

Abstract
This article introduces the concept of pulmonary vascular stiffness, discusses its increasingly recognized importance as a diagnostic marker in the evaluation of pulmonary vascular disease, and describes methods to measure and model it clinically, experimentally, and computationally. It begins with a description of systems-level methods to evaluate pulmonary vascular compliance and recent clinical efforts in applying such techniques to better predict patient outcomes in pulmonary arterial hypertension. It then progresses from the systems-level to the local level, discusses proposed methods by which upstream pulmonary vessels increase in stiffness, introduces concepts around vascular mechanics, and concludes by describing recent work incorporating advanced numerical methods to more thoroughly evaluate changes in local mechanical properties of pulmonary arteries.
Introduction
The arteries of the lung may be broadly categorized into three groups: the proximal elastic arteries, which provide a Windkessel (buffer) function that reduces right heart work and helps maintain forward flow during diastole; the arterioles, which regulate and direct local flow due to changing oxygen demands of the body or in response to environmental conditions such as hypoxia; and the capillaries, where the primary function of the lung occurs, gas exchange. These groups may also be described by their primary hemodynamic functions: compliance (the proximal elastic arteries), and resistance (arterioles and capillaries).
Earlier study of pulmonary hypertension (PH) has focused heavily on pulmonary vascular resistance (PVR), which quantifies mean hemodynamic parameters (mean flow and mean pressure). Indeed, the pulmonary circulation chapter in the 1983 edition of the APS Handbook of Physiology discussed only resistance and other mean measures of pulmonary vascular function (63). This focus was mostly reasonable; clinically, the clearest hemodynamic manifestation of PH is an increase in mean pulmonary artery pressure (mPAP), the primary result of an increase in PVR. Since that article, diagnostic and treatment paradigms focused on PVR and its reactivity to vasodilators have markedly decreased morbidity and mortality for the disease (4, 6, 31, 142).
However, the pulmonary vascular circuit is composed not only of the distal resistance vessels but also the proximal vessels as well and it is becoming increasingly evident that these proximal vessels are in fact much more than simple conduits that connect the right ventricle to the lungs. Although changes in pulmonary vascular stiffness of the proximal elastic arteries had also been recognized at the time of the previous Handbook as intrinsic to the disease state (36, 59) and as a determinant of flow propagation and pulsatility, the contribution of vascular stiffening to the derangement of vascular function was not well understood and had been mostly neglected in both the basic science and clinical settings. The primary reasons for this were the lack of direct clinical data documenting the relationship between clinical outcomes and PVS diagnostics, and the difficulties in measuring PVS within the routine evaluation of PH. This is beginning to change. Recent clinical work has highlighted the importance of PVS in the progression of PH (55, 84, 110), mechanical studies have begun to elucidate the gross vascular changes responsible for stiffening, and existing and novel studies of cellular mechanotransduction suggest PVS may play a role in pulmonary disease pathogenesis (104,105). Many of these concepts have been well-adopted by investigators studying the systemic vascular circuit in health and disease. Here we propose that proximal vascular stiffening in the pulmonary vessels constitutes an equally important aspect of evaluating pulmonary vascular disease.
The Right Ventricle and Its Circulation
The right heart moves deoxygenated blood returning from the body through the pulmonary circuit. Long thought of as the “less important” side of the heart (143), its ventricle requires one-fifth of the energy of the left ventricle to move the same amount of blood through the much lower resistance offered by the lung vasculature (65). Also in contrast to the left heart, the right side is less capable of acutely or chronically increasing its volume and/or pressure output.
In the simplest functional sense, the heart is a pump that uses energy (oxygen and glucose) to mechanically increase the pressure of its working fluid (blood). This naturally leads to the idea of pump efficiency, which can be esoterically described as actual work divided by some theoretically maximum possible work, or more practically as a cost function of actual work per energy input (171). This definition is not used in current clinical evaluation of right heart function, where the focus is on right ventricular afterload, as has been historically quantified by PVR.
Pulmonary hypertension
Elevations in PVR have long been considered to be a defining attribute of PH (4, 6), given that increases in PVR due to distal vasoconstriction, remodeling, proliferation, or other effects (183,184) are primarily responsible for increased mPAP. These pathologies all change distal vascular diameter and/or overall flow area of the distal pulmonary circulation and thus strongly affect resistance; these will be reviewed briefly below.
