

Abstract
Defining the ultrastructure of endocytic sites and localization of endocytic proteins in Saccharomyces cerevisiae by immunoelectron microscopy is central in understanding the mechanisms of membrane deformation and scission during endocytosis. We show that an improved sample preparation protocol based on high-pressure freezing, freeze substitution, and low-temperature embedding allows us to maintain the cellular fine structure and to immunolabel green fluorescent protein–tagged endocytic proteins or actin in the same sections. Using this technique we analyzed the stepwise deformation of endocytic membranes and immuno-localized the endocytic proteins Abp1p, Sla1p, Rvs167p, and actin, and were able to draw a clear ultrastructural distinction between endocytic sites and eisosomes by immunolocalizing Pil1p. In addition to defining the geometry and the fine structure of budding yeast endocytic sites, we observed associated actin filaments forming a cage-like meshwork around the endocytic membrane.
Keywords: yeast, endocytosis, trafficking, high-pressure freezing, freeze substitution, transmission electron microscopy, electron tomography, immunogold labeling, actin filaments, anti-GFP immunolabeling
Introduction
Endocytosis is the process by which cells internalize extracellular solutes and receptors from the plasma membrane. While mammalian cells utilize a variety of endocytic pathways (Doherty & McMahon, 2009), the yeast Saccharomyces cerevisiae primarily uses clathrin-mediated endocytosis. Although there are differences in geometry and mechanism, the majority of effectors of clathrin-mediated endocytosis are highly conserved (Engqvist-Goldstein & Drubin, 2003; Boettner et al., 2012). This high conservation, the lack of alternate endocytic routes, and the ability to routinely integrate sequences encoding fluorescent protein tags at designated genomic loci to avoid protein overexpression make S. cerevisiae an ideal organism for studies of the endocytic machinery. Live-cell fluorescence microscopy has been used extensively to establish the order of sequential recruitment of endocytic proteins and to identify their mechanistic contribution to the process (Kaksonen et al., 2003, 2005; Newpher et al., 2005; Kim et al., 2006). In budding yeast, clathrin-mediated endocytosis occurs through a predictable sequence of protein recruitment events (Weinberg & Drubin, 2012). The individual functions of many of the 60 or so proteins involved in this process have been genetically tested. Like in mammalian cells, vesicle formation involves a burst of actin assembly (Merrifield et al., 2002; Kaksonen et al., 2003; Smythe & Ayscough, 2006), which in yeast is essential for membrane invagination and vesicle scission. Understanding how actin and the various other proteins mediate the steps of endocytic vesicle formation depends on an ultra-structural understanding of endocytic membrane geometry and of where specific proteins localize on the subdomains of the invagination and the forming vesicle.
Imaging the ultrastructure of yeast endocytic sites and the localization of endocytic proteins by immunogold labeling is challenging due to technical difficulties in sample preparation and in epitope preservation. Immuno-electron microscopy (immuno-EM) of chemically fixed S. cerevisiae cells by Mulholland et al. (1994) showed that actin patches characterized by fluorescence microscopy (Adams & Pringle, 1984; Kilmartin & Adams, 1984) coincide with invaginations of the plasma membrane in chemically fixed cells, and that actin patches also contain Abp1p and cofilin. A similar preparation and labeling technique has recently been used to localize a number of additional components of the endocytic machinery to invaginated membrane profiles (Idrissi et al., 2008; Idrissi et al., 2012). While these studies have provided valuable information, there is room for improvement in the procedures utilized because they relied on chemical fixation protocols to immobilize the specimen and prevent rearrangement of the cellular architecture during the following dehydration and infiltration with resin. While being well established for antibody labeling of HA tags, this fixation technique is known to result in changes to cellular structures, especially in the geometry of membranes as described for tubular sorting sites in endosomes (Murk et al., 2003). An alternative approach is to use freezing methods, which immobilize the sample within milliseconds without requiring perfusion of a fixative, and which thus dramatically improve the preservation of cellular structures. Cryo-fixed cells are either directly imaged in the frozen state by cryo-electron microscopy or after freeze-substitution (FS) and low-temperature embedding (LTE) in a resin. While cryo-electron microscopy allows imaging of cells in the most native state, low-temperature embedding enables postprocessing of sections for immunogold labeling while still preserving most structural details. We have demonstrated previously that the presence of water during FS is important in maintaining the cellular structures by preventing extraction of cellular components, especially membranes (Walther & Ziegler, 2002; Buser & Walther, 2008). Here we present evidence that water is also beneficial for preserving green fluorescent protein (GFP) antigenicity for immunolabeling, which has been difficult to achieve reproducibly.
Recently, Kukulski et al. (2011, 2012) established an alternative approach to define the mechanistic contribution of endocytic proteins by preserving the fluorescent signals of GFP and RFP in resin sections, which are then imaged by fluorescence microscopy and electron tomography to correlate individual endocytic sites and thus define their stage in the dynamic pathway and their protein composition. This method appears more sensitive due to its ability to detect fluorescent tags within the sections, while immunolabeling only detects antigens at the section surface. However, the protein localization accuracy is limited by the resolution limit of the fluorescence microscope.
Here we used high-pressure freezing (HPF) followed by FS and LTE in the presence of water in HM20 resin at − 50°C (HPF-FS-LTE) for studies of yeast endocytic sites. We performed anti-actin and anti-GFP immunolabeling on serial sections to localize GFP fusions of the endocytic proteins Abp1p, Sla1p, and Rvs167p, and to define the morphological stages of membrane deformation and the surrounding fine structure, including associated actin filaments. We also performed immunolabeling of the eisoso-mal component Pil1-GFP to draw a clear distinction between eisosome and endocytic site morphology in exponentially growing yeast cells.
Materials and Methods
Yeast Growth and High-Pressure Freezing
S. cerevisiae cells were grown to early log phase in standard YPD medium containing 1% glucose, concentrated by syringe filtration, aspirated in cellulose capillaries, and high-pressure frozen in a Wohlwend HPF Compact 01 high-pressure freezer (Engineering Office M. Wohlwend, Switzerland) or a HPM 010 high-pressure freezer (ABRA Fluid AG, Switzerland) as described previously (Buser & McDonald, 2010; Kishimoto et al., 2011).
Freeze Substitution and Embedding
For purely structural imaging, FS was performed in acetone containing 2% glutaraldehyde, 0.1% uranyl acetate, and 5% water (Walther & Ziegler, 2002; Buser & Walther, 2008) using a Leica AFS freeze-substitution system (Leica Microsystems, Vienna, Austria), and embedded in Epon (Kishimoto et al., 2011).
For immuno-EM, FS was done in acetone containing 0.1% uranyl acetate and 2% water in a Leica AFS2 (Leica Microsystems). The samples were kept at − 90°C for 3 h, then warmed at 2°C/h to − 50°C and washed 3× 15 min with acetone containing 2% water. Infiltration with HM20 resin diluted in acetone containing 2% water was done at − 50°C as follows: 25% HM20 for 4 h, 50% HM20 for 4 h, and two changes of pure HM20 for 24 h each. The resin was ultraviolet (UV) polymerized at − 50°C for 24 h.
Sectioning and Immunogold Labeling
Samples were serially sectioned at 50 to 100 nm thickness depending on the application and picked up on formvar-coated 1×2 mm nickel slot grids, blocked for 15 min in blocking buffer, i.e., PHEM buffer supplemented with 1% BSA (# A2153, Sigma-Aldrich, St. Louis, MO, USA) and 1% cold-water fish skin gelatin (# G7765, Sigma-Aldrich). Primary goat anti-GFP antibody (# 70R-GG001, Fitzgerald Industries International, Acton, MA, USA) was diluted 1:200 in blocking buffer and pre-adsorbed against an acetone powder prepared from a non-GFP expressing yeast strain (see below) at approximately 20 mg/mL over night at 4°C. After centrifugation at 14,000 rpm for 5 min, the supernatant was used to label the blocked sections for 45 min, followed by five washes for 5 min each in PHEM buffer, then incubated with a 10 nm gold-coupled secondary antibody (rabbit anti goat, #15796, Ted Pella, Inc., Redding, CA, USA), diluted 1:50 in blocking buffer for 45 min, followed by three washes for 5 min each in PHEM buffer and two washes for 2 min each in double-distilled water. The antibody-labeled sections were fixed in 2% glutaraldehyde in double-distilled water for 2 min and briefly washed three times in water.
