (e.g. MDA-MB-231, MDA-MB-468), and therefore did not require activation with DAG-lactone. The intrinsic motility of these metastatic cells was inhibited by 60–75% following either expression of phosphorylation-resistant S165N-α6-tubulin or treatment with BIM, and correlated well with decreased levels of endogenous phospho-α-tubulin under these conditions.
Taken together, our previous findings implicate Ser-165 in α6-tubulin as a functionally significant phosphorylation site recognized by DAG-sensitive PKC isoforms that, upon phosphorylation, results in the acquisition of movement by non-motile MCF-10A cells. However, the specific mechanism by which phospho-α6-tubulin produces this phenotype remains unknown. Our hypothesis is that the phosphorylation state of Ser-165 in α-tubulin alters the properties of MTs in MCF-10A cells. The present study investigates the impact of Ser-165 mutants or DAG-lactone on the dynamic behavior and stability of MTs, and explores whether MT behavior is similarly perturbed in metastatic human breast cells. In addition, the small GTPases are investigated in MCF-10A cells as downstream components of the pathway being driven by phosphorylated α-tubulin.
RESULTS
Quantitation of DAG-lactone-induced MT dynamics with EGFP-α-tubulin in MCF-10A cells
Experiments were performed with MCF-10A cells that had been transiently transfected with human EGFPα-tubulin (TUBA1B; Kα1) in order to produce uniformly fluorescent MTs and to analyze their advancing and receding movements. Cells that expressed a moderate level of EGFP-α-tubulin fluorescence were selected for time-lapse confocal microscopy. For measuring MT dynamic parameters, plus-ends of MTs arriving at the cell periphery were tracked for 30 sec, and changes in MT length were plotted against time to generate life history plots for an individual MT.
DAG-lactone, a cell permeable PKC activator, was used to phosphorylate intracellular α-tubulin via activation of endogenous DAG-sensitive PKC isoforms, and the dynamic behavior of MTs infiltrating the leading edges was investigated. A comparison of the life history plots of three representative MTs from control- (DMSO) and DAG-lactone-treated cells (Fig. 1A) illustrates certain key responses by MTs to stimulation by DAG-lactone. Control cells exhibited prolonged pauses, while cells treated with the PKC activator displayed longer intervals of growth interrupted by brief shortening episodes. Cells treated with DAG-lactone exhibited increased rates of growth coupled with a significant increase in the time spent growing, that enabled MTs to exhibit continuous growth to the cell margin. This response provided a major contribution to the enhanced dynamicity (Fig. 1B). A notable observation was that, following treatment with DAG-lactone, MTs in membrane lamellipodia tended to be longer and curved, indicating a reversal in direction upon contact with the cell periphery (inset in Fig. 1C). This phenomenon suggested that DAG-sensitive PKC isoforms promoted increased longevity of MTs [Chan et al., 2011].
FIGURE 1.

Fluorescent live cell imaging of MT dynamics in MCF-10A cells expressing EGFP-α-tubulin. Transfectants were treated with either 10 μM DAG-lactone (DAG) or DMSO as vehicle control (0.05% v/v) for 1 h at 37°C and 5% CO2 prior to imaging (as described in the Methods) (A) Life history plots of three representative MTs in these cells treated with or without DAG-lactone. (B) Quantitation of life history plots of MTs show that treatment with 10 μM DAG-lactone (DAG) increases the dynamicity of MTs (n = 54) in these cells. Statistical significance was determined by the Student’s t-test (*, p<0.0001). (C) A representative cell expressing EGFP-α-tubulin shows that following DAG-lactone treatment, growing MTs reverse direction upon contact with the cell periphery (inset). All results are representative of three independent experiments. Scale bar, 10 μm.
Table I presents the results of extensive quantitation of life history plots for 42–62 MTs of the kind shown in Fig. 1A. Compared with control cells, DAG-lactone treatment gave rise to an overall 2-fold higher dynamicity due to shifts in those parameters that favored net growth (Fig. 1B). The most pronounced effect that resulted from DAG-lactone treatment was a 3-fold increase in the duration of MT growth with shorter periods spent either shrinking or pausing. Moreover, MTs grew at a 1.4-fold higher rate while the frequency of catastrophe was decreased by 2-fold. In contrast, MTs in control cells, either DMSO-treated (Table I) or untreated parental cells (S. De, unpublished data), spent more than half the observed time in a paused state that corresponded to an overall lower dynamicity. This sedentary feature of MTs is consistent with the observed non-motile behavior of MCF-10A cells [Sun and Rotenberg, 1999]. Videos depicting EGFP-α-tubulin signals in control MCF-10A cells (Movie 1) and in cells treated with DAG-lactone as in Fig. 1B (Movie 2), can be found in the “Supplemental Materials”.
Table I. I. Quantitation of MT dynamic parameters in MCF-10A cells.
Dynamic parameters of MTs in non-transformed human breast cells. MCF-10A cells were treated with DAG-lactone (10 μM) or DMSO (0.05% v/v) for 1 h at 37°C, or transfected with a plasmid expressing a mutant of α6-tubulin or the vector control (VC).
| Dynamic parameter | Treatment
|
Transfection
|
|||
|---|---|---|---|---|---|
| DMSO | DAG-lactone | VC | S165N-α-Tubulin | S165D-α-Tubulin | |
| Dynamicity (μm/min) | 3.56 ± 2.94 | 7.05 ± 2.46a | 3.59 ± 2.31 | 3.87 ± 1.9 | 6.03 ± 2.05a |
|
| |||||
| Growth rate (μm/min) | 5.16 ± 1.23 | 7.5 ± 1.87a | 5.78 ± 1.66 | 5.68 ± 1.04 | 7.63 ± 1.50a |
|
| |||||
| Shortening rate (μm/min) | 10.75 ± 2.9 | 13.0 ± 4.7e | 9.22 ± 2.47 | 9.44 ± 3.36 | 11.61 ± 4.68d |
|
| |||||
| Time spent growing (%) | 25.74 ± 3.23 | 77.36 ± 2.78a | 32.57 ± 7.37 | 33.72 ± 4.48 | 62.29 ± 7.7 |
|
| |||||
| Time spent shortening (%) | 22.15 ± 4.06 | 10.98 ± 4.54d | 16.45 ± 5.16 | 20.22 ± 3.28 | 8.77 ± 1.17e |
|
| |||||
| Time spent pausing (%) | 52.11 ± 6.36 | 11.66 ± 1.80b | 50.98 ± 12.53 | 46.06 ± 3.28 | 28.94 ± 8.12e |
|
| |||||
| Catastrophe frequency (min−1) | 1.25 | 0.57 | 1 | 1.37 | 0.67 |
|
| |||||
| Rescue frequency (min−1) | NS | NS | NS | 2.17 | NS |
|
| |||||
| Number of microtubules | 54 | 54 | 40 | 63 | 61 |
Values are the mean ± s.d. except for percentages which represent the entire observed population of MTs. NS, not significant (fewer than 10 events observed for all MTs). Statistical significance relative to the appropriate control (DMSO or vector control) was determined by the Student’s t-test: (a) p<0.0001, (b) p<0.001, (c) p<0.01, (d) p<0.05 and (e) p<0.1. For all t-test calculations, n = total number of MTs was used except for percentages for growth/shortening/pause where n = 3, corresponding to the number of independent experiments (described in the ‘Methods’).
PKC phosphorylation site mutations in Ser-165 of α6-tubulin modulate MT dynamics of MCF-10A cells
Previous findings from this laboratory identified Ser-165 as the site in α6-tubulin (TUBA1C) that undergoes phosphorylation by PKC in DAG-lactone-activated MCF-10A cells, and is co-incident with the acquisition of motility [Abeyweera et al., 2009]. As shown in this earlier study, non-motile MCF-10A cells also acquired this phenotype with a pseudo-phosphorylated α6-tubulin that had been mutated at Ser-165 [Ser165→Asp (D)], thereby recapitulating the outcome produced by PKC activation. In contrast, the motility phenotype was not observed when MCF-10A cells expressed the phosphorylation-site resistant mutant [Ser165→Asn (N)] of α6-tubulin. In that previous study it was documented that the ratio of mutant protein to native α-tubulin was consistently 1:3.
