

Abstract
The mucin MUC1 is overexpressed and aberrantly glycosylated by many epithelial cancer cells manifested by truncated O-linked saccharides. Although tumor-associated MUC1 has generated considerable attention because of its potential for the development of a therapeutic cancer vaccine, it has been difficult to design constructs that consistently induce cytotoxic T-lymphocytes (CTLs) and ADCC-mediating antibodies specific for the tumor form of MUC1. We have designed, chemically synthesized, and immunologically examined vaccine candidates each composed of a glycopeptide derived from MUC1, a promiscuous Thelper peptide, and a TLR2 (Pam3CysSK4) or TLR9 (CpG-ODN 1826) agonist. It was found that the Pam3CysSK4-containing compound elicits more potent antigenic and cellular immune responses, resulting in a therapeutic effect in a mouse model of mammary cancer. It is thus shown, for the first time, that the nature of an inbuilt adjuvant of a tripartite vaccine can significantly impact the quality of immune responses elicited against a tumor-associated glycopeptide. The unique adjuvant properties of Pam3CysSK4, which can reduce the suppressive function of regulatory T cells and enhance the cytotoxicity of tumor-specific CTLs, are likely responsible for the superior properties of the vaccine candidate 1.
Keywords: adjuvants, cancer, carbohydrates, mucins, peptides, vaccines
Introduction
A large number of carcinomas of breast, ovary, colon, rectum, pancreas, and prostate exhibit striking overexpression of the mucin MUC1, resulting in a loss of polarized expression.[1] Furthermore, tumor-associated MUC1 is often aberrantly glycosylated, due to an increase in sialylation[2] or a lack of core 1,3-galactosyltransferase (T-synthase),[3] resulting in the expression of truncated carbohydrate structures such as Tn (αGalNAc-Thr) and STn (αNeu5Ac-(2,6)-αGalNAc-Thr) antigens.
MUC1 is immunogenic, with both humoral and cellular immune responses against tumor-associated MUC1 having been observed in cancer patients. The presence of circulating antibodies against MUC1 at the time of cancer diagnosis has been correlated with a favorable disease outcome in breast cancer patients.[4] Furthermore, cytotoxic T-lymphocytes (CTLs) isolated from patients with breast carcinoma can recognize epitopes present on MUC1 tandem repeat peptide.[5] It has been proposed that T-cell epitopes from the MUC1 core domain are packaged within tumor cells in their truncated glycosylation state into major histocompatibility complex (MHC) class I molecules, leading to natural MHC-restricted recognition of hypoglycosylated epitopes.[6] Several MUC1-derived HLA-A2-binding peptides have been identified; they include STAP-PAHGV, SAPDTRPAPG, STAPPVHNV, LLLLTVLTV, ALGSTAPPV, and NLTISDVSV,[5, 7] although only STAPPVHNV, LLLLTVLTV, and NLTISDVSV have been directly eluted from the HLA-A2 molecules from cancer patients or cancer cell lines.[7b,8]
The inherent immunological properties of tumor-associated MUC1 have stimulated the development of cancer immune therapies; however, it has been difficult to design therapeutic vaccines that can elicit IgG antibodies and CTLs against tumor-associated MUC1. Recently, we addressed this deficiency by identifying the minimum structural requirements for consistent induction of CTLs and ADCC-mediating (ADCC: antibody-dependent cell-mediated cytotoxicity) antibodies specific for the tumor form of MUC1, resulting in a therapeutic response in a mouse model of mammary cancer.[9] The lead vaccine is composed of the immunoadjuvant Pam3CysSK4, a peptide Thelper epitope derived from polio virus,[10] and an aberrantly glycosylated MUC1 peptide (compound 1, Scheme 1, below). The vaccine produced CTLs that recognized both glycosylated and nonglycosylated peptides, whereas a similar nonglycosylated vaccine gave CTLs that recognized only nonglycosylated peptide. Antibodies elicited by the glycosylated tripartite vaccine were significantly more lytic than those elicited by the nonglycosylated control. As a result, immunization with the glycosylated tripartite vaccine was superior in tumor prevention in a mouse model of breast cancer. Furthermore, we established that covalent attachment of the three components is critical for achieving optimal immune responses. Subsequently, several other multicomponent MUC1-based cancer vaccines that exhibit some of the properties of compound 1 have been described.[11]
Scheme 1.

Chemical structures and synthesis of tripartite vaccine candidates 1 and 3, control compounds 2 and 4, and synthetic intermediates 5–8.