For anti-actin immunolabeling a previously described polyclonal guinea pig anti-actin antibody (Mulholland et al., 1994) was used as above, except for being diluted 1:150 and only briefly pre-adsorbed against a yeast acetone powder for 5 min before centrifugation to reduce cell wall background staining.
All sections were stained with 2% aqueous uranyl acetate for 10 min and 2% lead citrate for 1 min, observed at 120 kV in a Tecnai 12 (FEI, Netherlands) and recorded with an Ultrascan 1000 CCD camera (Gatan, Inc., Pleasanton, CA, USA). The resulting images were not software-processed except for brightness/contrast adjustments and image rotation. All adjustments and measurements were done using ImageJ (Collins, 2007) and Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA). Immuno-EM background was quantified as described in Supplementary Table 1.
Yeast acetone powder was prepared from an overnight culture of a wild-type strain grown in YPD medium. Fifty milliliters of this culture were pelleted, resuspended vigorously in 4X volume of acetone precooled to −20°C, and incubated at 4°C for 30 min. The cells were pelleted again, resuspended in fresh 4X volume of precooled acetone, and incubated at 4°C for 30 min. After pelleting again, the pellet was completely air dried in a fume hood, ground to a fine powder, and stored in the dark at room temperature.
Electron Tomography
Tilt series were acquired using a Tecnai 12 microscope (FEI, Netherlands) at 120 kV with SerialEM software (Mastronarde, 2005) on a dual axis tomography holder (E.A. Fi-schione Instruments, Inc., Export, PA, USA). Images were recorded with an Ultrascan 1000 charge-coupled device (CCD) camera (Gatan, Inc.) with 2048 × 2048 pixels at a tilt range of ±50° in 1° increments and on two orthogonal axes. The resulting dual axis datasets were reconstructed and visualized with the IMOD software package (Kremer et al., 1996; Mastronarde, 1997).
Results
Ultrastructure of Budding Yeast Endocytic Sites
Imaging endocytic sites in yeast by HPF-FS provides improved structural preservation compared to chemically fixed specimens and allows a detailed description of the ultrastructure and membrane geometry of endocytic sites (Kishimoto et al., 2011; Kukulski et al., 2011,2012). The process of membrane deformation begins with a shallow, dome-shaped indentation of the plasma membrane (Fig. 1A), which then further invaginates (Fig. 1B) and begins to constrict at the base (Fig. 1C). Further invagination to a length of 50 to 80 nm and constriction of the tubule creates the characteristic flask-shaped membrane profile of typical intermediate endocytic sites in yeast (Fig. 1D), as we described previously (Kishimoto et al., 2011). In wild-type cells the membrane invaginates perpendicularly to the plasma membrane with less than 30° deviation (Smaczynska-de Rooij et al., 2010; Kishimoto et al., 2011). The constriction of the tubule region brings the opposing membranes within fusion range and at the same time creates the lumen of the future endocytic vesicle. When the tip of the invagination is approximately 80 to 90 nm from the plasma membrane, scission seems to occur rapidly, since longer invaginations are rarely observed in wild-type cells. During the scission process the tubule is further constricted (Fig. 1E) and the vesicle is pinched off (Fig. 1F), leaving behind a membrane stem, which rapidly relaxes back toward the plasma membrane.
Figure 1.
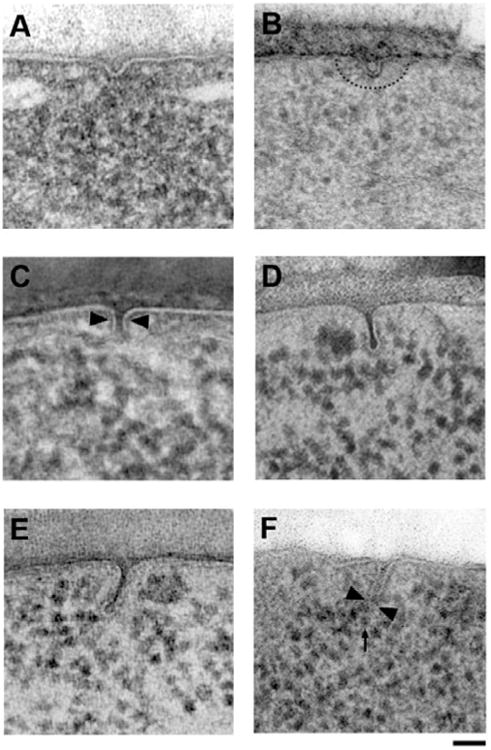
Progressive deformation of the plasma membrane during endocytosis. A: Earliest morphologically detectable stage of membrane deformation into a shallow dome. B: Dome internalization and formation of a faint ribosomal exclusion zone (dashed line). C: Further internalization and onset of membrane constriction (arrowheads). D: Typical intermediate endocytic membrane with characteristic flask-shape, tightly constricted neck, and visible coat. E: Rare pre-scission stage with tightly constricted tubule and formation of a rounded vesicle. F: Early post-scission stage in a bzz1Δ strain showing the point of scission (arrowheads) and the newly formed vesicle (arrow). Scale bars = 50 nm.
Simultaneous Preservation of the Cellular Ultrastructure and GFP Antigenicity
Sample preparation by HPF-FS-LTE is used with great success for structural imaging of biological samples. In contrast, immunolocalization on the same specimens has proven difficult, especially when attempting to label the most widely used fluorescent protein tag GFP. We found that FS in acetone containing 5% water and 0.1% uranyl acetate followed by three washes in pure ethanol or acetone, a short infiltration in LR-Gold (2 h 50% ethanol/LR-Gold; 2 h 100% LR-Gold), and overnight UV polymerization at − 18°C allowed labeling of GFP (Buser & McDonald, 2010), but later experiments showed that simultaneous labeling and adequate infiltration were poorly reproducible. In our first attempt to address the possible interference of the resin with the labeling, we performed FS-LTE in the same FS mixture, but with three pure acetone washes at − 50°C, followed by infiltration and UV polymerization in HM20, which resulted in a very well-preserved structure. However, immunolabeling of GFP again failed (Fig. 2A). We then speculated that the reported loss of GFP fluorescence after washes in pure solvent (Sims & Hardin, 2007) might also disrupt GFP epitopes. Indeed, saturating the acetone used during washing and infiltration steps with water resulted in successful labeling of GFP in HM20 (acetone with 2% water content; HM20 polymerization at − 50°C) (Figs. 2B-2F). Randomized evaluation of the background labeling showed low background of 0.50 gold/μm2 caused by the primary antibody, with overall cytoplasmic signal levels of 1.75 gold/μm2 for Abp1-GFP (Supplementary Table 1). We confirmed the specificity and accuracy of our labeling reaction by performing anti-GFP labeling of the endocytic coat component Sla1p-GFP, which localized to the tip of endocytic sites (Fig. 2B) and Rvs167p-GFP, which localized to neck (Fig. 1C), as previously reported in chemically fixed samples (Idrissi et al., 2008). We also compared this commercially available antibody with two previously published anti-GFP antibodies with comparable immunostaining results on an Abp1-GFP strain (Figs. 2D-2F). The present preparation protocol simultaneously preserves the cellular fine structure and the epitopes required for anti-GFP immunolabeling with a variety of antibodies and thus allows precise localization of GFP-tagged endocytic proteins within their ultrastructural context.