To investigate whether phosphorylation of Ser-165 directly impacts MT dynamic behavior, live cell imaging was performed with these α6-tubulin mutants. For this purpose, each myc-tagged mutant was encoded from a bi-cistronic pBI-CMV4-DsRed plasmid that expressed DsRed and the mutant as separate proteins. The use of a bi-cistronic vector enabled fluorescence detection of those cells that were successfully transfected and therefore circumvented the need for a large fluorescent fusion protein of α6-tubulin. Cells were transfected with this bi-cistronic vector along with the vector encoding EGFP-α-tubulin so as to uniformly label MTs. Time lapse studies of MT dynamics were performed on individual MCF-10A cells that expressed both DsRed and EGFP signals, and dynamic parameters of MTs were determined for each α6-tubulin mutant in comparison with the empty vector (Table I).
As was observed with DAG-lactone-treated MCF-10A cells, MTs in cells expressing S165D-α6-tubulin showed a significantly higher dynamicity as compared with the S165N mutant, or cells that received the empty vector (VC) (Table I). This effect was dominated by a longer duration of the growth phase (by 2-fold) (Fig. 2) and a higher growth rate (by 1.3-fold) and accompanied by a decreased frequency of catastrophe. In cells expressing the phosphorylation-resistant S165N mutant, the percentage of time MTs spent growing was unchanged from values obtained for the vector control (Fig. 2) but was associated with an increased frequency of catastrophe (Table I). It became apparent that expression of S165D-α6-tubulin evoked quantitatively similar responses from MTs as those induced by DAG-lactone, while the S165N mutant caused only minor variation in the basal values of untreated MCF-10A cells. It is notable that the impact of S165D-α6-tubulin on the percentage of the total time spent growing (62%) is significantly less than the DAG-lactone effect on this parameter (77%) (Table I). Although pseudo-phosphorylation of α-tubulin at Ser-165 accounts for almost 80% of the DAG-lactone effect on this parameter, there may be additional PKC substrates, presently unknown, whose phosphorylation by DAG-sensitive PKC isoforms contributes to MT dynamics.
FIGURE 2.

Phosphorylation or pseudo-phosphorylation of α6-tubulin increases the percentage of the total time that MTs spend in the growth phase. Phosphorylation-site mutants of α6-tubulin impact the percentage time that MTs spent growing (positive values) and decrease the time spent shrinking (negative values) in non-motile MCF-10A (light and dark grey bars bars) and metastatic human breast MDA-MB-231 (black and stippled bars) cells. Cells were transfected with DsRed plasmids encoding the S165D or S165N mutants of α6-tubulin, or the empty DsRed vector control (VC), and their impact was determined by live-cell imaging, as described in ‘Methods’. Addition of 10 μM DAG-lactone (DAG) to MCF-10A cells or 10 μM BIM to MDA-MB-231 cells were compared with their respective vehicle control (DMSO, 0.05% v/v). The results are based on a total of 42–62 MTs examined for each condition divided by the total time MTs spent growing or shortening. Statistical significance was evaluated by the Student’s t-test whereby *, p<0.1; **, p<0.05; ***, p<0.01; and ****, p<0.0001.
Phosphorylation of α6-tubulin impacts MT dynamicity in triple-negative metastatic breast epithelial cells (MDA-MB-231 cells)
The estrogen-progesterone-EGF-independent (triple negative) MDA-MB-231 human breast cancer cell line is highly invasive and expresses high endogenous levels of active PKC isoforms, of which PKCα is the most abundant [Gauthier et al., 2003]. Down-regulation or inhibition of PKCα in MDA-MB-231 cells results in decreases in both proliferation and migration [Lonne et al., 2010]. Since expression of S165N-α6-tubulin was found to inhibit the intrinsic motility of these cells by 60% [Abeyweera et al., 2009], it was of interest to compare MT dynamic parameters in MDA-MB-231 cells with those values found for non-transformed, non-motile MCF-10A cells.
Table II displays the quantitation of life history plots of MTs observed in MDA-MB-231 cells. In these highly motile cells, MTs spent more than 60% of their lifetime in the growth phase. The dynamic behavior exhibited by these MTs was characterized by random MT movements near the cell periphery, followed by sudden catastrophe and steep shortening intervals (plot not shown). It is notable that the chaotic behavior of MTs in these cells contrasted with DAG-lactone-treated MCF-10A cells in which MTs were more uniformly polarized even when the dynamic parameters of the two cell types had comparable values.
Table II. II. Quantitation of MT dynamic parameters in MDA-MB-231 cells.
Dynamic parameters of MTs in metastatic human breast cells. MDA-MB-231 cells were treated with BIM (10 μM) or DMSO (0.05% v/v) for 1 h at 37°C, or transfected with a plasmid expressing a mutant of α6-tubulin or the vector control (VC).
| Dynamic parameter | Treatment
|
Transfection
|
|||
|---|---|---|---|---|---|
| DMSO | BIM | VC | S165N-α-Tubulin | S165D-α-Tubulin | |
| Dynamicity (μm/min) | 7.07 ± 2.99 | 4.94 ± 2.40a | 5.23 ± 2.78 | 4.15 ± 2.82d | 5.40 ± 2.1 |
|
| |||||
| Growth rate (μm/min) | 7.97 ± 2.08 | 6.14 ± 1.71a | 7.08 ± 1.55 | 6.02 ± 1.17b | 7.35 ± 0.97 |
|
| |||||
| Shortening rate (μm/min) | 11.77 ± 5.02 | 9.11 ± 2.61c | 12.19 ± 3.75 | 8.16 ± 6.21d | 10.98 ± 4.41 |
|
| |||||
| Time spent growing (%) | 64.97 ± 8.71 | 51.67 ± 3.69e | 47.47 ± 8.28 | 23.99 ± 5.04d | 56.97 ± 0.25 |
|
| |||||
| Time spent shortening (%) | 15.42 ± 2.43 | 19.27 ± 2.83e | 15.9 ± 2.82 | 27.17 ± 7.62e | 10.63 ± 0.94 |
|
| |||||
| Time spent pausing (%) | 19.61 ± 6.36 | 29.06 ± 6.42e | 36.64 ± 10.95 | 48.84 ± 5.35e | 32.41 ± 0.78 |
|
| |||||
| Catastrophe frequency (min−1) | 1.05 | 1.31 | 0.88 | 1.33 | 1.08 |
|
| |||||
| Rescue frequency (min−1) | 2.15 | 2.53 | NS | NS | NS |
|
| |||||
| Number of microtubules | 66 | 58 | 44 | 42 | 42 |
Values represent the mean ± s.d. NS, not significant (fewer than 10 events observed for all MTs). Statistical significance relative to the appropriate control (DMSO or vector control) was determined by the Student’s t-test: (a) p<0.0001, (b) p<0.001, (c) p< 0.01, (d) p<0.05 and (e) p<0.1. For all t-test calculations, n = total number of MTs except for percentages for growth/shortening/pause where n = 3, corresponding to the number of independent experiments,
Analysis of phosphorylation site mutants of α6-tubulin in MDA-MB-231 cells (Table II) showed that the phosphorylation-resistant S165N mutant substantially decreased the rates and time spent growing (Fig. 2), and produced longer pause intervals. It was noted that treatment of MDA-MB-231 cells with the PKC inhibitor BIM produced the same direction of change in these parameters, and was as effective as the phosphorylation-resistant mutant in decreasing the overall dynamicity. Accordingly, the altered rates and duration of growth and shrinking collectively led to slower growing MTs, similar to control MCF-10A cells. In contrast with MCF-10A cells, more modest effects were associated with the expression of the S165D mutant in MDA-MB-231 cells, such as an increase from 47% to 57% in the duration of the growth phase. Since MDA-MB-231 cells are intrinsically motile and express abundant levels of PKCα, the muted impact by the pseudo-phosphorylated mutant is consistent with α-tubulin that is already highly phosphorylated at Ser-165, presumably due to endogenously active PKC. By comparing the duration of growth and shortening intervals associated with each mutant or treatment, we conclude that for both MCF-10A cells and MDA-MB-231 cells, the duration of the growth phase was most responsive to the phosphorylation state of Ser-165. In Fig. 2, this parameter was used as the basis for comparing the two cell lines, and reveals the impact of phosphorylation or its inhibition.
Phosphorylation of α-tubulin promotes partitioning into MTs
A central question was whether site-specific phosphorylation facilitates the incorporation of α6-tubulin into endogenous MTs. This matter was addressed by expressing each myc-tagged α6-tubulin mutant and determining its distribution between intact MTs and unincorporated α-tubulin. Whole cell lysates prepared from MCF-10A cells that had been transfected with either the myc-tagged wildtype (WT) or mutant α6-tubulin were fractionated into intact MT polymers (insoluble pellet) and unincorporated α/β–tubulin dimers (soluble fraction) for assay by Western blot with either anti-myc or anti-α-tubulin.