Persistent distal vasoconstriction has been long noted as a feature of some patients with idiopathic pulmonary arterial hypertension (IPAH) (186), and likely results from changes in vasoactive mediators due to endothelial dysfunction (21). Distal vascular reactivity to vasodilators such as inhaled nitric oxide, determined by acute reduction in PVR, remains an important part of clinical diagnosis (5), although this constriction becomes less important in the more advanced disease state (139). Hypoxic animal models of PH all display persistent distal vasoconstriction (113, 124, 141, 165) that can be acutely reduced by vasodilators (44).
Structural changes to the distal vasculature, collectively known as vascular remodeling, involve the thickening of all three layers of the vessel wall. At the lumen, endothelial and myofibroblast cell proliferation is associated with concentric or obliterative plexiform lesions (30, 68, 170), again a likely result of endothelial dysfunction (21). A “distal extension of smooth muscle” or neomuscularization also results, due to precursor cell differentiation into vascular smooth muscle cells (SMCs) in the partially muscularized and non-muscular distal vasculature (116). Finally, medial and adventitial thickening occurs due to increased accumulation of extracellular matrix (ECM) proteins, as well as increased accumulation of SMCs (media) and fibroblasts and myofibroblasts (adventitia) (34, 91, 140, 164). Together, these structural alterations act to occlude the vessel lumen and to collectively reduce the overall vascular bed area (known as vessel rarefaction or pruning) (72, 73, 91, 117, 132), thus increasing PVR.
The Biology of Pulmonary Vascular Stiffness
Stiffening is an increasingly important component of systemic vascular disease in humans and is associated with significant morbidity and mortality. While the natural history of pulmonary vascular stiffening in a population due to pulmonary arterial hypertension (PAH) is unknown, there have been many studies of vascular stiffness in the setting of systemic vascular disease. Stiffening of the great systemic arteries appears to be an age-associated change in older adults resulting in increases in ECM, specifically elastin and collagen, and is related to systemic hypertension, diabetes, atherosclerosis, and chronic renal failure (33, 102, 103, 106). Aortic stiffening has also been shown to be predictive of cardiovascular events (37, 101, 122, 137, 160, 162). Interestingly, it is yet unknown whether stiffening is a cause or effect of these diseases (58); however, stiffening contributes to increased left ventricular workload and likely contributes to heart failure.
As will be shown below, it is established that the stiffness of the great pulmonary arteries is elevated in several forms of PH, and there is increasing clinical awareness that this change affects disease course. Elevated PVS essentially reduces the Windkessel, or buffering, effect of the proximal arteries. This in turn adds further to right heart afterload—beyond that already caused by pathological PVR—and increases both pressure and flow pulsatility, which may have implications to hemodynamic mechanotransduction events. At least three mechanisms have been cited for increasing PVS: acute high-strain-induced passive stiffening; chronic passive stiffening due to vascular remodeling; and acute active stiffening due to SMC response.
Elastic tissues
The ECM of connective tissues consists of four major, mechanically relevant, passive constituent materials: collagen, proteoglycans, elastin, and glycoproteins. These materials act in concert to provide connective tissues with unique material properties required for their homeostatic function and are the subject of significant research on connective tissue pathology. Material properties such as tensile strength, compliance, friction coefficient, viscosity, resistance to wear and fracture are all largely determined by ECM constituent materials and have either a direct correlation with, or causality to, numerous pathologic conditions.
Conduit pulmonary arteries are composed of tissues whose homeostatic function depends on an elastic response to applied loads. As such, the behavior of these tissues is largely determined by the mechanics of the elastin and collagen present within arterial ECM. These structural proteins produce a common stress-strain response of elastic arterial tissues to applied load that can be categorized by a low-strain, linear, elastin-dominant region, a highly nonlinear transition region, and a second high-strain, linear, collagen-dominant region (Fig. 1). Therefore, in conduit pulmonary arterial (PA) tissues, elastin is the material primarily responsible for the ability to repeatedly deform under load and to return to the original shape with minimal dissipative (viscous) losses, while collagen provides strength and limits the degree of deformation of elastic tissue thereby preventing damage or rupture. It is useful to discuss these important structural proteins briefly.
Figure 1.

Typical pressure-diameter curve for elastic tissues.