Figure 2.
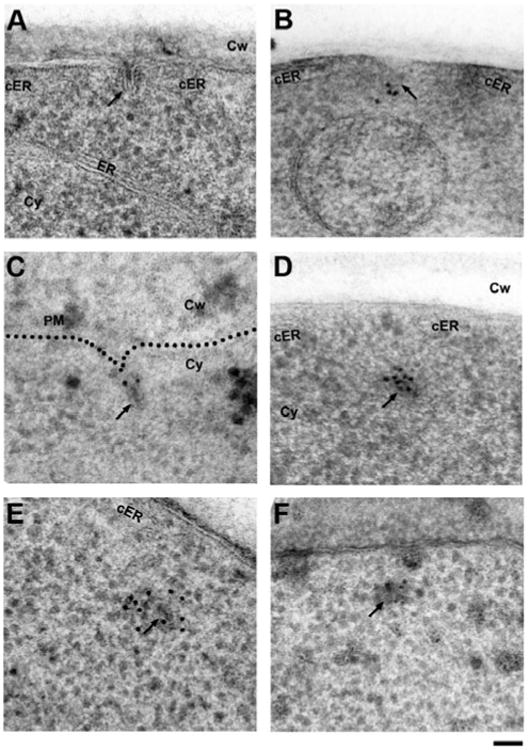
Establishing freeze-substitution and low-temperature embedding conditions for anti-GFP immunolabeling of endocytic proteins in S. cerevisiae. A: Freeze-substitution in acetone containing 2% water and 0.1% uranyl acetate followed by washes in pure acetone and embedding in HM20 results in excellent structural preservation of typical intermediate endocytic sites (arrow), but anti-GFP immunolabeling fails. Note the cortical ER cisternae (cER) flanking the endocytic site. B: Maintaining hydration of GFP by including 2% water in all acetone solutions (washes and infiltration) allows detection of the coat protein Sla1p-GFP at the tip of endocytic membranes (arrow) with a commercial primary anti-GFP antibody (Fitzgerald Industries), which was used throughout the study, except for panels E and F. C: Labeling of a strain expressing the BAR protein Rvs167p-GFP detects the protein at the neck region as previously described (Idrissi et al., 2008). D: The same antibody intensely labels the primary endosome (arrow) in a strain expressing Abp1p-GFP (see background quantification in Supplementary Table 1). The opening of the cortical ER (cER) is transiently maintained after scission. E: Labeling of the same Abp1p-GFP strain with a different, previously published anti-GFP antibody (Seedorf et al., 1999) also labels the post-scission primary endosomes (arrow). F: Identical but less efficient labeling of Abp1p-GFP using a different antibody (Collins et al., 2005). ER, endoplasmic reticulum; cER, cortical endoplasmic reticulum; Cy, cytoplasm; Cw, cell wall. Scale bar = 50 nm.
Anti-Actin Immunolabeling
To further verify that these invaginations represent bona fide endocytic sites, we performed anti-actin immunolabeling on 100 nm thick sections. Light microscopy studies have established that cortical actin patches are endocytic sites (Kaksonen et al., 2003). Actin label was found at various positions within the ribosomal exclusion zone on invaginations in both pre- (Fig. 3A) and post-scission stages (Supplementary Fig. S1), confirming that the structures observed above are endocytic sites. Tomographic reconstruction of the invagination shown in Figure 3A confirms the typical flask-shaped membrane invagination and a distinct coat density on the tip of the invagination, which ends above the tubule (Fig. 3B; Supplementary Movie S1). Interestingly, tomographic Z-slices through the bud show a slightly ellipsoid rather than spherical membrane profile (Fig. 3B, inset). In addition, after a close inspection of the projection image, we noticed faintly visible traces of actin filaments. Digital filtering and enhancement of the image reveal a cage-like meshwork of actin filaments surrounding the endocytic membrane (Fig. 3C). Furthermore, the anti-actin gold particles were observed in the same area and appeared to label the filaments where they are accessible at the section surface (Fig. 3D), thus confirming that these are actin filaments.
Figure 3.
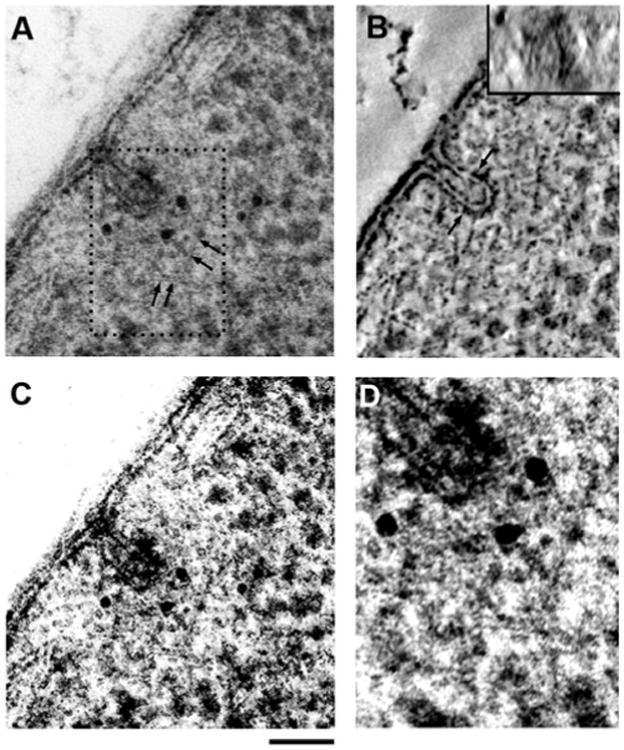
A: Anti-actin immunolabeling of a pre-scission endo-cytic site in a 100 nm thick section, with actin label spread within the ribosomal exclusion zone. Faint traces of actin filaments (arrows) can be seen within the exclusion zone and especially in the region at the top of the endocytic membrane, coinciding with most of the gold label. The ribosomal exclusion zone spreads laterally to both adjacent cER cisternae and approximately 100 nm into the cytoplasm (see Fig. 7). B: Central XY slice through the corresponding tomographic reconstruction (see Supplementary Movie S1) showing the typical flask-shaped membrane and the coat density covering the top portion of the invagination. A vertical Z-slice through the bud tip (inset) shows the ellipsoid shape of the membrane in cross section. C: Image shown in panel A digitally filtered and contrast-enhanced to increase the visibility of actin filaments. D: Magnification of the boxed area in panel A with enhanced contrast to highlight the cage-like organization of actin filaments around the tip of the endocytic site, as postulated previously (Kaksonen et al., 2006). Scale bars = 50 nm.
In addition to the deformation of the plasma membrane, endocytic sites create a zone devoid of ribosomes extending approximately 100 nm from the membrane tip (Fig. 3A), which was suggested to be formed by the actin meshwork (Mulholland et al., 1994). We also frequently observed cisterns of the cortical endoplasmic reticulum (ER) laterally surrounding endocytic sites as if creating an opening to allow the plasma membrane to internalize (Figs. 2A, 3A; Supplementary Fig. S1).