While the total pool of native α-tubulin was distributed between the insoluble and soluble fractions in a constant ratio of 55:45 (insoluble:soluble) for all samples (S. De, unpublished data), the distribution of myc-tagged α6-tubulin was found to vary with its phosphorylation state. The results (Fig. 3A) showed that myc-tagged WT-α6-tubulin isolated from cells treated with DAG-lactone or myc-tagged S165D-α6-tubulin mutant expression both resulted in a far greater extent of incorporation when compared with the WT-α6-tubulin in untreated cells or expression of the S165N mutant. Hence, in control cells expressing myc-WT-α6-tubulin, the insoluble fraction displayed 41% of the total myc signal (i.e. soluble plus insoluble), whereas in cells expressing myc-WT-α6-tubulin and treated with DAG-lactone, or cells that solely expressed the myc-tagged S165D mutant, the insoluble fraction incorporated 58% and 60% of the total myc signal, respectively. In view of the fact that the pseudo-phosphorylated mutant cannot undergo dephosphorylation, the close similarity of the two values is interesting as it suggests that phosphatase activity is only weakly reversing the DAG-stimulated phosphorylation of WT-α6-tubulin. In stark contrast, expression of the myc-tagged S165N mutant displayed only 31% of the myc signal in the insoluble fraction, which represented an almost 30% lower incorporation than the S165D mutant, and a decrease by 10% when compared to the myc-tagged WT-α6-tubulin in control cells. Therefore, phosphorylation (or pseudo-phosphorylation) of α-tubulin is sufficient to promote its incorporation into growing MTs.
FIGURE 3.

Phosphorylation of α6-tubulin increases its partitioning into MTs (insoluble fraction). (A) Western blot showing the level of myc-tagged WT-α6-tubulin from MCF-10A cells treated with DAG-lactone (WT + DAG) or DMSO (WT), or α6-tubulin mutants isolated in insoluble (25 μg protein per lane) and soluble fractions (100 μg protein per lane). Each sample represented 25% of the fraction volume. Myc-tagged α6-tubulin proteins were detected with anti-myc and the total level of α-tubulin present in each set of samples was detected by Western blot with anti-α-tubulin, as described in ‘Methods’. β-actin levels served as the loading control for the soluble fraction. The results are representative of three independent experiments that gave similar results. (B) From Western blots, the distribution of myc-α6-tubulin signals between the insoluble (dark gray bars) and soluble (light gray bars) fractions were quantitated by Image J. The values are represented as a fractional distribution of the total myc signal calculated for insoluble and soluble preparations. The results are the average ± s.d. of three independent experiments (n=3). Statistical significance was evaluated by the Student’s t-test: *, p<0.05; **, p<0.005.
The pattern of MT incorporation of phosphorylated α-tubulin was visualized by immunofluorescence of intact cells (Fig. 4). In these experiments, only the incorporated myc-tagged α-tubulin was visualized since any unincorporated monomer/heterodimeric species were removed from the fixed cells by multiple wash steps. DAG-lactone treatment induced the incorporation of myc-WT α6-tubulin (green signals) to an extent that was comparable to that of the endogenous α-tubulin (red signal). Under these conditions, myc-WT-α6-tubulin was evenly distributed along the entire length (from base to tip) of MTs growing into membrane protrusions (Fig. 4A), as shown by the yellow signals and the alternating red-green coloration of highly elongated MTs (inset). In contrast, control-treated cells displayed very weak incorporation of myc-WT-α6-tubulin, and was consistent with the slight myc-WT signal incorporated into the insoluble fraction, as found by Western blot (Fig. 3). The incorporation of each myc-α6-tubulin mutant into MTs was addressed in parallel. As was found for DAG-lactone-treated cells, myc signals (green) were observed in MTs for the myc-S165D-α6-tubulin. This observation implied a high degree of incorporation that was evenly distributed along MTs, including those extending into cell protrusions. In contrast, only slight incorporation of the phosphorylation-resistant myc-S165N mutant was observed; for this mutant, the myc signal was primarily localized to MT structures in the cell interior. Nonetheless, MTs continued to elongate by incorporating the native α-tubulin protein (red signals). Further treatment of these cells with DAG-lactone did not improve the incorporation of the myc-S165N mutant into MTs (S. De, unpublished data). These results implied that when phosphorylation at Ser-165 was blocked, there was very limited incorporation of this α6-tubulin mutant into growing MTs.
FIGURE 4.

Immunofluorescence of MCF-10A cells expressing myc-tagged wildtype or mutant α6-tubulin. (A) Incorporation of myc-tagged WT α6-tubulin was compared in cells pretreated for 1 h with 10 μM DAG-lactone or DMSO (0.05% v/v), and in cells transfected with S165D or S165N α6-tubulin mutants. Following fixation with 4% paraformaldehyde, cells were stained with myc antibody to delineate each myc-tagged α6-tubulin (mutant or WT) (green) and counter-stained with α-tubulin antibody to display total α-tubulin (red). Scale bar, 10 μm. A small area (red box) is enlarged in the adjacent image. (B) Co-localization of myc-tagged tubulin with endogenous MTs in immunochemically stained MCF-10A cells were analyzed by Pearson’s index (rp) (‘Methods’). Results are the average of signal intensities from 20–24 cells for each condition from three independent experiments. (*, p<0.0001) (C) Quantitation of the number of myc-tagged signals at the cell periphery was performed with Image J in cells either transfected with the indicated mutant α6-tubulin, or transfected with WT and treated with or without DAG-lactone treatment. Each value was normalized to the WT (minus DAG-lactone) condition. To assess MT stability under these conditions, a similar measurement of myc signals was performed with cells treated with DMSO (dark grey bars) or 300 nM nocodazole (light grey bars) for 1 h, followed by fixation and staining, as described in (A). (*, p<0.0001)
The cell images obtained by immunofluorescence were analyzed by estimating Pearson’s correlation coefficient (rp) [Bolte and Cordelieres, 2006] that describes the degree of co-localization of myc-α6-tubulin signals and native α-tubulin in MTs (Figure 4B). In control and DAG-lactone-treated cells, values of rp = 0.7 and 0.85, respectively, implied that DAG-lactone induced 21% higher co-localization of myc-tagged WT-α6-tubulin (green) and endogenous MTs (red). This finding is in good agreement with the 17% higher incorporation of the WT protein in DAG-lactone-treated cells found in isolated MTs by Western blot (Figs 3A, 3B). Similarly, S165D-α6-tubulin-expressing cells gave rp = 0.81, while cells expressing the S165N mutant gave rp = 0.59. Thus, the pseudo-phosphorylated mutant produced a 37% higher co-localization of the two signals than the phosphorylation-resistant mutant (Fig. 4C). The rp value for S165N mutant was approximately 16% less than that for WT-α6-tubulin in control cells, which indicated that the phosphorylation-resistant mutant was less frequently incorporated into MT polymers than WT-α6-tubulin in the (untreated) basal condition. The decreased incorporation corresponds well with the fractionation assay (Fig. 3) where myc-tagged S165N-α6-tubulin was weakly detectable in the insoluble fraction.
Next we investigated whether cells expressing myc-tagged WT and treated with DAG-lactone, or expressing myc-tagged S165D-α6-tubulin conferred stability to MTs, as suggested by the ‘curved structures’ near the membrane periphery observed repeatedly in live cell imaging (Fig. 1C) and immunofluorescence (Fig. 4A) experiments. Results obtained by immunofluorescence (Fig. 4C) showed that the average intensity of myc signals incorporated by MTs was consistently higher when α-tubulin was phosphorylated (DAG-lactone treatment) or pseudo-phosphorylated (S165D expression) (Fig. 4C, dark grey bars). To assess MT stability under conditions of α-tubulin phosphorylation, cells were treated with 300 nM of the MT depolymerizing drug nocodazole for 1 h (Liao et al., 1995). The average intensity of myc-signals associated with MTs near the cell periphery was used to obtain an index of MT stability, i.e., the higher the retention of signals in elongated MTs, the greater the stability. It was observed that cells treated with nocodazole exhibited higher average intensity (Fig. 4C, light grey bars) when treated with DAG-lactone or expressing the S165D mutant, as compared to their respective negative controls. Thus, phosphorylation of α-tubulin enhances not only its incorporation but also the relative stability of the resulting MT polymers.