Arterial elastin
Elastin is an insoluble protein largely found in connective tissues in which the ability to stretch is a functional requirement. The elastic tissue found within the lamellae of the arterial tunica media is composed of a high-density network of elastin fibers. Elastin fibers are the morphologic subunit, consisting of cross-linked elastin and a microfibrillar core, which are bound together to form elastic tissues. These elastic tissues imbue the arteries with the ability to reversibly stretch (elastic behavior), meaning that under load the tissue will deform and upon release of the applied load the tissue will return to its original shape.
Arterial elastic fibers are composed of tropoelasin deposited within a microfibrillar bundle. Tropoelastin is the soluble precursor to the insoluble elastin and is believed to be the moiety that is produced within the cell for transportation to, and incorporation into, an elastic fiber under construction. Elastic fiber construction begins with the deposition of microfibrils by fibroblastic cells. These microfibrils direct elastin fiber growth and may help align tropoelastin molecules in the orientation needed for cross-link formation (69, 145). Tropoelastin is deposited within the microfibrillar bundles and the lysyl oxidase-derived crosslinks, desmosine, and isodesmosine act to bind the tropoelastin molecules into a stable insoluble elastin core (2, 93, 209). These individual elastic fibers are arranged in a network and bound together to form fenestrated elastic sheets that surround the artery in concentric layers and work to uniformly distribute the tensile stress to which the artery is subjected (153). While the individual elastic fibers do not have a linear stress-strain relationship, network effects allow the resulting elastin tissue to obey Hooke’s law up to large strains (209). The resulting fenestrated elastic sheets, which are bound to non-elastin ECM components and SMCs through glycoprotein and integrin bonds, imbue the artery with a Hookean load-carrying elastin component with near perfect elastic behavior (1).
Amino acid analysis indicates that tropoelastin has a repeating domain structure, alternating between highly non-polar and cross-linking domains. Nearly 80% of the tropoelastin molecule is composed of the non-polar, hydrophobic, amino acid residues of proline (P), valine (V), glycine (G), leucine (L), isoleucine (I), and alanine (A) (155). Nearly one-third of all tropoelastin residues are glycine, a particularly hydrophobic amino acid. Typical amino acid sequences found within mammalian tropoelastin include PGGV, PGVGV, PGVGVA, and GGLGV (145, 155). When no axial loads are present, these amino acid sequences are folded within the hydrophobic core of the tropoelastin molecule. Upon application of an axial load, the coiled structure of the tropoelastin molecule becomes deformed and these hydrophobic sequences are brought into direct contact with the surrounding solvent which results in an entropic change in the system and concomitant tangential reactive force generated within the elastin (145, 155).
Arterial collagen
Collagen is a heterogeneous class of molecules that share common chemical and physical properties but are often disparate in their mechanical or structural attributes. Collagenous proteins occur frequently in eukaryotic phyla, sometime constituting more than half of the total protein of adult organisms, and are a major constituent of ECM. The collagen molecule provides support and tensile strength to the skin, bone, tendon, elastic arteries, and cornea of vertebrates. In addition to its structural role, collagen has numerous developmental and physiological functions and is involved either directly or indirectly in cell attachment and differentiation, as a chemotactic agent for macrophages and fibroblasts, as an antigen in immunological processes, and as a causal agent in certain pathological conditions (69).
Arterial collagen works in tandem with elastin to define the mechanical response of vascular tissues to applied loads as detailed in Figure 1. As is typical in large elastic arteries, the initial force-deformation response follows a linear relationship in which the applied loads are largely carried by elastin. At some intermediate level of vessel wall deformation, collagen begins to carry a measureable amount of the applied load and the force-deformation response of the material becomes significantly nonlinear, as is evidenced by the transition region detailed in Figure 1. This transition region is the result of arterial collagen naturally occurring as a loosely connected network of collagen fiber bundles integrated within the arterial ECM and largely co-localized with elastin lamellar sheets (99, 158). In unloaded arterial tissues, the collagen fiber bundles exhibit a tortuous microstructure which has been described as “wavy” (35, 46, 203). The ability of collagen fiber bundles to exhibit very little mechanical stiffness over a low-deformation range while contributing significantly to mechanical stiffness at larger deformations is a result of both the microscopic collagen fiber structure as well as the molecular structure of the collagen protein itself.