Immunolabeling of Abp1p-GFP Marks Scission and Post-Scission Endocytic Sites
We chose to focus on Abp1p-GFP to optimize GFP immuno-EM because this protein is expressed highly and because of our expectation that it would associate with endocytic sites throughout the entire membrane deformation process (Kaksonen et al., 2003). Unfortunately, we did not succeed in detecting labeling at early endocytic sites, indicating that the signal was too low to be detected reliably by our protocol, which prevented us from determining Abp1p's exact arrival time. We observed the highest Abp1p-GFP-labeling intensity at what appeared to be the scission and post-scission stages in serial sections, with one pool of label on the endocytic vesicle and a second transverse pool remaining at the site of scission (Fig. 4). The transverse Abp1p-GFP signal was only present when the endocytic vesicle was still within approximately 300 nm of the plasma membrane, whereas heavily labeled vesicles could be found deeper within the cytoplasm (Fig. 4A, middle panel). In a tomogram of an Abp1p-GFP strain immunolabeled for GFP, two gold particles label a transverse actin filament (Figs. 4C, 4D; Supplementary Movie S2), which is in agreement with the actin-binding properties of Abp1 and its suggested role in disassembly of the endocytic machinery (Goode et al., 2001; Toret et al., 2008). Furthermore, the apparent presence of transverse actin filaments could hint at a direct role for actin in constriction or scission. Comparison of the different distributions of Abp1p-GFP and actin suggests that Abp1p-GFP either binds to a subset of actin filaments or to a specific location on actin filaments.
Figure 4.
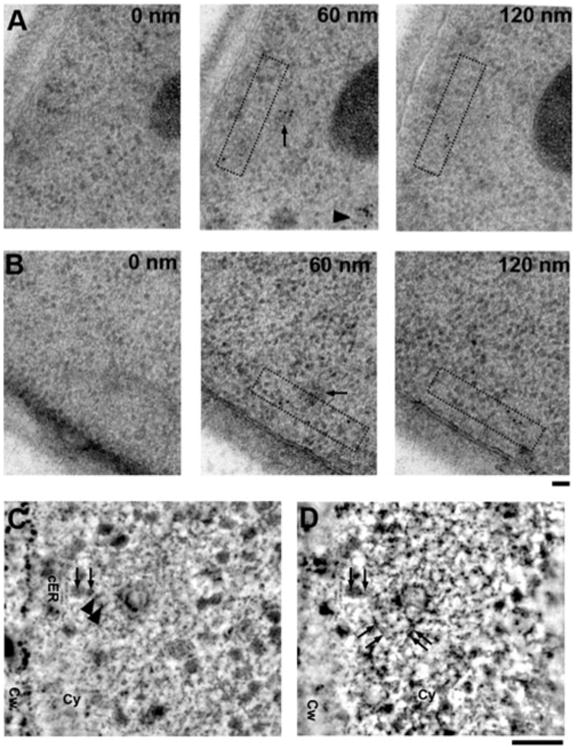
Abp1p-GFP label splits into two distinct populations and appears to mark transverse actin filaments (see Supplementary Table 1 for background quantification). A: 60 nm thin serial sections of a strain expressing Abp1p-GFP labeled using anti-GFP antisera showing redundant labeling of post-scission endocytic sites in adjacent sections. Intense Abp1p-GFP labeling of the primary endosomes immediately post-scission (arrow) is maintained on en-dosomes more than 500 nm into the cytoplasm (arrowhead). A second pool of label is seen crossing below the primary endosome (boxed area). B: 60 nm thin serial sections of a strain expressing Abp1p-GFP labeled using anti-GFP antisera with a primary endosome (arrow) showing a clear pool of Abp1-GFP running parallel to the plasma membrane (boxed area) and crossing the presumed point of scission. C: Slice through a tomographic reconstruction of a 100 nm thick section of a newly-formed primary endosome labeled using anti-GFP antisera to detect Abp1p-GFP (see Supplementary Movie S2). The arrowheads mark the positions of two gold particles labeling what appears to be a kinked, transverse actin filament (arrows). D: Montage of a tilted section averaged over seven adjacent image planes shows several actin filaments (arrows) surrounding the primary endosome. cER, cortical ER; Cw, cell wall; Cy, cytoplasm. Scale bars = 50 nm.
Distinguishing Eisosomes from Endocytic Sites
Eisosomes were originally proposed to be static sites of endo-cytosis (Walther et al., 2006) and were subsequently shown to coincide with long furrows in the plasma membrane in stationary yeast cultures (Stradalova et al., 2009). To distinguish eisosomal furrows from the endocytic sites studied here, we performed immuno-EM on the abundant eisosomal component Pil1p-GFP in our exponentially growing yeast cultures. We observed few furrow-like membrane invaginations in serial sections. Most such furrows were between 200 and 300 nm in length, and they mostly were shallow (less than 50 nm deep). Anti-GFP labeling of Pil1p-GFP frequently showed intense clusters of label up to 300 nm away from the plasma membrane, which were not connected to furrows (Figs. 5A, 5C). In some cases, we observed similar clusters that were in close proximity to the tip of shallow furrows as previously reported (Stradalova et al., 2009) (Fig. 5B). On very rare occasions, 100 nm deep, tightly apposed straight membranes were labeled at the tip (Figs. 5D, 5E), which probably represent obliquely sectioned furrows. In all instances these furrows can be readily distinguished from endocytic sites based on their sheet-like structure, characterized by the tightly apposed straight membranes, and the absence of a ribosomal exclusion zone.
Figure 5.

Identification of eisosomes by anti-GFP labeling of a strain expressing GFP-tagged Pil1p, the most abundant eisosomal/MCC component. A: A 60 nm section series showing the most commonly observed pattern of Pil1p-GFP labeling under our conditions of exponential growth, where dense clusters of label are neither at the plasma membrane nor associated with any visible deformation of the plasma membrane. B: A 60 nm section series showing a cluster of Pil1p-GFP label in proximity to a shallow furrow (arrow). C: Cluster of Pil1p-GFP label 300 nm away from the plasma membrane. D: Pil1p-GFP label associated with the tip of a rare, 100 nm deep, sheet-like furrow, as previously described (Stradalova et al., 2009). E: Pil1p-GFP label at the tip of another obliquely sectioned, sheet-like furrow. Scale bars = 50 nm.
Endocytic Mutants
In an attempt to understand the mechanistic function of key endocytic components, we ultrastructurally analyzed some knockout mutants (sla2Δ, sac6Δ, sla1Δ bbc1Δ, and cap1Δ) that were previously reported to be prominently defective in actin organization and/or dynamics at endocytic sites when analyzed by live-cell fluorescence microscopy (Kaksonen et al., 2003, 2005).
Deletion of the gene encoding the coat component Sla2p causes massive alterations of the actin cytoskeleton, the emergence of actin comet tails, and a defect in fluid phase endocytosis (Kaksonen et al., 2003). Transmission electron microscope (TEM) imaging of a sla2Δ strain showed many irregularly shaped cells, which contained fewer endocytic sites, most of which were shallow domes (Fig. 6A). Occasionally, very long, unconstricted tube-like invaginations could be observed (Fig. 6B).
Figure 6.