DAG-lactone increases the number of EB1-EGFP-bound MT plus-ends in live MCF-10A cells
In order to determine whether DAG-lactone increased the number of growing MTs, cells were transfected with the plus-end binding protein EB1-EGFP. This protein can be detected by fluorescence microscopy due to the comet-like fluorescence track of EB1-EGFP proteins bound at growing MT plus-ends [Matov et al., 2010]. The fluorescent live cell images shown in Fig. 5A exhibit EB1-EGFP signals polarized towards flat membrane protrusions in control and DAG-lactone-treated cells. When EB1-EGFP-expressing cells were analyzed for MT comets approaching the cell periphery, the activity maintains a significantly higher number of growing MTs that, as a result of the prolonged growth phase, continue to elongate all the way to the periphery. Videos depicting EB1-EGFP signals in control (Movie 3) and DAG-lactone-treated MCF-10A number of EB1-EGFP-bound plus-ends at the cell periphery approximately doubled in response to DAG-lactone (Fig. 5B). This finding implied that DAG-sensitive PKC cells (Movie 4) can be found in the “Supplemental Materials”.
FIGURE 5.

Quantitation of MT growing ends with EB1-EGFP in MCF-10A cells. (A) Images of live cells depict the arrival of EB1-EGFP comets near the cell periphery following a 1 h treatment with 10 μM DAG-lactone or DMSO (0.05% v/v). (B) Quantitative analysis of EB1-EGFP signals per 100 μm2 area near the cell periphery following treatment with DAG-lactone (n = 1800 signals, 14 cells) or DMSO (n = 1112 signals, 16 cells). Scale bar, 10 μm. The results are represented as the average number of EB1 comets from three independent experiments. (*, p<0.0001) (C) After treating cells with 6.6 μM nocodazole followed by washout of the drug (see ‘Methods’), the appearance of EB1-EGFP comets bound to nascent MTs was measured after incubation with DAG-lactone (10 μM) or DMSO (0.05% v/v) for 0.5 min, followed by cell fixation and detection by fluorescence microscopy. (D) In cells treated with 10 μM DAG-lactone (dark grey bars) or DMSO (light grey bars), the number of nascent MTs in 18–55 cells treated with DAG-lactone for the indicated time interval was determined by fluorescence microscopy and the signals were quantified with Image J and numerically averaged. (*, p<0.0001; **, p<0.05)
The effect of DAG-lactone on the formation of nascent MTs was assessed with nocodazole to eliminate all pre-existing MTs, followed by wash-out of the drug, and addition of DAG-lactone. Cells were fixed at selected time points following DAG-lactone treatment (0.5–5.0 min), and viewed under the fluorescence microscope. As early as 0.5 min post-treatment, the appearance of EB1-EGFP signals could be detected and signified the presence of nascent MTs (Fig. 5C). At the 0.5 min time point, DAG-lactone treatment significantly increased the number of EB1-EGFP signals relative to control cells that rapidly increased for up to 5 min (Fig. 5D). The statistically significant higher numbers of EB1-EGFP signals (Fig. 5D) was evident throughout this 5-min growth period. This observation was consistent with increased MT stability by DAG-lactone (Figure 4C) that becomes apparent even during this earliest phase of the MT lifespan. However, DAG-lactone had no effect on the number of nucleation sites of MTs (S. De, unpublished results).
Phosphorylation of α6-tubulin promotes Rac1 activation and Rac1-driven cell movement
The small GTPases are known to participate in mechanisms that drive cell movement [Ridley et al., 1992; Waterman-Storer et al., 1999; Banyard et al., 2000; Hanna and El-Sibai, 2013]. Since an earlier report from this laboratory implicated Rac1 in PKC-driven motility of MCF-10A cells, the activation of Rac1 to its GTP-bound state was analyzed in cells treated with DAG-lactone. In Fig. 6A, cells treated with or without DAG-lactone were probed with an antibody that recognizes the active form of Rac1 (New East Biosciences). The results indicated that activated Rac1 signals were increased overall following DAG-lactone treatment, and could be detected in membrane ruffles. To analyze the effect of α-tubulin phosphorylation on the activation state of each of the small GTPases (Rac1, Cdc42, and RhoA), a pull-down assay was performed. MCF-10A cells expressing the pseudo-phosphorylated mutant of α6-tubulin (S165D) or phosphorylation-resistant mutant (S165N) were prepared into lysates. Each lysate was incubated with either PAK-PBD-agarose to pull-down activated Rac1 and Cdc42, or Rhotekin-agarose to pull-down activated RhoA. Each reagent bound its target only if the G-protein had undergone activation to the GTP-bound state, whereupon the extent of activation was detected by western blot with a G-protein-specific antibody. As shown in Fig. 6B, cells expressing the S165D mutant resulted in stronger signals for GTP-Rac1 than did cells expressing the S165N mutant or vector control. In contrast, GTP-Cdc42 and GTP-RhoA signals were not affected by expression of either α6-tubulin mutant. Thus, Rac1 undergoes activation by treatment with DAG-lactone or expression of the S165D mutant.
FIGURE 6.

Rac1 undergoes activation in cells expressing S165D-α6-tubulin. (A) Evidence of activated Rac1 in membrane ruffles of MCF-10A cells. Immunofluorescence of cells treated either with DMSO (0.05% v/v, control) or DAG-lactone (10 μM, 1h) was performed followed by fixation with 4% PFA and addition of anti-active Rac1 antibody (1:100). With DAG-lactone treatment, Rac1 localized to membrane ruffles (arrows). Scale bar, 10 μm. (B) Western blot analysis of the level of GTP-bound Rac1, Cdc42, or RhoA in response to S165D or S165N α6-tubulin mutants, or the vector control (VC). The immunoblot shows the results of pull-down assays with whole cell lysates (600 μg per sample), as described in ‘Methods’. The results are representative of three independent experiments. (C) Motility of MCF-10A transfectants expressing wildtype PKCα, S165D-α6-tubulin, or the empty vector (VC) was measured with or without the Rac1 inhibitor NSC23766 (75 μM) (in water). Measurements show the area occupied by the cells in triplicate samples after 8 h of treatment (‘Methods’). (*, p<0.0001) The results are representative of three independent experiments.
The significance of GTP-Rac1 to the motility phenotype actuated by the S165D mutant was analyzed by use of a Rac1 inhibitor (NSC23766). This cell-permeable molecule prevents Rac1 activation by inhibiting binding of Rac1-specific guanine nucleotide exchange factors (GEFs) without affecting activation of either Cdc42 or RhoA [Nassar et al., 2006]. As shown in Fig. 6B, cells transfected with either WT-PKCα or S165D-α6-tubulin, exhibited motility that was strongly elevated (by 4-fold), as previously reported [Abeyweera et al., 2009]. In each condition, motility could be inhibited by 75% by treatment with NSC23766. Taken together, our findings describe a model (Fig. 7) in which PKC-mediated phosphorylation of α-tubulin promotes more numerous, elongating MTs that by an unknown mechanism lead to the activation of Rac1, consequently eliciting downstream events that promote cell movement.
FIGURE 7.
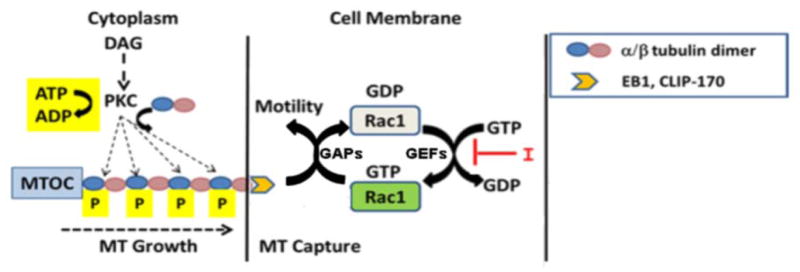
Model describing how PKC-mediated phosphorylation of α-tubulin impacts motility. Upon phosphorylation of α-tubulin (blue circles), elongating MTs promote the activation of Rac1 via a plus-end binding protein (EB1 or CLIP-170). Rac1 cycles between GTP-bound (active) and GDP-bound (inactive) states due to active GEFs and GAPs. A Rac1-specific inhibitor I (NSC23766) interferes in GEF-mediated GDP-GTP exchange thus blocking the formation of GTP-Rac which is required for driving downstream events that support motility.