Collagen molecules are rod-like structures, 3000 Å long and 15 Å in diameter (69). This rod-like shape of the collagen molecule is due to the triple helical region that comprises approximately 95% of the collagen molecule. Blood vessel walls are rich in both type I and type III collagen. Type I collagen contains two genetically distinct α-chains: one called α1 type I or α1(I) and the second called the α2(I) chain. Each type I molecule is composed of two α1(I) chains and one α2(I) chain, the complete molecule being abbreviated as [α1(I)]2 α2(I). In both α1(I) and α2(I), glycine accounts for one-third of the total amino acids, which reflects the presence of a significant number of G-X-Y triples present in the α-chains. Type III collagen molecules are composed of three molecules of a single type of α-chain termed α1(III). This collagen type shows an amino acid composition of high 4-hydroxyproline concentration and the presence of half-cystines, which participate in the intramolecular disulfide cross-links between adjacent α1(III) chains unique to this collagen type. Further, the high glycine concentration of type III collagen indicates that some of the G-X-Y triplets have and additional glycine at the X or Y position, which has been suggested as a possible locus of helix instability. Arterial collagen fiber bundles are composed largely of both type I and III collagen molecules that are bound together through strong intermolecular bonds to for a stiff and resilient collagen fiber capable of carrying large, tangential, mechanical loads while retaining the ability to bend or compress with little applied force.
Stiffening Modalities
The elastic pulmonary vessels may increase and decrease in stiffness acutely or chronically in response to loading. We broadly categorize these changes as due purely to (i) geometric factors, where passively attained gross changes in the vascular diameter alter the specific ECM proteins responsible for carrying wall stress (strain stiffening); (ii) SMC activity, in response to changing oxygen demand or supply or due to chronic increases in SMC number or activation status; and (iii) proximal vascular remodeling, where changes in wall thickness, ECM quantity and/or cross-linkage, SMC proliferation, and other factors combine to change the geometric and continuum stiffness of the vascular wall.
Strain stiffening
Studies that have compared the material properties of fresh arterial and purified elastin tissues suggest that at low strains collagen does not greatly contribute to artery mechanics (99, 144, 153). This, combined with the relatively large modulus of collagen in comparison to elastin, results in the characteristic bi-linear mechanical response of arterial tissues shown in Figure 1. Morphologic evidence suggests that the inability of collagen to carry significant load within the low-strain elastin-dominant region is due to the collagen fiber alignment in vivo (144, 153, 158). While medial arterial collagen is deposited with a roughly circumferential orientation, these collagen fibers demonstrate appreciable waviness and distributed alignment in the circumferential direction (35, 46, 203). Given that arterial collagen does not contribute significantly to tissue modulus during alignment or straightening, elastin is likely the dominant load-bearing ECM component for strains that fall below the onset of collagen engagement. Collagen engagement begins with the transition strain where the first collagen fibers become straightened and begin to actively carry load. As strain increases beyond the transition strain and through the transition region, more collagen fibers become progressively aligned and straightened in the direction of the applied load and are thereby recruited to carry additional tensile loads within the tissue sample. This causes the overall tissue modulus to increase sharply over the transition strain range, leading to the larger modulus seen in the collagen-dominant region. Therefore, while elastin provides the elastic behavior associated with arterial tissue, it is the collagen-based strain stiffening that prevents excessive deformation and possible damage at high pressures.
Myogenic response
In a recent series of studies, PA pressure-diameter (PD) measurements were performed directly in “active” and “passive” forms of acute PH in sheep obtained through phenylephrine injection and left PA occlusion, respectively (14, 15, 61, 62, 149). The results from this sheep model suggest that SMC activity strongly contributes to vascular mechanics in response to acute pressure increases, in that SMC activation maintained or slightly reduced the diameter at which vascular buffering function occurred (Fig. 2, left plot). Thus, SMC activation may prevent or reduce the impacts of collagen engagement (149) during acute pressure overload. In contrast, maintenance of PA diameter due to SMC effects was not evident in healthy (control) calves acutely exposed to hypoxia (217); rather, their main PAs significantly increased in diameter and stiffened greatly (presumably due to collagen engagement), which suggests a delay in SMC activation absent vasoconstrictor treatment (Fig. 2, right). Neonatal calf PAs exposed to 2 weeks of chronic hypoxia—which have been shown to chronically stiffen due to hypoxic exposure (99)—also did not appear to maintain their diameters due to SMC activation in acute hypoxia. However these chronically hypoxic animals displayed PD responses different from the control response in that their diameter increases were smaller and they did not appear to stiffen greatly at higher pressures (i.e., engage collagen) (217). Together, these animal models suggest that the SMC response in the proximal PAs does not occur in acute pressure overload.