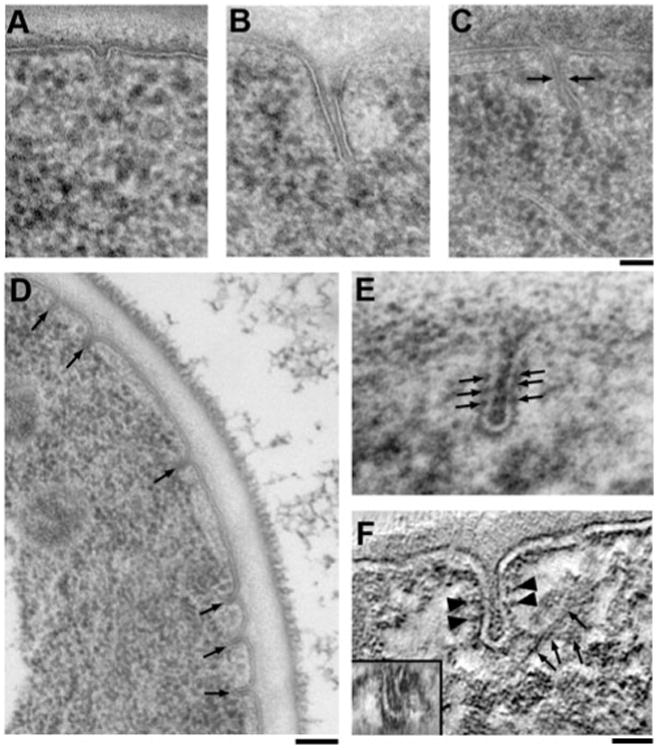
Ultrastructural phenotypes of endocytic knockout mutants. A: Thin section through an sla2Δ strain where mainly shallow endocytic domes can be observed. B: Rare aberrant invaginations in an sla2Δ strain show that occasionally internalization can proceed to form deep, tubular membrane profiles. C: Aberrant endocytic site in a sla1Δ bb c1Δ double knockout strain showing that the increased actin polymerization observed by fluorescence microscopy translates into endocytic profiles extending further into the cytoplasm than in wild-type cells. Interestingly, the point of constriction (arrows) is at the same distance from the plasma membrane as in wild-type cells. The tip of the site is elongated and often tilted laterally compared to the profile base. D: Overview image of a cap1Δ cell lacking functional capping protein and showing an unusual accumulation of intermediate endocytic profiles (arrows). E: Higher magnification of a cap1Δ endocytic profile. The membrane invagniated with a slight defect in constriction, thus appearing more tubular in shape. Note the appearance of repetitive membrane densities (arrows). F: Slice through a tomographic reconstruction of a cap1Δ strain (see Supplementary Movie S3) tilted and averaged over seven adjacent image planes showing lateral, intersecting actin filaments (arrows) and the repetitive densities mentioned above (arrowheads). Furthermore, a Z-slice through the bud tip shows an exaggerated, sheetlike cross section, confirming a defect in constriction (inset). Scale bars: A-C = 50 nm; D = 100 nm; E, F = 25 nm.
Similarly, deletion of SAC6, which encodes the actin filament-cross-linking protein fimbrin, appeared to completely block the invagination process at the shallow dome stage (not shown). However, in contrast to sla2Δ cells, no long invaginations were observed.
By fluorescence microscopy, sla1Δ bbc1Δ cells show extended bursts of actin polymerization suggesting unusually high levels of actin polymerization at endocytic sites (Kaksonen et al., 2005). When imaged by TEM, the invaginations were markedly longer than in wild-type cells (up to 300 nm) but were constricted at a similar position, approximately 50 nm away from the plasma membrane (Fig. 6C). In addition, the invagination tip was elongated and frequently tilted away from the perpendicular axis, while the invagination base remained perpendicular to the plasma membrane.
cap1Δ knockout cells showed a surprisingly strong phenotype in the TEM. When observed by live-cell fluorescence microscopy, no major irregularities are seen, except for a slightly delayed internalization of coat markers (Kaksonen et al., 2005). But when imaged by TEM, a strikingly high number of intermediate stage endocytic sites were observed (Fig. 6D). Furthermore, these invaginations seem slightly more tubular than flask-shaped, and repetitive densities could be seen on the invaginated membrane (Fig. 6E). Such repetitive densities have been observed in wild-type cells previously (Smaczynska-de Rooij et al., 2010). In our hands these densities were only clearly visible in the cap1Δ knockout strain, possibly due to accumulation of endocytic invaginations in a specific stage of internalization. Tomographic reconstruction revealed a pronounced ellipsoid shape of the invagination cross section and long adjacent filaments, presumably actin (Fig. 6F; Supplementary Movie S3). This phenotype suggests an important role for actin filament capping in shaping the endocytic membrane.
Discussion
Simultaneous Maintenance of Cellular Fine Structure and GFP Antigenicity
Preparation of resin embedded samples for immuno-EM by chemical fixation leads to the classical dilemma: stronger fixation yields better structural preservation but disrupts antigenic sites on the target proteins. This problem is aggravated in yeast because the fixatives need to penetrate the cell wall, resulting in long fixation times during which the ultrastructure deteriorates, although antigenicity can be preserved (Mulholland et al., 1994; Idrissi et al., 2008). High-pressure freezing followed by freeze substitution and low-temperature embedding offers a solution to this dilemma. Fixation is achieved solely by rapid cooling, and the following dehydration and embedding can be performed without chemical cross-linking of the sample by embedding at low temperatures. The result is a sample with excellent structural preservation and unfixed antigenic sites. However, reports of successful post-embedding immunolabeling of GFP-tagged proteins in resin sections are rare relative to the widespread use of GFP in biological research, especially in HPF-FS samples. There has been some success with Tokuyasu cryosections, where chemically fixed cells are cryo-protected in sucrose, immersion-frozen and cryosectioned. These sections are then thawed and picked up in methyl cellulose and immunolabeled using the same protocols as for resin sections (Tokuyasu, 1973; Henne et al., 2010). The key to successful immunolabeling might lie in the hydration state of GFP molecule: successful FS and resin embedding is traditionally thought to require a fully dehydrated sample. Therefore, efforts have typically been made to remove any residual water in FS solvents (Humbel & Mueller, 1985). Contrary to this assumption, it has been shown that the presence of water during FS helps to preserve cellular fine structure (Walther & Ziegler, 2002; Buser & Walther, 2008). We succeeded in retaining GFP antigenicity early on using FS with water followed by low-temperature embedding (Buser & McDonald, 2010), but later modifications showed that the protocol was unreliable in preserving antigenicity. Based on observations that retention of GFP fluorescence in sections requires the presence of water during embedding (Sims & Hardin, 2007), we speculated that the same might be true for GFP antigenicity. Indeed, maintaining minimal hydration of the specimens throughout FS, washes, and infiltration by using acetone containing 2% (vol./vol.) water at − 50°C results in efficient labeling of GFP with different antibodies (Fig. 2). The requirement for water for the stability of GFP and its maintenance throughout freeze substitution and embedding has implications in resin development (Carlemalm et al., 1982). The rationale in the development of the Lowicryl resins was that an ideal resin should closely mimic the biophysical properties of water (i.e., polarity and hydrogen bonding) while being polymerizable at the same time. Here we hypothesize that maximal hydrophilicity might not be required for molecular preservation. Instead, an ideal FS solvent and resin should have an intermediate polarity and be saturated with water. This should ensure proper replacement of the bulk water by the solvent during FS followed by replacement of the solvent with the resin without extracting the structural water during the process. Contrary to our hypothesis, other groups have reported successful preservation of GFP fluorescence but not antigenicity without water (Kukulski et al., 2011, 2012), and preservation of fluorescence and antigenicity with water present during FS only (Nixon et al., 2009). The reason for these discrepancies is not clear.
An inherent problem associated with immunolabeling is the low labeling efficiency and thus poor detection limits due to the requirement that the epitope be accessible at the section surface. The correlative fluorescence and electron microscopy approach by Kukulski et al. uses fluorescent signals in the section to define the protein location in the corresponding electron tomogram. This solution has several advantages over immunogold labeling: it does not require preservation and labeling of epitopes (thus avoiding the problems inherent in antibody labeling), and it is able to detect the majority of fluorophores within the entire section (and not only on the surface). But due to the resolution limit of the fluorescence microscope, the precision of the localization is roughly 10× lower and assumes a point source, which is not the case for a diffusely distributed protein such as actin or Abp1p, but works well for a highly localized protein like Rvs167p. In conclusion, a combination of these two methods will still be necessary until new correlative methods using clonable metal-binding tags are firmly established (Mercogliano & DeRosier, 2007; Wang et al., 2011).