DISCUSSION
This report is the first to identify PKC-mediated phosphorylation of α-tubulin as a means of regulating MT dynamics. It was shown that phosphorylation of α-tubulin at Ser-165 favors its incorporation into MTs that in turn prolongs the duration of MT growth. This outcome results in relatively longer and more numerous MTs that extend into membrane protrusions of the cell cortex, and consequently elicit downstream events, namely Rac1 activation and cell movement, that were previously attributed to PKC activity in MCF-10A cells [Sun and Rotenberg, 1999; Abeyweera et al., 2009]
How phosphorylation of Ser-165 in α-tubulin promotes MT growth may lie in the strategic location of this phosphorylation site at the interface of an α/β-heterodimer. The physical site of Ser-165 is found at the surface of an α-tubulin subunit of an α/β-heterodimer that docks with the plus-end β-tubulin subunit [Abeyweera et al., 2009]. In view of the preferential incorporation of pseudo-phosphorylated S165D-α-tubulin into growing MTs (Figs. 3, 4), we propose that DAG-lactone stimulates PKC to phosphorylate α-tubulin as a monomer or when complexed with β-tubulin. The resulting negatively-charged phosphate group at Ser-165 may strengthen binding and electrostatic interactions between successively added α/β-heterodimers, thereby supporting MT stabilization and persistent growth. However, the precise mechanism by which phosphorylation (or pseudo-phosphorylation) promotes this phenomenon is unknown at this time. Further study of MT polymerization testing each α-tubulin mutant as part of a heterodimer with β-tubulin will require kinetic analysis in vitro with highly pure components.
Dynamic parameters that sustain MT growth were observed to be augmented by either a membrane-permeable PKC activator (DAG-lactone) or by expression of α6-tubulin bearing a pseudo-phosphorylated mutation at Ser-165. The increased dynamicity produced by α-tubulin phosphorylation or pseudo-phosphorylation arose from an increased rate and duration of MT growth coupled with decreased duration of pause intervals and a lower incidence of catastrophe. Furthermore, our findings showed that these activated conditions correlated with a 2-fold increase in the number of EB1-EGFP-bound MTs arriving at the cell margin (Figs. 5A, B, D), having enhanced MT stability (Figs. 4C). The dramatic impact by PKC activity may be a general phenomenon since similar observations of PKC-associated effects on MT dynamics were reported during outgrowth of axonal cells treated with PDBu (a phorbol ester PKC activator) [Kabir et al., 2001]. However, the identity of the PKC substrate involved and the dynamic parameters affected by PDBu were not explored by that study.
A comparison of MT dynamic parameters operating in non-transformed, non-motile MCF-10A cells (Table I) and triple negative, metastatic MDA-MBA-231 cells (Table II) revealed complementary profiles. Among the parameters analyzed, the percentage of the total observation time that MTs were growing was most dramatically influenced by the state of Ser-165 phosphorylation in α-tubulin and therefore provided a basis for comparing the two cell lines (Fig. 2). In terms of this parameter, the results portrayed clear differences in a non-motile cell (MCF-10A) and an aggressively moving cell (MDA-MB-231) whose intrinsic motility is being driven to some extent by PKC-mediated phosphorylation of α-tubulin [Abeyweera et al., 2009]. In metastatic cells, expression of the S165N mutant (which blocks phosphorylation of Ser-165) corrected the intrinsically high rate and duration of MT growth to values found for non-motile MCF-10A cells, and correlated with the previously reported inhibition of motility of MDA-MB-231 cells (by 60%) by this mutant [Abeyweera et al., 2009]. This effect by the S165N mutant is consistent with making MTs intrinsically less stable upon its incorporation. Additionally, when expressed in MDA-MB-231 cells in which PKC activity is high, it is possible that the mutant, acting as a substrate mimic, exerts a direct inhibitory effect on PKC. The fact that the S165N mutant only partially inhibited motility and that PKC inhibitor BIM was modestly inhibitory (Table II) underscores the likelihood that additional mechanisms are modulating MT dynamics in these metastatic breast cells.
In a moving cell, elongating MTs are directed toward membrane protrusions such as lamellipodia [Komarova et al., 2002] through cortical membrane interactions afforded by the presence of a binding protein such as EB1, CLIP-170, or CLASP at the MT growing end (plus-end). The capture of MTs at the leading edge, is thought to be mediated by an interaction of the plus-end binding protein with a membrane-associated GTPase (Rac1 or Cdc42) [Fukata et al., 2002; Wittman and Waterman-Storer, 2005; Waterman-Storer et al., 1999]. In the activated (GTP-bound) state, Rac1 and Cdc42 each forms a complex with IQGAP [Fukata et al., 2002; Noritake et al., 2005], and thereupon collaborates in a series of unknown events in the actin cytoskeleton and membrane [Wittman et al., 2003], leading to polarized cell movement. In an earlier finding from this laboratory, we found that a dominant negative form of Rac1 inhibited PKC-mediated motility of MCF-10A cells, whereas dominant negative forms for Cdc42 or RhoA had no effect [Sun and Rotenberg, 1999]. Our current findings show that the level of activated Rac1 in MCF-10A cells is elevated in membrane ruffles following treatment with DAG-lactone (Fig. 6A) and also undergoes activation following expression of S165D-α-tubulin (Fig. 6B). Furthermore, Rac1 inhibition by NSC23766 (which inhibits GEF-mediated GDP–GTP exchange on Rac1) was shown to be critical to the mechanism of motility promoted by MTs bearing S165D-α6-tubulin (Fig. 6C). Taken together, these observations suggest that only Rac1 undergoes activation during constitutive expression of the S165D mutant, leading to cell movement. Our findings are consistent with a model (Fig. 7) in which phosphorylation of α6-tubulin by DAG-sensitive PKC isoforms results in an increased number of elongated MTs. It is proposed that the higher number of MTs reaching the membrane increases the probability of productive MT capture by activated Rac1, and consequently triggers Rac1-mediated signaling and cell movement. Although the precise means by which growing MTs lead to Rac1 activation is not known at this time, we speculate that elongating MTs interact directly or indirectly via a plus-end protein(s) with GEFs/GAPs resulting in elevation of local GTP-Rac1 levels. The significance of Rac1 cycling to cell movement, particularly in cancer cells, offers an avenue for further study [Wertheimer et al., 2012].
The α-tubulin mutants used in these studies were constructed with the α6-tubulin isotype which was previously identified from MCF-10A cells in its phosphorylated form [Abeyweera et al., 2009]. However, there are eight known isotypes of human α-tubulin, all having a Ser residue at a PKC consensus site analogous to Ser-165 in α6-tubulin. Therefore, all of the native α-tubulin isotypes can be regarded as potential substrates of PKC. While it is presently unknown which isotypes, other than α6-tubulin, are expressed in MCF-10A cells, it is not likely that they would have interfered with the α6-tubulin mutants being tested. An important reason is that these cells produce undetectable levels of endogenous phospho-α-tubulin (at PKC consensus sites) due to low PKC activity [Abeyweera et al., 2009]. Against this negligible background of phosphorylated α-tubulin, the S165D mutant produced the same impact on MT dynamics as DAG-lactone-stimulated PKC activity that presumably would have phosphorylated any available α-tubulin isotype. In contrast, MDA-MB-231 cells produce high levels of phospho-α-tubulin at PKC consensus sites [Abeyweera et al., 2009] due to endogenously active PKC, and consequently the S165D mutant predictably exhibited a more muted effect on MT dynamics.
Previous studies reported that mutations occurring in α- or β-tubulin consequently impact MT stability and cell motility [Hari et al., 2003; Ganguly et al., 2012]. Such anomalies have been implicated in different disease states [Tischfield et al., 2011] and taxol resistance [Verdier-Pinard et al., 2003; Wiesen et al., 2007]. These mutations affect the binding sites of microtubule-associated proteins (MAPs) and GTP molecules, lateral contacts between adjacent microfilaments, and longitudinal interactions within and between heterodimers. Our findings suggest that mutation of Ser-165 in α-tubulin to a residue that could serve as a phosphate mimic (Asp or Glu) could provide an important predictive marker for metastatic breast cancer.
This investigation focused on the role of PKC as the initiating agent that stimulates MT dynamics. However, it is possible that other protein kinases (e.g. cAMP-dependent protein kinase) with similar requirements for their consensus site will also phosphorylate Ser-165 or a nearby Ser or Thr residue(s) and thereby impact MT dynamics and cell movement. Furthermore, in light of signaling mechanisms known to regulate microtubule structural dynamics –either by other modes of covalent modification (Chao et al., 2012) or MAPs- phosphorylation represents only one mechanism to which MTs respond. The sheer diversity of pathways that communicate with MTs highlights the complex regulation and exquisite sensitivity of MTs to the moment-by-moment conditions of the intracellular environment.