The inbuilt immunoadjuvant Pam3CysSK4, which is a potent agonist of Toll-like receptor 2/6 (TLR2/6),[12] induces the production of cytokines and chemokines that promote the expression of a number of costimulatory proteins that are required for optimum interactions between antigen-presenting B- and T-cells. In addition, some cytokines and chemokines are responsible for overcoming suppression mediated by regulatory T-cells. Other cytokines are important for directing the effector T-cell response towards a T-helper-1 (Th-1) or T-helper-2 (Th-2) phenotype,[13] which in turn is critical for the mechanism by which microbes or cells are neutralized.
In addition to Pam3CysSK4, several other TLR agonists have received attention as vaccine adjuvants: examples include monophosphoryl lipid A, flagellin, and CpG oligodeoxynucleo-tides (CpG-ODNs).[12] Compounds of the last type are short single-stranded synthetic unmethylated DNA fragments, the natural counterparts of which are abundant in microbial genomes but rarely observed in vertebrate ones.[14] The immunomodulatory activity of CpG is mediated by the pattern recognition receptor TLR9. Several studies have shown that CpG-ODNs can augment the immunity of MUC1-based experimental cancer vaccines.[15] Furthermore, conjugation of a CpG-ODN to a protein antigen can result in the induction of more prominent T-cell responses than are obtained with administration of free CpG mixed with a protein antigen.[16] Covalent attachment of CpG to an antigen results in a more efficient cellular uptake, and this in turn facilitates the presentation of MHC-I and II epitopes.[17] Interestingly, cross-presentation of OVA-linked CpG occurs independently of TLR9 expression, but TLR9 expression is essential for activation of the dendritic cells (DCs). On the basis of these observations, we felt compelled to investigate immune responses of a tripartite vaccine candidate composed of a CpG-ODN, a Thelper epitope, and a MUC1 glycopeptide and to compare the responses elicited by such a vaccine candidate with those elicited by a similar and previously reported compound[9b] with Pam3CysSK4 as an inbuilt adjuvant.
Results and Discussion
Chemical synthesis
Tripartite vaccine 3 (Scheme 1) contains CpG ODN 1826, which is a CpG-ODN type B specific for mouse TLR9. It is based on nuclease-resistant phosphorothioate linkages, thereby increasing in vivo stability, and it enhances immune-stimulating properties. A CpG-ODN can exhibit antitumor activity on its own, so compound 4, lacking the MUC1 glycopeptide, was also prepared as a control. Furthermore, compounds 1 and 2 were synthesized to examine possible differences in activity between CpG- and Pam3CysK4-containing vaccine candidates.[9b]
Compound 3 was prepared by conjugation of glycopep-tide 7, derivatized at its N terminus with an electrophilic bromoacetyl moiety, with 3′-SH-modified CpG (5). Glycopeptide 7 was synthesized by a linear SPPS protocol with a Rink amide AM resin, Fmoc-protected amino acids, Fmoc-Thr-(3,4,6-tri-O-acetyl-α-D-GalNAc), and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetra-methyluronium hexafluorophosphate (HBTU) and N-hydroxy-benzotriazole (HOBt) as coupling agent. After the last amino acid coupling step, the N-terminal Fmoc group was removed with piperidine in DMF, and this was followed by saponification of the acetyl esters of the sugar residue (hydrazine in MeOH). Next, an N-terminal bromoacetyl moiety was installed by treatment with bromoacetic acid N-hydroxysuccinimide ester. The fully assembled compound was treated with tri-fluoroacetic acid (TFA), triisopropylsilane (TIS), and H2O to remove the side chain protecting groups and to release the compound from the resin. Homogenous glycopeptide was obtained after purification by C18 column chromatography.
CpG modified with a C-3′ thiol (compound 6) was obtained by treatment of commercially available CpG-S-S-propyl alcohol (5) with Cleland’s Reductacryl reagent,[18] an immobilized form of DTT on polyacrylamide resin that has the advantages that it can easily be removed by filtration and its use does not require compound purification prior to the conjugation step. Next, bromoacetyl-modified glycopeptide 7 was exposed to 6, and, after a reaction time of 48 h, MALDI-TOF mass spectrometry indicated completion of the reaction. The compound was purified by reversed-phase HPLC with a C18 column, and its structural identify was confirmed by mass spectrometry and UV spectroscopy. Reference compound 4 was prepared by a similar methodology starting from peptide 8 and CpG derivative 6. The Pam3CysSK4-containing compounds 1 and 2 were prepared by previously reported methodologies.[9]
Glycolipopeptide 1 and lipopeptide 2 were incorporated into phospholipid-based small unilamellar vesicles (SUVs) by hydration of thin films of the synthetic compounds, egg phos-phatidylcholine, phosphatidylglycerol, and cholesterol in HEPES buffer (10 mμ, pH 6.5) containing NaCl (145 mμ), followed by extrusion through a 100 nm Nuclepore polycarbonate membrane. Compounds 3 and 4 were mixed with incomplete Freund’s adjuvant (IFA) to create emulsions.