Figure 2.
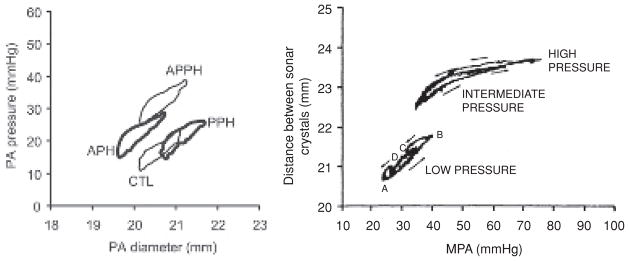
Typical conduit artery pressure-diameter (PD) response in acute and chronic models of hypertension. Left figure (149), diameter on abscissa: phenylephrine injection (active PH, APH), left PA occlusion (passive PH, PPH), or both (APPH) in an acute sheep model. Note that injection of a vasoconstrictor (phenylephrine) yields “active PH” along with reduced conduit artery diameter, while “passive PH” does not display conduit artery vasoconstriction. All the curves display roughly the same slope, suggesting that SMCs change the operating diameter but do not strongly affect the stiffness and that no strain-induced stiffening has occurred. Right figure (217), pressure on abscissa: (A) minimum diastolic pressure; (B) peak systolic pressure; (C) beginning of diastole; (D) later part of diastole, in a chronic hypobaric hypoxia calf model. In the calf, pressures are substantially higher which cause the diameter-pressure response to have a J-shape typically associated with collagen engagement. There also appears to be little to no acute SMC contribution to the mechanics of the proximal vasculature in the calf; such contributions would cause the PD curve to move downward and rightward from the low pressure condition without a significant change in slope. [Figures used with permission]
The sheep PD results under vasoconstrictor treatment (“active” PH) are consistent with results from a study of the carotid arteries of rats in systemic hypertension (53). This systemic study found that the SMCs attempted to maintain smaller (normal) carotid artery diameters under chronic pressure overload. Further, the carotid artery response was tracked over 8 weeks after initiation of systemic hypertension, during which time the SMC contribution to mechanics decreased while chronic changes to carotid mechanics occurred through vascular remodeling. Such a long-term study has not apparently been performed in the pulmonary circulation, although chronic stiffening due to remodeling has been reported in many studies, as noted subsequently.
Vascular remodeling
Histological and morphological studies have long indicated that ECM accumulation yields thickening of the proximal pulmonary vessels (34, 91, 140, 164) in chronic PH. The mechanical impacts of these changes have been quantified in both large and small animal models of chronic PH. Interestingly, histological evidence has already indicated that the remodeling response of the PAs in PH from large and small mammals is different (165), but no studies have established if these varied responses have different mechanical consequences.
Extracellular matrix remodeling
Both the structure and physiology of the ECM proteins, elastin, and collagen, present in the large conduit arteries, have been shown to change as a result of hypertension. A number of studies have examined various biological mechanisms involved in ECM remodeling as a result of PH (79, 165, 166, 202). However, much more research has been conducted regarding ECM remodeling of the conduit systemic vasculature including investigating the effect of inflammatory factors, upregulation of proliferative catalysts, and phenotypic changes in resident cells (2, 12, 13, 18, 86–89, 90). Other studies have shown increased collagen content and vascular tone in stiffer arteries of both the systemic (25, 44, 87) and pulmonary vasculature (40, 85, 95, 125, 126) and ECM degradation in atrophic remodeling of cuffed carotid arteries (7). In elastin specific studies, elastolytic activity has been shown to increase during PH (112), and inhibition of this pathway by the serine elastase inhibitor elafin mitigates hypoxia-induced elastin remodeling in PH (213). In recent work from the Shandas group coupling a novel orthotropic, hyperelastic, microstructural model of the PA wall to biomechanical studies of PAs from normotensive and hypertensive rats, we suggested that increased ECM structural protein crosslinking may be one mechanism by which these vessels stiffen (214, 215). Structurally, PH results in an increase in elastin content, but typically the ratio of collagen to elastin remains unchanged (2, 146). Physiologically, the three-dimensional (3D) architecture of the ECM proteins (elastin, collagen, proteoglycans and structural glycoproteins), which are deposited during fetal development, are optimized for efficient structure/function relationship within the systemic arteries (89, 90), and presumably that structure-function efficiency is maintained within the pulmonary circulation as well. However, the relative efficiencies between fetal ECM and that which is deposited during PH vascular remodeling have not been studied explicitly. In healthy arteries, the turnover rate for collagen and elastin is low, but vascular pathology upsets the regulatory pathway which maintains this balance, resulting in an increase in both the synthesis and destruction of the matrix proteins, suggesting an overall reconstruction of the arterial ECM in both systemic (2) and pulmonary (165) arteries. However, these adult-synthesized proteins have a 3D architecture, which may be functionally less optimal then the proteins deposited during fetal development.