METHODS
Materials
Human mammary epithelial cells (MCF-10A) were obtained from The Barbara Ann Karmanos Cancer Center (Detroit, MI), and metastatic human breast MDA-MB-231 cells were from the ATCC (Manassas, VA). The bi-cistronic DsRed plasmid was purchased from Clontech Laboratories (Mountain View, CA), and pIRESneo-EGFP-α-tubulin (human TUBA1B; #12298) and human EGFP-EB1 (#17234) plasmids were obtained from Addgene (Cambridge, MA). Cell culture media, antibiotics, serum, EGF, insulin, DABCO, and secondary antibodies (FITC-conjugated goat anti-rabbit and Alexa Fluor 594-conjugated goat anti-mouse) were purchased from Life Technologies (Carlsbad, CA). HRP-conjugated secondary antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). PolyExpress transfection reagent was obtained from Excellgen, Inc. (Rockville, MD), bis-indoleylmaleimide (BIM) from EMD Biosciences (La Jolla, CA), oxyrase from Oxyrase, Inc. (Ontario, OH), and restriction enzymes were from New England BioLabs (Ipswich, MA). Hydrocortisone, sodium lactate, protease inhibitors, phosphatase inhibitors, and α-tubulin antibody (DM1A) were purchased from Sigma-Aldrich (St. Louis, MO). Myc and β-actin antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). The pull-down assay reagent for RhoA and related buffers were purchased from Cytoskeleton, Inc. (Denver, CO). PAK-PBD-agarose and the Rac1 inhibitor NSC23766 were acquired from Calbiochem-Millipore Corp. (Billerica, MA), and the active Rac1 antibody was from New East Biosciences (Malvern, PA). Polylysine-coated 35-mm glass-bottom dishes were purchased from MatTek (Ashland, MA). DAG-lactone (JH-131E-153) was a gift from V. Marquez (NCI-Frederick).
Plasmid construction
Human α6-tubulin (TUBA1C) myc-tagged constructs (wild-type, pseudo-phosphorylated S165D and phosphorylation-resistant S165N mutants) were expressed either from a pCDNA3.1-myc vector, [Abeyweera et al., 2009], or a pCMV4-DsRed vector. By a PCR extension method, a SalI restriction site was introduced near the 3′ end of the myc-α6-tubulin constructs, followed by in-frame cloning into BamHI and SalI sites of the pCMV4-DsRed vector. The entire sequence was verified by DNA sequencing (Macrogen, Inc., Rockville, MD).
Cell culture and transfection
MCF-10A cells were cultured in 10-cm plates (BD Falcon) at 37°C and 5% CO in DMEM:F12 media supplemented with 5% equine serum, insulin (10 μg/ml), epidermal growth factor (20 ng/ml), cholera toxin (100 ng/ml), and hydrocortisone (0.5 μg/ml), and maintained with antibiotics [1% penicillin/streptomycin, and fungizone (0.5 μg/ml)] [Soule et al., 1990]. MDA-MB-231 cells were grown in Iscove’s Modified Dulbecco’s medium with L-glutamine, 10% fetal bovine serum, and antibiotics. Cells were recovered from the plates by trypsin-EDTA (0.25%) treatment and sub-cultured in a 1:3 ratio every 3–4 days.
Transient transfections were performed with PolyExpress, followed by a 48-h incubation period prior to experimental analysis. At 44–46 hours, the transfection efficiencies were typically 70–80% (single transfection) and 60–65% (co-transfection).
Isolation of MTs and Western blot analysis
Separation of cellular tubulin into a soluble fraction (unincorporated tubulin monomers and heterodimers) and an insoluble fraction (MT polymers) was performed with MCF-10A cells. Cells were grown in 6-well Falcon plates, transfected with myc-tagged α6-tubulin constructs (wildtype or mutants) or empty vector (control), and processed as previously described [Vogelsberg-Ragaglia et al., 2000; Fourest-Lieuvin et al., 2006]. Cells were washed with phosphate-buffered saline (PBS) and lysed in pre-warmed (37 °C) high salt RAB buffer [0.1 M MES (pH 6.8), 0.5 mM Mg2SO4, 1 mM EGTA, and 2 mM dithiothreitol (DTT)] supplemented with 0.1% Triton X-100, 40 μM Taxol, 2 mM GTP, 10 μM BIM, serine/threonine phosphatase inhibitors and protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 10 ng/ml leupeptin, 10 ng/ml soybean trypsin inhibitor). Ultracentrifugation of cell lysates (Beckman TLA 100.2) for 1 h at 288,000 × g resulted in a soluble fraction (supernatant) and an insoluble fraction containing MT polymers. The pellets were resuspended in 200 μl 1X RAB buffer following brief sonication. Total protein content of each fraction was determined by colorimetric assay (Bio-Rad). Twenty-five percent of each fraction volume was resolved by 8% SDS-PAGE followed by immunoblotting with either rabbit monoclonal anti-myc or mouse monoclonal anti-α-tubulin (DM1A). β-Actin antibody was used to judge equivalent loading of samples from the soluble fractions. HRP-conjugated secondary antibodies were used for detection by chemiluminescence with West Pico Super Signal reagents (Pierce Biotechnology). Band intensities were analyzed using Image J software (version 1.41a).
Live cell image acquisition and data analysis
To perform live cell imaging, cells were initially grown on 60-mm culture dishes and at 24 h post-transfection the cells were replated into 35-mm polylysine-coated glass-bottom dishes. Before imaging, the cell growth medium was replaced with phenol-red (PR)-free complete DMEM/F12 medium containing 15 mM HEPES, 0.6 units/ml oxyrase and 10 mM sodium lactate (oxyrase substrate) to reduce photo-bleaching and photo-toxicity [Salaycik et al., 2005]. Where indicated, either 10 μM DAG-lactone or DMSO (0.05% v/v) was added to cells, followed by incubation for 1 h (37°C, 5% CO2) prior to imaging.
Live cell images were collected using an inverted TCS-SP5 laser-scanning confocal microscope (Leica Microsystems) with a 63x 1.4NA oil-immersion objective lens. Laser intensities were kept at 25% and the pinholes were typically set to 1–1.5 Airy units. For time-lapse studies, images were acquired at 2-sec intervals at ambient temperature from a single Z-plane while applying a 32-line mean average. The nominal observation window was 30 sec due to constraints imposed by photobleaching. Images were analyzed with MetaMorph™ software (Molecular Devices, version 7.7.6) for determining dynamic parameters with EGFP-α-tubulin, or with Image J software for image quantitation of results obtained with EB1-EGFP, as described below.
Measurement of MT growth rate and dynamic instability with EGFP-α-tubulin
MT growth and shortening rates were measured by tracking MT ends using the ‘track-points’ function of Metamorph™ linked to an Excel spreadsheet with a customized macro [Wittmann et al., 2003]. MTs that remained visible for 30 sec and approached the cell periphery were chosen for analysis. For accurate measurements, a static point in the culture dish was adopted as a fiduciary mark relative to which the MT ends were analyzed for changes in length. The lengths of individual MTs were graphed as a function of time (life history plot). Only MT length changes exceeding the optical resolution limit of 0.1 μm per frame (~3 μm per min) were considered as growth or shortening events [Wittmann et al., 2003]. Dynamicity was calculated as the sum of the distance of the growth and shortening events divided by the total observation time. The percentage of total time that MTs spent growing, shortening or pausing was determined, as were the transition events from pause or growth to shortening (catastrophes), and from shortening to growth (rescues). The catastrophe frequency (or rescue frequency) was defined as the number of catastrophe events (or rescue events) divided by the time of growth and pause (or time of shortening).
The average rates of growth, shortening and dynamicity were presented as the mean ± s.d. A minimum of 40 MTs were analyzed for each condition over three independent experiments. The remaining dynamic parameters (i.e. percentage time spent growing, shortening, or pausing, and frequencies of catastrophes and rescues) are defined on the basis of the entire life history of all MTs for the indicated condition on a given day. Due to the 30-sec observation time constraint imposed by photo-bleaching, these parameters were not all attainable for individual MTs, especially the relatively low frequency of rescue. As a result, the percentages were calculated on an aggregate basis [Kamath et al., 2006] from each independent experiment and presented as the mean ± s.d. of three independent experiments conducted on different days. Since very few rescue and catastrophe events were observed for a single experiment, these quantities were reported on an aggregate basis over all three experiments. Events observed fewer than 10 times were not reported.