Immunizations and immunology
Groups of MUC1.Tg mice (C57BL/6; H-2b) that express human MUC1[19] were immunized three times intradermally at the base of the tail at biweekly intervals with compounds 1–4.[20] After 35 days, the mice were challenged with 1 0 106 MT.MUC1 mammary tumor cells (positive for MUC1 and Tn), followed by one more boost after one week. One week after the last immunization, the mice were sacrificed, and the efficacies of the candidate vaccines were determined by tumor weight. Furthermore, the robustness of humoral immune responses was assessed by titers of MUC1-specific antibodies and the ability of the antisera to lyse MUC1-bearing tumor cells. In addition, cellular immune responses were evaluated by determining the lytic activity of CD8+ T-cells.
As expected, immunization with tripartite vaccine candidate 1 led to a significant reduction in tumor burden relative to the control compound 2, which does not contain a MUC1 glyco-peptide epitope (Figure 1). Surprisingly, the CpG-containing multicomponent vaccine candidate 3 did not exhibit a significant improvement in anticancer properties relative to control immunization with 4.
Figure 1.
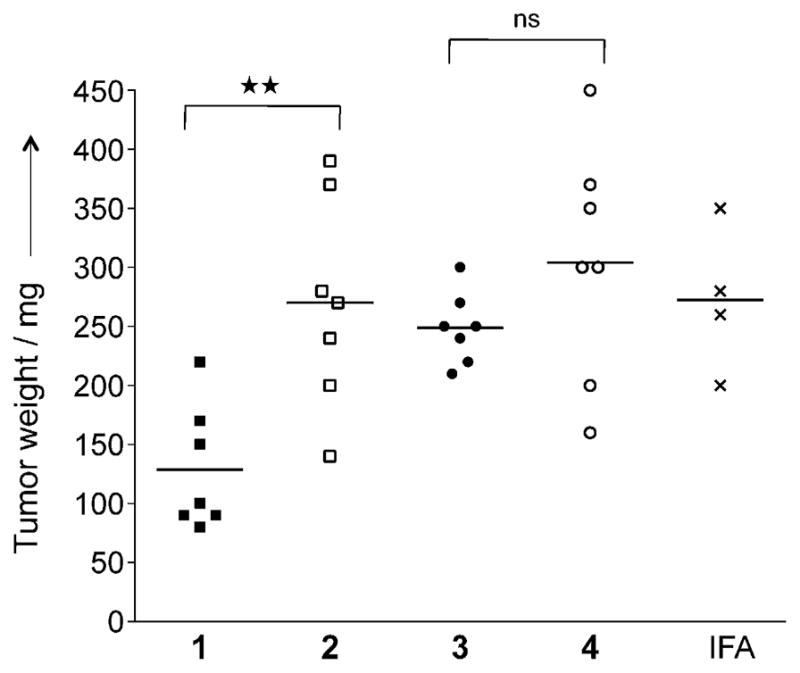
Glycosylated multicomponent vaccine reduces MT.MUC1 tumor burden in MUC1.Tg mice. MUC1.Tg mice were immunized with IFA as control, 1 and 2 in liposomes, or 3 and 4 together with IFA. Three biweekly immunizations were given prior to a tumor challenge with MUC1-expressing MT tumor cells (1 0 106 cells), followed by one boost one week after. The animals were sacrificed seven days after the last injection, and tumor wet weight was determined. Each data point represents an individual mouse, and the horizontal lines each indicate the mean for the group of mice. Asterisks indicate statistically significant differences (** P <0.01), and ns indicates no significant difference.
Anti-MUC1 antibody titers were determined by coating mi-crotiter plates with the MUC1-derived glycopeptide CTSAPDT-(αGalNAc)RPAP conjugated to maleimide-modified BSA. Compound 1 had elicited robust IgG antibody responses, and subtyping of the antibodies indicated a mixed Th1/Th2 response (Table 1 and Figure S1 in the Supporting Information). Compound 3 had elicited substantially lower titers of antibodies, thus highlighting the importance of the nature of the inbuilt adjuvant for robust antigenic responses. As expected, the controls 2 and 4, containing no MUC1-derived epitope, did not elicit substantial anti-MUC1 antibody responses.
Table 1.