Structure of Endocytic Membrane Invagination and Constriction
Our observations of endocytic sites show a tightly controlled and uniform membrane deformation process (Figs. 1, 7). The first morphologically distinct stage is a less than 20 nm deep, domed membrane deformation. This initial deformation has been suggested to be actin independent in mammalian cells (McMahon & Boucrot, 2011). It is not clear if this is the case for the same stage in yeast before deep invagination and constriction occur (Fig. 1A). The relatively small ribosomal exclusion zone, minimal actin label, and the observation that sac6Δ cells still form these early stages even if they are defective in converting actin turnover into coat motility provide contradictory evidence and do not allow us to reach a conclusion on the importance of actin in the very early stages. The situation in sla2Δ cells is more complicated because in addition to shallow invaginations, long aberrant invaginations do form occasionally (Figs. 6A, 6B). Recently, Kukulski and coworkers (2012) showed that no invaginations form at Sla1-GFP positive sites in cells treated with the actin-depolymerizing drug latrunculin A, thus suggesting that actin is also essential for the initial bending of the plasma membrane.
Figure 7.
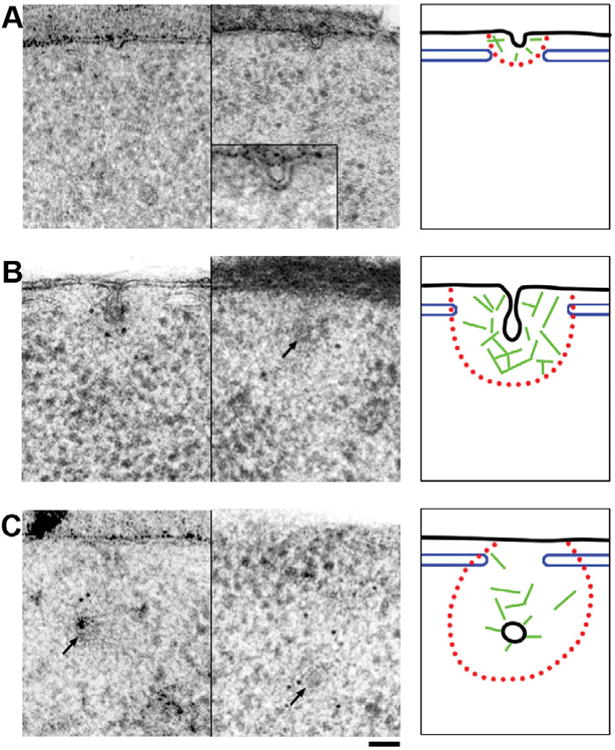
Two representative images and schematic interpretation of early, pre- and post-scission endocytic membranes and the associated structures. A: Early endocytic sites are characterized by a <30 nm deep, dome-shaped indentation of the plasma membrane. Frequently, the endocytic site is flanked by cisterns of the cortical ER (blue), which extend to the boundary of the ribosomal exclusion zone (red). While these early sites do not show significant anti-actin gold label, there is a faintly visible ribosomal exclusion zone (middle panel and inset). Thus the presence of short actin filaments (green) remains a possibility. B: Intermediate stage, pre-scission endocytic sites (arrow) show a typical flask-shaped membrane profile and a large ribosomal exclusion zone extending approximately 100 nm from the invaginated membrane and laterally ending at the flanking cER cisterns. Anti-actin label is found throughout the ribosomal exclusion zone and actin filaments can be seen forming a cage-like meshwork around the endocytic membrane. At this point a spiked coat density extending to the site of scission is also observed at the endocytic tip. C: Post-scission endocytic sites with the primary endosome (arrows) within <300 nm of the plasma membrane have a less clear ribosomal exclusion zone but still show strong anti-actin labeling both around the vesicle and diffusely within the area of scission (middle panel). Abp1p-GFP labeling was observed on the endosome and on transverse filaments crossing the site of scission (left panel). The coat completely encloses the endosome and the openings of the cER remain in place, but appear to narrow. Black membrane, blue cortical ER, red ribosomal exclusion zone, green actin filaments. Scale bar = 50 nm.
After the initial bending of the plasma membrane, the characteristic ribosomal exclusion zone appears, the coat is driven inward, and the membrane is progressively constricted (Figs. 1B–1D). As we have shown previously, constriction is achieved by the concerted action of Bzz1p and Rvs167p, and probably actin-derived forces (Kishimoto et al., 2011). The involvement of actin in constriction and scission is supported by the presence of transverse actin filaments (Fig. 3), which would not be required if the sole purpose of the filament network would be to drive coat internalization (Kaksonen et al., 2006). Additional indirect evidence for a role of actin in membrane constriction comes from the accumulation of weakly constricted invaginations in cap1 D cells, which lack a major actin filament barbed end capping complex (Figs. 6D-6F). In wild-type cells, the tubule is already highly constricted when the invaginations are 60 nm deep, and the distance between the two apposed membranes is reduced below 5 nm without immediately triggering scission. At this stage the endocytic coat can be seen enclosing the bud tip down to the approximate point of scission (Figs. 1D, 3C). All of the above-mentioned intermediate stages are observed frequently, and the geometry described here rarely varies. Furthermore, the membrane invaginates perpendicular to the plasma membrane with little deviation from 908. Even outliers never tilt more than 308 from the perpendicular axis, with the exception of knockout mutants like bzz1Δ cells (Kishimoto et al., 2011).
An interesting feature is the flanking cortical ER cisterns, which are commonly observed surrounding endocytic sites from the very earliest stages of membrane invagination to the late post-scission stages. The dimensions of the opening in the ER seem to correlate with growth and subsequent shrinkage of the ribosomal exclusion zone (Fig. 7). The frequency of this feature and the precise dimensions of the opening (Figs. 2, 3A; Supplementary Fig. S1) suggest direct communication between endocytic sites and the cortical ER. A similar correlation has been made in the context of the overall organization of the plasma membrane (Stradalova et al., 2012).
Pre- and Post-Scission Stages
Endocytic invaginations deeper than roughly 80 nm were very rarely observed, but in such cases the membrane was even further constricted along most of the tubule (Fig. 1E). At this point it appears that the vesicle is pinched off by membrane fusion at the interface of the vesicle and the tubule (Fig. 1F), which corresponds to the point of closest membrane proximity and the end of the coat density in wild-type cells. This position also coincides with the inter-facial region described by theoretical modeling (Liu et al., 2006, 2009). Both actin and the actin-binding protein Abp1p are likely involved in scission and post-scission processes. Abp1p was previously implicated in the disassembly of the endocytic machinery by recruiting the kinases Ark1p and Prk1p, which phosphorylate key components of the endocytic machinery (Fazi et al., 2002), and by recruiting the synaptojanin Sjl2p (Stefan et al., 2005). Abp1p also binds to preexisting actin filaments, is a weak Arp2/3 activator, and is internalized with the endocytic vesicle (Goode et al., 2001; Sun et al., 2006). A previous localization study of HA-tagged Abp1p in chemically fixed cells found it predominantly on intermediate and deep endocytic invaginations, but noted a shift from the tip to the base as the internalization process continued (Idrissi et al., 2008). In our hands, anti-GFP labeling of Abp1p-GFP intensely labeled post-scission endocytic sites (Fig. 4). One pool of label was tightly associated with the endocytic vesicle far into the cytoplasm, while a second pool labeled the site of scission parallel to the plasma membrane, including transverse actin filaments (Figs. 4C, 4D). In contrast, actin labeling was found on intermediate and post-scission sites, with label spread over the entire ribosomal exclusion zone (Fig. 3A) including a possible transverse pattern on post-scission sites (Supplementary Fig. S1). This transverse organization of actin filaments is consistent with a direct role for actin filaments in constriction and scission. Our identification of these structures as actin filaments is based on three considerations: (1) Their thickness and linearity correspond to previous TEM and scanning electron microscope images of actin filaments (Svitkina & Borisy, 1999; Koestler et al., 2008; Walther, 2008). (2) They precisely label with antibody-gold colloids directed against actin (Fig. 3) and against the actin-binding protein Abp1p-GFP (Figs. 4C, 4D; Supplementary Movie S2). (c) They are organized in the predicted geometry to drive internalization of the endocytic coat (Figs. 3, 7) (Kaksonen et al., 2006). The different distribution of actin and Abp1p-GFP immunostaining suggests that Abp1p might bind to a subset of actin filaments crossing the site of scission, which could be part of an actin-based constriction apparatus as recently proposed for mammalian endocytosis (Collins et al., 2011). The presence of Abp1p-GFP labeling of these transverse actin filaments and the primary endosome on post-scission sites supports the idea that Abp1p is involved in disassembly of the endocytic coat (through protein kinase and lipid phosphatase recruitment) and the actin network.