Quantitation of EB1-EGFP comets in live cells
The peripheral regions of MCF-10A cells, treated with 10 μM DAG-lactone or 0.05% DMSO (v/v) (1 h, 37°C) were sub-divided into regions of known dimensions by Image J. The number of EB1-EGFP comets in each region was counted manually and the average number of signals per unit area (μm2) was determined.
Immunofluorescence and image analysis
MCF-10A cells grown on glass-coverslips were transfected with the indicated plasmid(s) and incubated in complete medium for 48 h. Cells were washed once with PBS, and incubated for 1 h with complete medium containing either DAG-lactone (10 μM) or DMSO (0.05 % v/v). Cells were fixed in 4% paraformaldehyde at room temperature for 10 min. Coverslips were incubated for 30 s in microtubule-stabilizing and extraction (MSE) buffer (80 mM PIPES, 1 mM MgCl2, 4 mM EGTA, 0.5% Triton X-100), followed by washing with PBS and incubation in immunoblocker (2% BSA, 0.1% sodium azide) for 15 min at 37°C. Fixation followed by permeabilization and extraction gave consistently high imaging quality with these cells. After blocking, cells were incubated overnight at 4°C with mouse monoclonal DM1A anti-α-tubulin and rabbit monoclonal anti-myc followed by FITC-conjugated goat anti-rabbit (green) and Alexa Fluor 594-conjugated goat anti-mouse secondary antibodies (red). Coverslips were mounted on clean glass slides with DABCO and sealed with clear nail polish.
For measuring co-localization of myc-tagged α6-tubulin with endogenous MTs in MCF-10A cells, the red and green signal intensities of individual cells were analyzed by Pearson’s index in Image J [Bolte and Cordelieres, 2006; Dunn et. al., 2011]. Pearson’s index (rp) is a normalized cross correlation coefficient between two channels (red and green), and is averaged across the total number of pixels. As given by the equation below, the intensities of channel A and channel B in the ith pixel are denoted as Ai and Bi, respectively, and the corresponding average intensities over the full image are denoted as a and b. The resulting rp value falls within a range of −1 (complete exclusion) to 1 (complete co-localization).
To quantify the myc signals (green) for α6-tubulin at the cell periphery, images of MCF-10A cells were analyzed digitally by Image J with 160 μm2 boxes at 2–3 places along the leading edge of a cell, and the average intensities were determined for three independent experiments.
Measurement of MT plus-ends with EB1-EGFP in fixed MCF-10A cells
Experiments were conducted with MCF-10A cells in which MTs were eliminated with nocodazole (NOC) and analyzed at different time points for the appearance of nascent MTs after removal of the drug. At 42–44 h post-transfection with EB1-EGFP, cells were treated with 6.6 μM NOC for 5 h at 37°C to eliminate all microtubules. Following NOC treatment, cells were rinsed once with PHEM buffer [60 mM piperazine-N, N′-bis (2-ethanesulfonic acid), 25 mM HEPES, pH 6.9, 10 mM EGTA, and 4 mM Mg2SO4], treated at t=0 with either DAG-lactone (10 μM) or DMSO (0.05% v/v), followed by fixation at the indicated times for 5–10 min in ice-cold methanol (−20°C) [Tulu et al., 2006]. The samples were incubated in 1X MSE buffer for 30-sec and washed 3 times with PHEM buffer before mounting. Imaging was performed with a Plan-Neofluor 100X oil immersion objective lens on a Zeiss Axio Imager-M2 upright fluorescence microscope. EB1 signals were counted by the particle detector program of Image J [Sbalzarini and Koumoutsakos, 2005]. Threshold values were selected that maximized the number of EB1-GFP comets in each cell (i.e., the object size was adjusted to a radius of 3-pixels, and the circularity setting was optimized [Gusnowski and Srayko, 2011]. A minimum of 18 cells from different time points of each treatment were used for quantitation. Results were reported as an average number of EB1 signals from three independent experiments.
Pull-down assay of activated small GTPases
Activation of small GTPases was measured in lysates of MCF-10A cells using commercially available buffers (Cytoskeleton, Inc. Denver, CO). Briefly, cells (transfected with either S165D or S165N-α6-tubulin) were lysed in a lysis buffer (50 mM TRIS, pH 7.5, 10 mM MgCl2, 0.5 M NaCl, 2% Igebal) supplemented with phosphatase and protease inhibitors followed by centrifugation at 13,000 × g at 4 °C for 10 min. Agarose beads conjugated to either PAK-binding domain (PBD) (Millipore Corp.) or Rho-binding domain (RBD) (Cytoskeleton, Inc.) were used in a pull-down assay to isolate each activated G-protein. Cell lysates with a total protein content of 600 μg of protein were incubated with PAK-PBD beads (10 μg) or RBD (25 μg) beads at 4°C for 1 h. The beads were washed once with wash buffer (25 mM TRIS, pH 7.5, 30 mM MgCl2 and 40 mM NaCl) followed by centrifugation at 4°C. The entire sample was loaded and the bound proteins were separated by 12% SDS-PAGE and immunoblotted with anti-Rac or anti-Cdc42 (Cell Signaling Technology), or anti-Rho (Cytoskeleton).
Motility assay
MCF-10A cells were plated onto a 10-well slide through a cell sedimentation manifold (CSM, Inc., Phoenix, AZ) as concentric circles followed by incubation at 37°C and 5% CO2. After removal of the manifold (t=0), images were recorded with a camera attached to an inverted Nikon Diaphot microscope. After 16 h, during which cells radiated outwardly, images were recorded and motility was calculated by the change in total area (in μm2) occupied by the cells (Motic Image Plus 2.0). Each reported value is the average of triplicate measurements.
Statistical analysis
Each experiment was performed a minimum of three times, and differences between groups were determined using the unpaired Student’s t test. The data were evaluated as the mean ± 1 standard deviation (s.d.)
Supplementary Material
Acknowledgments
We thank Torsten Wittman (University of California, San Francisco) for providing the algorithm for image analysis of MT dynamics, and David Szent-Györgyi (BioVision Technologies) for customizing it for use with Metamorph™. We thank our colleagues at Queens College (Karl Fath, Nathalia Holtzmann, Cathy Savage-Dunn, and Corinne Michels), and The College of Staten Island (Jimmie Fata and Shawon Debnath) for insightful discussions and technical advice. The DAG-lactone reagent was a gift of Victor Marquez (NCI-Frederick), the EGFP-EB1 plasmid was provided by Lynne Cassimeris (Lehigh University), and the pIRESneo-EGFP-α-tubulin plasmid was made available by Patricia Wadsworth (University of Massachusetts). Live cell imaging and fluorescence microscopy experiments were performed in the Core Facility for Imaging and Molecular Biology at Queens College.
This research was supported by the National Cancer Institute of the National Institutes of Health under award CA125632 (to S.A.R.).