ELISA anti-MUC1 and anti-Thelper antibody titers[a] after three and four immunizations with various preparations.
| Imm[b] | IgG total MUC1 Imm 3 | IgG total MUC1 EP | IgG1 MUC1 EP | IgG2a MUC1 EP | IgG2b MUC1 EP | IgG3 MUC1 EP | IgM MUC1 EP | IgG total Thelper EP |
|---|---|---|---|---|---|---|---|---|
| 1 | 28 500 | 59 100 | 17 800 | 7000 | 26 800 | 11 300 | 100 | 300 |
| 2 | 700 | 1000 | 400 | 0 | 600 | 200 | 50 | 1000 |
| 3 | 7800 | 10 600 | 9000 | 1000 | 6800 | 5300 | 50 | 50 |
| 4 | 0 | 0 | 0 | 0 | 0 | 0 | 50 | 50 |
| IFA | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 50 |
Anti-MUC1 and anti-Thelper antibody titers are presented as median values for groups of mice. ELISA plates were coated with BSA-MI-CTSAPDT-(αGalNAc)RPAP conjugate for anti-MUC1 antibody titers or NeutrAvidin-biotin-Thelper for anti-Thelper antibody titers. Titers were determined by linear regression analysis, with plotting of dilution versus absorbance. Titers are defined as the highest dilutions yielding optical densities of 0.1 or greater relative to normal control mouse sera. [b] For immunizations with 1 and 2 liposomal preparations were employed, whereas 3 and 4 were given together with IFA. MT.MUC1 tumors were induced between the third and the fourth immunization. EP indicates endpoint serum samples.
ADCC was examined by labeling MUC1-expressing mammary cancer cells with 51Cr, followed by the addition of antisera and cytotoxic effector cells (NK cells) and measurement of released 51Cr. As can be seen in Figure 2A, the antiserum obtained by immunization with 1 was able to increase cancer cell lysis significantly relative to the control compound 2, whereas this was not the case for CpG-containing compound 3 and control 4.
Figure 2.
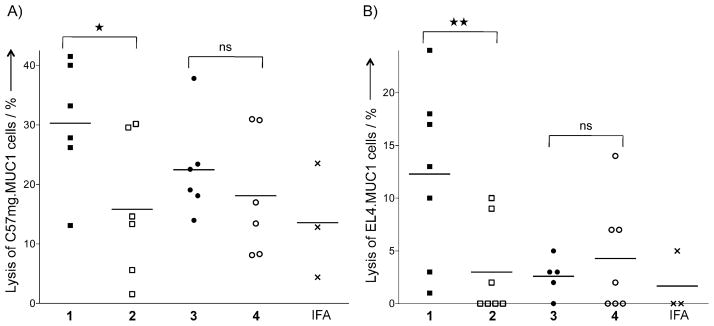
The tripartite Pam3CysSK4-containing vaccine generated both cellular and humoral responses. A) Induction of ADCC. Tumor cells (C57mg.MUC1) were labeled with chromium for 2 h and then incubated with serum (1:25 diluted) obtained from mice immunized with IFA, 1 and 2 in liposomes, or 3 and 4 together with IFA for 30 min at 37 °C. The tumor cells were then incubated with effector cells (NK cells, KY-1 clone) at an effector/target ratio of 50:1 for 4 h. Spontaneous release was 14 % of complete release. B) Induction of cytolytic T-cells in MUC1.Tg mice. CD62Llow T-cells—isolated from lymph nodes of mice immunized with IFA, 1 and 2 in liposomes, or 3 and 4 together with IFA and cultured for two weeks with DCs pulsed with glycopeptide SAPDT-(αGalNAc)RPAP for 1–4 or unpulsed for IFA—were subjected to a 51Cr-release assay with EL4.MUC1 cells as targets. Spontaneous lysis was less than 15 % of total lysis. Each data point represents an individual mouse, and the horizontal lines each indicate the mean for the group of mice. Asterisks indicate statistically significant difference (** P <0.01, *P <0.05), and ns indicates no significant difference.
The lytic activity of the isolated CD62Llow T-cells was examined by a 51Cr-release assay with EL4.MUC1 cells as the target. As can be seen in Figure 2B, CTLs activated by compound 1 exhibited significantly greater cytotoxicity than control 2. T-cells isolated from mice immunized with compound 3 exhibited low lytic activity, thus further demonstrating the importance of the nature of the inbuilt adjuvant for immunological responses.
There is emerging evidence that successful cancer vaccine development benefits from a multimodal treatment that activates several arms of the immune system at once.[21] Previously, we demonstrated that a tripartite vaccine composed of a glycopeptide derived from MUC1, a promiscuous Thelper peptide, and a TLR2 agonist[9] can elicit IgG antibodies that can lyse MUC1-expressing cancer cells and stimulate the cytotoxicity of T-lymphocytes, thereby reversing tolerance and generating a therapeutic response in a mouse model of mammary cancer.