Further indirect evidence for the presence of a tubule-stabilizing structure on the neck comes from the phenotype observed in a sla1Δ bbc1Δ strain. These cells show exaggerated long bursts of actin polymerization at endocytic sites by fluorescence microscopy (Kaksonen et al., 2005), corresponding to an elongated membrane profile in the TEM (Fig. 6C). Interestingly, the membrane profile still constricts at the same distance from the plasma membrane as in wild-type cells and the tip of the invagination is tilted, whereas the base remains perpendicular to the plasma membrane. The approximate distance at which the membrane tubule is bent corresponds to the position of the putative scission structure labeled by Abp1p (Fig. 4).
Unfortunately, only a subset of actin filaments is visible even under ideal conditions in our resin sections due to their weak staining with lead and uranium salts, which prevents reproducible tracing in three-dimensional datasets. Nevertheless, the clear splitting of Abp1p-GFP label into two distinct pools, i.e., the transient transverse actin filaments and the long-lived vesicular fraction, prompts us to speculate that actin could fulfill separate roles in the scission machinery and on the newly-formed endosome.
Synthesis of the collected data allows us draw a generalized picture that links the deformation of the endocytic membrane to the changes in the surrounding ultrastructure (Fig. 7).
Eisosomes Can Be Morphologically Distinguished from Endocytic Sites
A major issue in studying endocytosis in yeast is the danger of confusing endocytic sites with eisosomes or membrane compartments-containing Can1 (MCCs), which are a second class of invaginations of the plasma membrane, previously termed furrows (Takeo et al., 1976; Walther et al., 1984; Walther et al., 2006; Stradalova et al., 2009). Furrows are >200 nm long and are usually shallow invaginations of the plasma membrane, which are prominent in stationary yeast cells but disappear during exponential growth (Takeo et al., 1976; Walther et al., 1984). Recently, the eisosomal/MCC proteins Pil1p and Sur7p have been localized to plasma membrane invaginations in stationary, overnight cultures of yeast (Stradalova et al., 2009). To verify that the endocytic structures we studied here are not eisosomes, we immunolabeled Pil1p-GFP in our exponentially growing cells. Under our conditions, clusters of Pil1p-GFP are not associated with any deformations in the plasma membrane in most cases (Figs. 5A, 5C), but occasionally localize to shallow (less than 50 nm) and short (∼200 to 300 nm) furrows (Fig. 5B) and very rarely to deeper, tube-like invaginations without a ribosomal exclusion zone (Figs. 5D, 5E). Furthermore, small buds in S. cerevisiae, where actin-labeled endocytic sites can be found (Supplementary Fig. S1), are free of Pil1p (Moreira et al., 2009). We are therefore confident that under our conditions deep furrows are rare and cannot be confused with endocytic sites.
Conclusions
Endocytosis in yeast shares several characteristics with mammalian clathrin-mediated endocytosis, including many conserved proteins. The differences are that in yeast actin is absolutely essential for membrane deformation, whereas in mammalian cells actin is clearly involved in endocytosis, but the coat proteins alone appear sufficient to generate a clathrin-coated pit (Yarar et al., 2005; Boucrot et al., 2006), unless the membrane is under high tension (Boulant et al., 2011). This mechanistic difference is reflected in the shape of endocytic sites, with mammalian cells forming larger, omega-shaped structures and yeast forming smaller, extended flasks, and thus also smaller primary endosomes. Nevertheless, the high conservation of endocytic effectors and the emerging evidence for a conserved role of actin in scission shows the similarity of the underlying processes and reaffirms the role of S. cerevisiae as a valuable organism for studies of endocytosis.
Supplementary Material
Acknowledgments
We are indebted to Kent McDonald for the many fruitful discussions and suggestions, and for access to the collection of anti-GFP antibodies. We also thank Pam Silver and Bill Wickner for the anti-GFP antibodies. C.B. was supported by a fellowship for prospective researchers by the Swiss National Science Foundation (SNF). This research was supported by National Institutes of Health grant R01 GM50399 to D.G.D.
Footnotes
Supplementary Material To view Supplementary Materials (including a table, figure, and movie clips) for this article, please visit http://dx.doi.org/10.1017/S1431927612014304.
References
- Adams AE, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic-mutant Saccharomyces cerevisiae. J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner DR, Chi RJ, Lemmon SK. Lessons from yeast for clathrin-mediated endocytosis. Nat Cell Biol. 2012;14:2–10. doi: 10.1038/ncb2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucrot E, Saffarian S, Massol R, Kirchhausen T, Ehrlich M. Role of lipids and actin in the formation of clathrin-coated pits. Exp Cell Res. 2006;312:4036–4048. doi: 10.1016/j.yexcr.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirch-hausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol. 2011;13:1124–1131. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser C, McDonald K. Correlative GFP-immunoelectron microscopy in yeast. Methods Enzymol. 2010;470:603–618. doi: 10.1016/S0076-6879(10)70024-0. [DOI] [PubMed] [Google Scholar]
- Buser C, Walther P. Freeze-substitution: The addition of water to polar solvents enhances the retention of structure and acts at temperatures around −60 degrees C. J Microsc. 2008;230:268–277. doi: 10.1111/j.1365-2818.2008.01984.x. [DOI] [PubMed] [Google Scholar]
- Carlemalm E, Garavito RM, Villiger W. Resin development for electron-microscopy and an analysis of embedding at low-temperature. J Microsc-Oxford. 1982;126:123–143. doi: 10.1111/j.1365-2818.1982.tb00358.x. [DOI] [PubMed] [Google Scholar]
- Collins A, Warrington A, Taylor KA, Svitkina T. Structural organization of the actin cytoskeleton at sites of clathrin-mediated endocytosis. Curr Biol. 2011;21:1167–1175. doi: 10.1016/j.cub.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins KM, Thorngren NL, Fratti RA, Wickner WT. Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 2005;24:1775–1786. doi: 10.1038/sj.emboj.7600658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: From yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- Fazi B, Cope MJ, Douangamath A, Ferracuti S, Schir-witz K, Zucconi A, Drubin DG, Wilmanns M, Ce-sareni G, Castagnoli L. Unusual binding properties of the SH3 domain of the yeast actin-binding protein Abp1: Structural and functional analysis. J Biol Chem. 2002;277:5290–5298. doi: 10.1074/jbc.M109848200. [DOI] [PubMed] [Google Scholar]
- Goode BL, Rodal AA, Barnes G, Drubin DG. Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J Cell Biol. 2001;153:627–634. doi: 10.1083/jcb.153.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne WM, Boucrot E, Meinecke M, Evergren E, Vallis Y, Mittal R, McMahon HT. FCHo proteins are nucleators of clathrin-mediated endocytosis. Science. 2010;328:1281–1284. doi: 10.1126/science.1188462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbel BM, Mueller M. Freeze substitution and low temperature embedding. In: Mueller M, Becker RP, Boyde A, & Wolosewick JJ, editors. In Science of Biological Specimen Preparation. Chicago, IL: SEM Inc., AMF O'Hare; 1985. pp. 175–183. [Google Scholar]