ABBREVIATIONS
- PKC
protein kinase C
- MT
microtubule
- MTOC
microtubule organizing center
- DAG
diacylglycerol
- EGFP
enhanced green fluorescent protein
- BIM
bis-indoleylmaleimide
- NOC
nocodazole
- WT
wildtype
- VC
vector control
- s.d
standard deviation
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Abeyweera TP, Rotenberg SA. Design and characterization of a traceable protein kinase Cα. Biochemistry. 2007;46:2364–2370. doi: 10.1021/bi0622017. [DOI] [PubMed] [Google Scholar]
- Abeyweera TP, Chen X, Rotenberg SA. Phosphorylation of α6-tubulin by protein kinase Cα activates motility of human breast cells. J Biol Chem. 2009;284:17648–17656. doi: 10.1074/jbc.M902005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banyard J, Anand-Apte B, Symons M, Zetter BR. Motility and invasion are differentially modulated by Rho family GTPases. Oncogene. 2000;19:580–591. doi: 10.1038/sj.onc.1203338. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres F. A guided tour into subcellular colocalization analysis in light microscopy. J Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Chan A, Andreae PM, Northcote PT, Miller JH. Peloruside A inhibits microtubule dynamics in a breast cancer cell line MCF7. Invest New Drugs. 2011;29:615–626. doi: 10.1007/s10637-010-9398-2. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhao X, Abeyweera TP, Rotenberg SA. Analysis of substrates of protein kinase C isoforms in human breast cells by the traceable kinase method. Biochemistry. 2012;51:7087–7097. doi: 10.1021/bi300999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SK, Yang C-PH, Horwitz SB. Posttranslational modifications of tubulin. In: Kavallaris M, editor. Cytoskeleton and human disease. New York: Humana Press; 2012. pp. 241–258. [Google Scholar]
- Desai A, Mitchison T. Microtubule dynamics. Ann Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Dhamodharan R, Jordan MA, Thrower D, Wilson L, Wadsworth P. Vinblastine suppresses dynamics of individual microtubules in living interphase cells. Mol Biol Cell. 1995;6:1215–1229. doi: 10.1091/mbc.6.9.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draber P, Draberova E. Microtubules. In: Kavallaris M, editor. Cytoskeleton and human disease. New York: Humana Press; 2012. pp. 29–54. [Google Scholar]
- Dunn K, Kamocka M, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S. Microtubules in cell migration. Ann Rev Cell Dev Biol. 2013 doi: 10.1146/annurev-cellbio-101011-155711. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Fourest-Lieuvin A, Peris L, Gache V, Garcia-Saez I, Juillan-Binard C, Lantez V, Job D. Microtubule regulation in mitosis: tubulin phosphorylation by the cyclin-dependent kinase Cdk1. Mol Biol Cell. 2006;17:1041–1050. doi: 10.1091/mbc.E05-07-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP170. Cell. 2002;109:873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem. 2012;287:43359–43369. doi: 10.1074/jbc.M112.423905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermejo ML, Leskow FC, Fuji T, Wang Q, Blumberg PM, Ohba M, Kurok T, Han KC, Lee J, Marquez VE, Kazanietz MG. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J Biol Chem. 2002;277:645–655. doi: 10.1074/jbc.M107639200. Correction (2004) J. Biol. Chem. 279: 23846. [DOI] [PubMed] [Google Scholar]
- Gauthier ML, Torretto C, Ly J, Francescutti V, O’Day DH. Protein kinase Calpha negatively regulates cell spreading and motility in MDA-MB-231 human breast cancer cells downstream of epidermal growth factor receptor. Biochem Biophys Res Commun. 2003;307:839–846. doi: 10.1016/s0006-291x(03)01273-7. [DOI] [PubMed] [Google Scholar]
- Gusnowski EM, Srayko M. Visualization of dynein-dependent microtubule gliding at the cell cortex: implications for spindle positioning. J Cell Biol. 2011;194:377–386. doi: 10.1083/jcb.201103128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signalling. 2013 doi: 10.1016/j.cellsig.2013.04.009. in press. [DOI] [PubMed] [Google Scholar]
- Hari M, Wang Y, Veeraraghavan S, Cabral F. Mutations in α- and β-tubulin that stabilize microtubules and confer resistance to colcemid and vinblastine. Mol Cancer Ther. 2003;2:597–605. [PubMed] [Google Scholar]
- Honore S, Pasquier E, Braguer D. Understanding microtubule dynamics for improved cancer therapy. Cell Mol Life Sci. 2005;62:3039–3056. doi: 10.1007/s00018-005-5330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir N, Schaefer AW, Nakhost A, Sossin W, Forscher P. Protein kinase C activation promotes microtubule advance in neuronal growth cones by increasing average microtubule growth lifetimes. J Cell Biol. 2001;152:1033–1043. doi: 10.1083/jcb.152.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath K, Okouneva T, Larson G, Panda D, Wilson L, Jordan MA. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol Cancer Ther. 2006;5:2225–2233. doi: 10.1158/1535-7163.MCT-06-0113. [DOI] [PubMed] [Google Scholar]
- Komarova YA, Vorobjev IA, Borisy GG. Life cycle of MTs: persistent growth in the cell interior, asymmetric transition frequencies and effects of the cell boundary. J Cell Sci. 2002;115:3527–3539. doi: 10.1242/jcs.115.17.3527. [DOI] [PubMed] [Google Scholar]
- Larsson C. Protein kinase C and regulation of the actin cytoskeleton. Cell Signalling. 2006;18:276–284. doi: 10.1016/j.cellsig.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Liao G, Nagasaki T, Gundersen GG. Low concentrations of nocodazole interfere with fibroblast locomotion without significantly affecting microtubule level: implications for the role of dynamic microtubules in cell locomotion. J Cell Sci. 1995;108:3473–3483. doi: 10.1242/jcs.108.11.3473. [DOI] [PubMed] [Google Scholar]
- Lonne GK, Cornmark L, Zahirovic IO, Landberg G, Jirstrom K, Larsson C. PKCalpha expression is a marker for breast cancer aggressiveness. Mol Cancer. 2010;9:76–90. doi: 10.1186/1476-4598-9-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matov A, Applegate K, Kumar P, Thoma C, Krek W, Danuser G, Wittmann T. Analysis of microtubule dynamic instability using a plus-end growth marker. Nature. 2010;7:761–769. doi: 10.1038/nmeth.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar N, Cancelas J, Zheng J, Williams DA, Zheng Y. Structure-function based design of small molecule inhibitors targeting Rho family GTPases. Curr Top Med Chem. 2006;6:1109–1116. doi: 10.2174/156802606777812095. [DOI] [PubMed] [Google Scholar]
- Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. J Cell Sci. 2005;118:2085–2092. doi: 10.1242/jcs.02379. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Kavallaris M. Microtubules: A dynamic target in cancer therapy. IUBMB Life. 2008;60:165–170. doi: 10.1002/iub.25. [DOI] [PubMed] [Google Scholar]
- Piehl M, Cassimeris L. Organization and dynamics of growing microtubule plus ends during early mitosis. Mol Biol Cell. 2003;14:916–925. doi: 10.1091/mbc.E02-09-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Risinger AL, Mooberry SL. Microtubules as a target in cancer therapy. In: Kavallaris M, editor. Cytoskeleton and human disease. New York: Humana Press; 2012. pp. 203–221. [Google Scholar]
- Salaycik KJ, Fagerstrom CJ, Murthy K, Tulu US, Wadsworth P. Quantification of microtubule nucleation, growth and dynamics in wound-edge cells. J Cell Sci. 2005;118:4113–4122. doi: 10.1242/jcs.02531. [DOI] [PubMed] [Google Scholar]
- Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X-g, Rotenberg SA. Over-expression of PKCα in MCF-10A human breast cells engenders dramatic alterations in morphology, proliferation and motility. Cell Growth Differ. 1999;10:343–352. [PubMed] [Google Scholar]
- Tischfield MA, Cederquist GY, Gupta ML, Jr, Engle EC. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr Op Gen Devel. 2011;21:286–294. doi: 10.1016/j.gde.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulu US, Fagerstrom C, Ferenz NP, Wadsworth P. Molecular requirements for kinetochore-associated microtubule formation in mammalian cells. Current Biol. 2006;16:536–541. doi: 10.1016/j.cub.2006.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier-Pinard P, Wang F, Martello L, Burd B, Orr GA, Horwitz SB. Analysis of tubulin isotypes and mutations from taxol-resistant cells by combined isoelectrofocusing and mass spectrometry. Biochemistry. 2003;42:5349–5357. doi: 10.1021/bi027293o. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM. Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell. 2000;11:4093–4104. doi: 10.1091/mbc.11.12.4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nature Cell Biol. 1999;1:45–50. doi: 10.1038/9018. [DOI] [PubMed] [Google Scholar]
- Wertheimer E, Gutierrez-Uzquiza A, Rosemblit C, Lopez-Haber C, Sosa MS, Kazanietz MG. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell Signal. 2012;24:353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesen KM, Xia S, Yang CP, Horwitz SB. Wild-type class I beta-tubulin sensitizes Taxol-resistant breast adenocarcinoma cells harboring a beta-tubulin mutation. Cancer Lett. 2007;257:227–235. doi: 10.1016/j.canlet.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol. 2003;161:845–851. doi: 10.1083/jcb.200303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM. Spatial regulation of CLASP affinity for microtubules by Rac1 and GSk3β in migrating epithelial cells. J Cell Biol. 2005;169:929–939. doi: 10.1083/jcb.200412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon A-M, Wadsworth P, Jordan MA. Taxol suppresses dynamics of individual microtubules in living human tumor cells. Mol Biol Cell. 1999;10:947–959. doi: 10.1091/mbc.10.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.