CpG-ODN has received considerable interest as an immunoadjuvant,[14] and it has been shown that it can enhance immune responses of several MUC1-based experimental cancer vaccines.[15] We therefore felt compelled to investigate whether replacement of Pam3CysSK4 in vaccine candidate 1 by a CpG-ODN might further improve its properties. Surprisingly, it was found that compound 3, which is modified with CpG-ODN 1826, elicited weaker antigenic and cellular immune responses than compound 1, which contains Pam3CysSK4 as the inbuilt adjuvant, and it did not significantly reduce the tumor burden over control in a mouse model for mammary cancer.
The studies presented here demonstrate, for the first time, that the nature of an inbuilt adjuvant of a tripartite vaccine significantly impacts the quality of immune responses elicited against a tumor-associated glycopeptide. Specifically, it was found that Pam3CysSK4 is a superior adjuvant to CpG. A recent study demonstrated that TLR1/2 agonists have a unique ability to reduce the suppressive function of Foxp3+ regulatory T-cells (Tregs) and to enhance the cytotoxicity of tumor-specific CTLs in vitro and in vivo and have more favorable antitumor effects than other TLR agonists.[22] These adjuvant properties are likely responsible for the superior properties of vaccine candidate 1, which contains an inbuilt TLR1/2 agonist.
Although considerable efforts have been directed to the design of vaccines composed of tumor- or viral-derived proteins conjugated to a TLR9 ligand,[16] much less information is available in the case of similar vaccines containing antigenic peptide or glycopeptide components. A model vaccine composed of CpG conjugated to specific T-cell epitopes derived from immune-dominant antigens of HIV, which in turn were linked to a Thelper epitope, was able to elicit potent immune activity against vaccinia virus expressing the full-length HIV antigen.[23] In another study, it was shown that a synthetic long peptide conjugated to CpG greatly enhanced in vitro antigen presentation relative to a mixture of free TLR ligand and peptide.[24] This study, however, did not evaluate immunological properties of the conjugates in animal models. The underperformance of vaccine candidate 3 might be due to the fact that MUC1 is a self-antigen to which the mice are tolerant.
Conclusions
Fully synthetic multicomponent vaccines are receiving considerable interest, and the studies described here highlight the fact that appropriate selection of the inbuilt adjuvant is critical for eliciting optimal ADCC-mediating antibody and CTL responses, which in turn leads to superior therapeutic antitumor effects.
Experimental Section
Cell culture
Cell lines used in these studies include MT mammary gland tumor cells derived from MUC1.Tg mice crossed with MMTV-PyV MT mice[25] and transfected with full-length human MUC1, C57mg.MUC1 mammary gland tumor cells,[26] NK cells (KY-1 clone), and EL4.MUC1 cells. Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with FCS (5 %), penicillin (100 U ml−1), streptomycin (0.1 μg mL−1), L-glutamax (2 mμ), and G418 (150 μg mL−1). All cells are derived originally from C57BL/6 mice.
Immunizations and tumor palpation
MUC1.Tg mice (8 to 12 weeks old, C57BL/6; H-2b) that express human MUC1 at physiological level were immunized three times at biweekly intervals at the base of the tail intradermally with liposomal preparations of the three-component vaccine construct (containing 3 μg of carbohydrate) and its control lacking the tumor-associated MUC1 epitope or with nonliposomal three-component vaccine construct (3 μg of carbohydrate) and its control emulsified in an equal volume of IFA (100 μL total volume injected). After 35 days, the mice were challenged with transfected MMT mammary tumor cells (1 0 106 cells), which express MUC1 and Tn. On day 42, one more immunization was given. Palpable tumors were measured with calipers, and tumor weight was calculated according to the formula: grams = [(length) 0 (width)2]/2, where length and width are measured in centimeters. On day 49, the mice were sacrificed, the tumors were surgically removed, and tumor weight was determined. These animal studies have been approved by the Institutional Animal Care and Use Committee (IACUC) of the Mayo Clinic.