- Idrissi FZ, Blasco A, Espinal A, Geli MI. Ultra-structural dynamics of proteins involved in endocytic budding. Proc Natl Acad Sci USA. 2012;109(39):E2589–2594. doi: 10.1073/pnas.1202789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idrissi FZ, Grotsch H, Fernandez-Golbano IM, Presciatto-Baschong C, Riezman H, Geli MI. Distinct acto/myosin-I structures associate with endocytic profiles at the plasma membrane. J Cell Biol. 2008;180:1219–1232. doi: 10.1083/jcb.200708060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M, Sun Y, Drubin DG. A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell. 2003;115:475–487. doi: 10.1016/s0092-8674(03)00883-3. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. A modular design for the clathrin- and actin-mediated endocytosis machinery. Cell. 2005;123:305–320. doi: 10.1016/j.cell.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharo-myces. J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Galletta BJ, Schmidt KO, Chang FS, Blumer KJ, Cooper JA. Actin-based motility during endocytosis in budding yeast. Mol Biol Cell. 2006;17:1354–1363. doi: 10.1091/mbc.E05-10-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, Sun Y, Buser C, Liu J, Michelot A, Drubin DG. Determinants of endocytic membrane geometry, stability, and scission. Proc Natl Acad Sci USA. 2011;108:E979–988. doi: 10.1073/pnas.1113413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler SA, Auinger S, Vinzenz M, Rottner K, Small JV. Differentially oriented populations of actin filaments generated in lamellipodia collaborate in pushing and pausing at the cell front. Nat Cell Biol. 2008;10:306–313. doi: 10.1038/ncb1692. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Kaksonen M, Briggs JA. Plasma membrane reshaping during endocytosis is revealed by time-resolved electron tomography. Cell. 2012;150:508–520. doi: 10.1016/j.cell.2012.05.046. [DOI] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Welsch S, Picco A, Kaksonen M, Briggs JA. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J Cell Biol. 2011;192:111–119. doi: 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kaksonen M, Drubin DG, Oster G. Endo-cytic vesicle scission by lipid phase boundary forces. Proc Natl Acad Sci USA. 2006;103:10277–10282. doi: 10.1073/pnas.0601045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sun Y, Drubin DG, Oster GF. The mecha-nochemistry of endocytosis. PLoS Biol. 2009;7:e1000204. doi: 10.1371/journal.pbio.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. Dual-axis tomography: An approach with alignment methods that preserve resolution. J Struct Biol. 1997;120:343–352. doi: 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Mercogliano CP, DeRosier DJ. Concatenated me-tallothionein as a clonable gold label for electron microscopy. J Struct Biol. 2007;160:70–82. doi: 10.1016/j.jsb.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- Moreira KE, Walther TC, Aguilar PS, Walter P. Pil1 controls eisosome biogenesis. Mol Biol Cell. 2009;20:809–818. doi: 10.1091/mbc.E08-03-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland J, Preuss D, Moon A, Wong A, Drubin D, Botstein D. Ultrastructure of the yeast actin cytoskel-eton and its association with the plasma membrane. J Cell Biol. 1994;125:381–391. doi: 10.1083/jcb.125.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murk JL, Posthuma G, Koster AJ, Geuze HJ, Verkleij AJ, Kleijmeer MJ, Humbel BM. Influence of aldehyde fixation on the morphology of endosomes and lyso-somes: Quantitative analysis and electron tomography. J Mi-crosc. 2003;212:81–90. doi: 10.1046/j.1365-2818.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Smith RP, Lemmon V, Lemmon SK. In vivo dynamics of clathrin and its adaptor-dependent recruitment to the actin-based endocytic machinery in yeast. Dev Cell. 2005;9:87–98. doi: 10.1016/j.devcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Nixon SJ, Webb RI, Floetenmeyer M, Schieber N, Lo HP, Parton RG. A single method for cryofixation and correlative light, electron microscopy and tomography of ze-brafish embryos. Traffic. 2009;10:131–136. doi: 10.1111/j.1600-0854.2008.00859.x. [DOI] [PubMed] [Google Scholar]
- Seedorf M, Damelin M, Kahana J, Taura T, Silver PA. Interactions between a nuclear transporter and a subset of nuclear pore complex proteins depend on Ran GTPase. Mol Cell Biol. 1999;19:1547–1557. doi: 10.1128/mcb.19.2.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims PA, Hardin JD. Fluorescence-integrated transmission electron microscopy images: Integrating fluorescence microscopy with transmission electron microscopy. Methods Mol Biol. 2007;369:291–308. doi: 10.1007/978-1-59745-294-6_14. [DOI] [PubMed] [Google Scholar]
- Smaczynska-de Rooij I, Allwood EG, Aghamohammadza-deh S, Hettema EH, Goldberg MW, Ayscough KR. A role for the dynamin-like protein Vps1 during endo-cytosis in yeast. J Cell Sci. 2010;123:3496–3506. doi: 10.1242/jcs.070508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe E, Ayscough KR. Actin regulation in endo-cytosis. J Cell Sci. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- Stefan CJ, Padilla SM, Audhya A, Emr SD. The phosphoinositide phosphatase Sjl2 is recruited to cortical actin patches in the control of vesicle formation and fission during endocytosis. Mol Cell Biol. 2005;25:2910–2923. doi: 10.1128/MCB.25.8.2910-2923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradalova V, Blazikova M, Grossmann G, Opekarova M, Tanner W, Malinsky J. Distribution of cortical endoplasmic reticulum determines positioning of endocytic events in yeast plasma membrane. PLoS One. 2012;7:e35132. doi: 10.1371/journal.pone.0035132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradalova V, Stahlschmidt W, Grossmann G, Blazikova M, Rachel R, Tanner W, Malinsky J. Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J Cell Sci. 2009;122:2887–2894. doi: 10.1242/jcs.051227. [DOI] [PubMed] [Google Scholar]
- Sun Y, Martin AC, Drubin DG. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev Cell. 2006;11:33–46. doi: 10.1016/j.devcel.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 complex and actin depolymerizing factor/cofilin in dendritic organization and treadmilling of actin filament array in lamellipodia. J Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeo K, Shigeta M, Takagi Y. Plasma membrane ultrastructural differences between the exponential and stationary phases of Saccharomyces cerevisiae as revealed by freeze-etching. J Gen Microbiol. 1976;97:323–329. doi: 10.1099/00221287-97-2-323. [DOI] [PubMed] [Google Scholar]
- Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol. 1973;57:551–565. doi: 10.1083/jcb.57.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toret CP, Lee L, Sekiya-Kawasaki M, Drubin DG. Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic. 2008;9:848–859. doi: 10.1111/j.1600-0854.2008.00726.x. [DOI] [PubMed] [Google Scholar]
- Walther P. High-resolution cryo-SEM allows direct identification of F-actin at the inner nuclear membrane of Xenopus oocytes by virtue of its structural features. J Microsc. 2008;232:379–385. doi: 10.1111/j.1365-2818.2008.02109.x. [DOI] [PubMed] [Google Scholar]
- Walther P, Muller M, Schweingruber ME. The ultrastructure of the cell-surface and plasma-membrane of exponential and stationary phase cells of Schizosaccharomyces-Pombe, grown in different media. Arch Microbiol. 1984;137:128–134. [Google Scholar]
- Walther P, Ziegler A. Freeze substitution of high-pressure frozen samples: The visibility of biological membranes is improved when the substitution medium contains water. J Microsc. 2002;208:3–10. doi: 10.1046/j.1365-2818.2002.01064.x. [DOI] [PubMed] [Google Scholar]
- Walther TC, Brickner JH, Aguilar PS, Bernales S, Pan-toja C, Walter P. Eisosomes mark static sites of endocytosis. Nature. 2006;439:998–1003. doi: 10.1038/nature04472. [DOI] [PubMed] [Google Scholar]
- Wang Q, Mercogliano CP, Lowe J. A ferritin-based label for cellular electron cryotomography. Structure. 2011;19:147–154. doi: 10.1016/j.str.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Weinberg J, Drubin DG. Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol. 2012;22:1–13. doi: 10.1016/j.tcb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:964–975. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.