Serologic assays
Anti-MUC1 IgG, IgG1, IgG2a, IgG2b, IgG3, and IgM antibody titers were determined by enzyme-linked immunosorbent assay (ELISA) as described previously.[27] Briefly, ELISA plates (Thermo Electron Corp.) were coated with a conjugate of the MUC1 glycopeptide conjugated to BSA through a maleimide linker [BSA-MI-CTSAPDT(αGalNAc)RPAP]. Serial dilutions of the sera were allowed to bind to immobilized MUC1. Detection was achieved by the addition of alkaline phosphatase-conjugated anti-mouse antibodies and p-nitrophenyl phosphate (Sigma). To determine anti-body titers against the Thelper (polio) epitope, Reacti-bind NeutrAvidin coated and preblocked plates (Pierce) were incubated with biotin-labeled Thelper (10 μgmL−1, 100 μL per well) for 2 h. Next, serial dilutions of the sera were allowed to bind to immobilized Thelper epitope. Detection was achieved as described above. The antibody titer was defined as the highest dilution yielding an optical density of 0.1 or greater over that of normal control mouse sera.
Determination of ADCC
Tumor cells (C57mg.MUC1) were labeled with 51Cr (100 μCi) for 2 h at 37 °C, washed, and incubated with serum (1 in 25 dilutions) obtained from the vaccinated mice for 30 min at 37 °C. NK cells (KY-1 clone), which have high expression of CD16 receptor, were used as effectors. These cells were stimulated with IL-2 (200 units mL−1) for 24 h prior to assay. Effector cells were seeded with the antibody-labeled tumor cells in 96-well culture plates (Costar high binding plates) at an effector/target cell ratio of 50:1 for 4 h. Radioactive 51Cr release was determined with a Topcount Microscintillation Counter (Packard Biosciences). Spontaneous and maximum release of 51Cr was determined. The percentage of specific release was calculated according to the formula: (release–spontaneous release/maximal release–spontaneous release)0 100.
51Chromium (Cr) release assay
Cytolytic activity was determined by a standard 51Cr release method with CD62Llow cells from tumor-draining lymph nodes, which were cultured for two weeks with pulsed or unpulsed DCs, as effector cells. Target cells (EL4.MUC1) were loaded with 51Cr (Amersham Biosciences, 100 μCi per 106 target cells) for 2 h before incubation with effectors. Radioactive 51 Cr release was determined as described above.
Statistical analysis
Multiple comparisons were performed by use of one-way ANOVA and Bonferroni’s multiple comparison test. Differences were considered significant when P <0.05.
Supplementary Material
Acknowledgments
We thank the Mayo Clinic Natalie Schafer Animal Care Attendants for excellent animal care and Dr. Pinku Mukherjee for providing MUC1 transfected MT cancer cells. This research was supported by the National Cancer Institute of the US National Institutes of Health grant R01 CA88986 (to G.J.B.), the Mayo Breast Specialized Programs of Research Excellence (SPORE) grant P50 CA116201 (to S.J.G.) and the Mayo Pancreas SPORE grant P50 CA102701 (to P.A.C. and S.J.G.).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201402077.
Contributor Information
Prof. Dr. Sandra J. Gendler, Email: gendler.sandra@mayo.edu.
Prof. Dr. Geert-Jan Boons, Email: gjboons@ccrc.uga.edu.
References
- 1.a) Hanisch FG, Muller S. Glycobiology. 2000;10:439–449. doi: 10.1093/glycob/10.5.439. [DOI] [PubMed] [Google Scholar]; b) Tarp MA, Clausen H. Biochim Biophys Acta Gen Subj. 2008;1780:546–563. doi: 10.1016/j.bbagen.2007.09.010. [DOI] [PubMed] [Google Scholar]; c) Beatson RE, Taylor-Papadimitriou J, Burchell JM. Immunotherapy. 2010;2:305–327. doi: 10.2217/imt.10.17. [DOI] [PubMed] [Google Scholar]
- 2.Cazet A, Julien S, Bobowski M, Burchell J, Delannoy P. Breast Cancer Res. 2010;12:204. doi: 10.1186/bcr2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ju T, Cummings RD. Proc Natl Acad Sci USA. 2002;99:16613–16618. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) von Mensdorff-Pouilly S, Verstraeten AA, Kenemans P, Snijdewint FG, Kok A, van Kamp GJ, Paul MA, van Diest PJ, Meijer S, Hilgers J. J Clin Oncol. 2000;18:574–583. doi: 10.1200/JCO.2000.18.3.574. [DOI] [PubMed] [Google Scholar]; b) Blixt O, Bueti D, Burford B, Allen D, Julien S, Hollingsworth M, Gammerman A, Fentiman I, Taylor-Papadimitriou J, Burchell JM. Breast Cancer Res. 2011;13:R25. doi: 10.1186/bcr2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doménech N, Henderson RA, Finn OJ. J Immunol. 1995;155:4766–4774. [PubMed] [Google Scholar]
- 6.a) Haurum JS, Hoier IB, Arsequell G, Neisig A, Valencia G, Zeuthen J, Neefjes J, Elliott T. J Exp Med. 1999;190:145–150. doi: 10.1084/jem.190.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vlad AM, Muller S, Cudic M, Paulsen H, Otvos L, Jr, Hanisch FG, Finn OJ. J Exp Med. 2002;196:1435–1446. doi: 10.1084/jem.20020493. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Stepensky D, Tzehoval E, Vadai E, Eisenbach L. Clin Exp Immunol. 2006;143:139–149. doi: 10.1111/j.1365-2249.2005.02965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ninkovic T, Hanisch FG. J Immunol. 2007;179:2380–2388. doi: 10.4049/jimmunol.179.4.2380. [DOI] [PubMed] [Google Scholar]
- 7.a) Brossart P, Heinrich KS, Stuhler G, Behnke L, Reichardt VL, Stevanovic S, Muhm A, Rammensee HG, Kanz L, Brugger W. Blood. 1999;93:4309–4317. [PubMed] [Google Scholar]; b) Barnea E, Beer I, Patoka R, Ziv T, Kessler O, Tzehoval E, Eisenbach L, Zavazava N, Admon A. Eur J Immunol. 2002;32:213–222. doi: 10.1002/1521-4141(200201)32:1<213::AID-IMMU213>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; c) Tsang KY, Palena C, Gulley J, Arlen P, Schlom J. Clin Cancer Res. 2004;10:2139–2149. doi: 10.1158/1078-0432.ccr-1011-03. [DOI] [PubMed] [Google Scholar]; d) Ninkovic T, Kinarsky L, Engelmann K, Pisarev V, Sherman S, Finn OJ, Hanisch FG. Mol Immunol. 2009;47:131–140. doi: 10.1016/j.molimm.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, Hilf N, Schoor O, Fritsche J, Mahr A, Maurer D, Vass V, Trautwein C, Lewandrowski P, Flohr C, Pohla H, et al. Nat Med. 2012;18:1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- 9.a) Ingale S, Wolfert MA, Gaekwad J, Buskas T, Boons GJ. Nat Chem Biol. 2007;3:663–667. doi: 10.1038/nchembio.2007.25. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lakshminarayanan V, Thompson P, Wolfert MA, Buskas T, Bradley JM, Pathangey LB, Madsen CS, Cohen PA, Gendler SJ, Boons GJ. Proc Natl Acad Sci USA. 2012;109:261–266. doi: 10.1073/pnas.1115166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leclerc C, Deriaud E, Mimic V, van der Werf S. J Virol. 1991;65:711–718. doi: 10.1128/jvi.65.2.711-718.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Toyokuni T, Dean B, Cai SP, Boivin D, Hakomori S, Singhal AK. J Am Chem Soc. 1994;116:395–396. [Google Scholar]; b) Vichier-Guerre S, Lo-Man R, BenMohamed L, Deriaud E, Kovats S, Leclerc C, Bay S. J Pept Res. 2003;62:117–124. doi: 10.1034/j.1399-3011.2003.00074.x. [DOI] [PubMed] [Google Scholar]; c) Dziadek S, Hobel A, Schmitt E, Kunz H. Angew Chem Int Ed. 2005;44:7630–7635. doi: 10.1002/anie.200501594. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2005;117:7803–7808. [Google Scholar]; d) Renaudet O, Dasgupta G, Bettahi I, Shi A, Nesburn AB, Dumy P, BenMohamed L. PLoS One. 2010;5:e11216. doi: 10.1371/journal.pone.0011216. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Abdel-Aal AB, El-Naggar D, Zaman M, Batzloff M, Toth I. J Med Chem. 2012;55:6968–6974. doi: 10.1021/jm300822g. [DOI] [PubMed] [Google Scholar]; f) Huang ZH, Shi L, Ma JW, Sun ZY, Cai H, Chen YX, Zhao YF, Li YM. J Am Chem Soc. 2012;134:8730–8733. doi: 10.1021/ja211725s. [DOI] [PubMed] [Google Scholar]; g) Wang Q, Zhou Z, Tang S, Guo Z. ACS Chem Biol. 2012;7:235–240. doi: 10.1021/cb200358r. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Wilkinson BL, Day S, Chapman R, Perrier S, Apostolopoulos V, Payne RJ. Chem Eur J. 2012;18:16540–16548. doi: 10.1002/chem.201202629. [DOI] [PubMed] [Google Scholar]; i) Gaidzik N, Westerlind U, Kunz H. Chem Soc Rev. 2013;42:4421–4442. doi: 10.1039/c3cs35470a. [DOI] [PubMed] [Google Scholar]; j) Sarkar S, Salyer AC, Wall KA, Sucheck SJ. Bioconjugate Chem. 2013;24:363–375. doi: 10.1021/bc300422a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lahiri A, Das P, Chakravortty D. Vaccine. 2008;26:6777–6783. doi: 10.1016/j.vaccine.2008.09.045. [DOI] [PubMed] [Google Scholar]
- 13.Dabbagh K, Lewis DB. Curr Opin Infect Dis. 2003;16:199–204. doi: 10.1097/00001432-200306000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Krieg AM. Nat Rev Drug Discovery. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 15.a) Mukherjee P, Pathangey LB, Bradley JB, Tinder TL, Basu GD, Akporiaye ET, Gendler SJ. Vaccine. 2007;25:1607–1618. doi: 10.1016/j.vaccine.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ding C, Wang L, Marroquin J, Yan J. Blood. 2008;112:2817–2825. doi: 10.1182/blood-2008-05-157396. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Moreno M, Mol BM, von Mensdorff-Pouilly S, Verheijen RH, von Blomberg BM, van den Eertwegh AJ, Scheper RJ, Bontkes HJ. Cancer Lett. 2008;272:70–76. doi: 10.1016/j.canlet.2008.06.028. [DOI] [PubMed] [Google Scholar]; d) Pinkhasov J, Alvarez ML, Pathangey LB, Tinder TL, Mason HS, Walmsley AM, Gendler SJ, Mukherjee P. Cancer Immunol Immunother. 2010;59:1801–1811. doi: 10.1007/s00262-010-0906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Schettini J, Kidiyoor A, Besmer DM, Tinder TL, Roy LD, Lustgarten J, Gendler SJ, Mukherjee P. Cancer Immunol Immunother. 2012;61:2055–2065. doi: 10.1007/s00262-012-1264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Datta SK, Cho HJ, Takabayashi K, Horner AA, Raz E. Immunol Rev. 2004;199:217–226. doi: 10.1111/j.0105-2896.2004.00149.x. [DOI] [PubMed] [Google Scholar]; b) Zom GG, Khan S, Filippov DV, Ossendorp F. Adv Immunol. 2012;114:177–201. doi: 10.1016/B978-0-12-396548-6.00007-X. [DOI] [PubMed] [Google Scholar]
- 17.Khan S, Bijker MS, Weterings JJ, Tanke HJ, Adema GJ, van Hall T, Drijfhout JW, Melief CJ, Overkleeft HS, van der Marel GA, Filippov DV, van der Burg SH, Ossendorp F. J Biol Chem. 2007;282:21145–21159. doi: 10.1074/jbc.M701705200. [DOI] [PubMed] [Google Scholar]
- 18.Cleland WW. Biochemistry. 1964;3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 19.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Cancer Res. 1998;58:315–321. [PubMed] [Google Scholar]
- 20.Although we had previously reported the synthesis and immunological evaluation of compounds 1 and 2, these derivatives were reesynthe-sized and immunologically examined side by side with compounds 3 and 4.
- 21.Morse MA, Whelan M. Curr Opin Mol Ther. 2010;12:11–13. [PubMed] [Google Scholar]
- 22.a) Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, Chu Y. J Immunol. 2011;186:1963–1969. doi: 10.4049/jimmunol.1002320. [DOI] [PubMed] [Google Scholar]; b) Amiset L, Fend L, Gatard-Scheikl T, Rittner K, Duong V, Rooke R, Muller S, Bonnefoy JY, Preville X, Haegel H. Oncoimmunology. 2012;1:1271–1280. doi: 10.4161/onci.21479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daftarian P, Sharan R, Haq W, Ali S, Longmate J, Termini J, Diamond DJ. Vaccine. 2005;23:3453–3468. doi: 10.1016/j.vaccine.2005.01.093. [DOI] [PubMed] [Google Scholar]
- 24.van Duikeren S, Fransen MF, Redeker A, Wieles B, Platenburg G, Krebber WJ, Ossendorp F, Melief CJ, Arens R. J Immunol. 2012;189:3397–3403. doi: 10.4049/jimmunol.1201540. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee P, Madsen CS, Ginardi AR, Tinder TL, Jacobs F, Parker J, Agrawal B, Longenecker BM, Gendler SJ. J Immunother. 2003;26:47–62. doi: 10.1097/00002371-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee P, Ginardi AR, Tinder TL, Sterner CJ, Gendler SJ. Clin Cancer Res. 2001;7:848s–855s. [PubMed] [Google Scholar]
- 27.Buskas T, Li YH, Boons GJ. Chem Eur J. 2004;10:3517–3524. doi: 10.1002/chem.200400074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.