Small mammals
Tozzi et al. (182) demonstrated a 2-fold increase in hydroxyproline content in rat extrapulmonary arterial PA segments after 10 days of hypoxic exposure that was not seen in the smaller vessels; additionally, vessel stiffening was correlated with collagen content during the development of PH. Increases in the initial elastic modulus of the main PA and left PA in the rat due to hypoxic PH have also been demonstrated by Drexler et al. (38,39) and Wright et al. (208); no difference in the right PA elastic modulus was found. More interesting, however, was their finding that the hypoxic arteries underwent strain stiffening, that is, collagen engagement, at lower strains compared to the control group. Histology suggested that increases in adventitial collagen were primarily responsible for these changes. In a monocrotaline rat model, Madden et al. (108) found time-dependent increases in the slope of the PD response of PAs excised from 7-, 14-, and 21-day-treated PH animals, which is consistent with vessel stiffening due to vascular remodeling. The Chesler group have shown that PAs excised from hypoxic mice show significant increases in incremental elastic modulus that correlate with increases in collagen thickness (area fraction times thickness) as well as combined elastin and collagen thicknesses measured histologically (26, 95). Very recently, this group has also quantified gene expression and total protein in their hypoxic mouse model (43). This work indicates large increases in type I collagen expression as early as 6 days into hypoxic exposure, with type III and type IV basement membrane collagen expression increasing 4 days later. Thus, chronically hypoxic and monocrotaline rat models of PH and chronically hypoxic PH mouse models all demonstrate increased proximal PA stiffness, and existing evidence indicates this increase is primarily due to increased collagen accumulation.
Large mammals
Naeije and associates measured characteristic impedance in pigs three months after the animals underwent either a sham operation or one that created a ventricular shunt yielding a high-flow model of chronic PH (32, 148, 194). At baseline, the shunt group displayed impedance curves indicative of increasing PVR and stiffness, although only resistance (the zero harmonic of impedance) and characteristic impedance were significantly higher; Bosentan therapy applied to a third shunt group prevented these increases (148). The pigs were further tested in hyperoxia and hypoxia; no differences existed in the former, while the shunt group displayed greater sensitivity to hypoxia with significant elevations in PVR and characteristic impedance (194). Lammers et al. (99) conducted ex-vivo mechanical pull tests on strips of main, right, and left PA extracted from control and 2-week chronically hypoxic calves. All three PAs were significantly stiffer in the hypoxic animals; by normalizing the measured force to the cross-sectional area, it was also shown that the elastic modulus increased across the entire stretch range, indicating a fundamental change in the artery material properties. Finally, the physiological operating ranges for the animals were estimated from simple PD relationships and measured PAP. This analysis indicated that the increased stiffness was prominent in what is traditionally considered the elastin region with no change in the collagen engagement strain between animal groups, both suggesting that elastin was primarily responsible for the increases seen.
Measurement of Pulmonary Vascular Stiffness
Historical review
Parameters quantifying PVS describe force-distension relationships of the pulmonary vasculature. Site-specific and global stiffness measures were first obtained in intact animals and in man by the mid 1960s; however, these measurements were highly invasive and thus difficult to obtain in a clinical setting.
Global measurement
Early investigations quantified the global in vivo stiffness of the pulmonary vascular tree with the total volumetric compliance (DV/DP, where V is total pulmonary tree volume and P is pressure) in both animal models (42, 52, 133) and in man (121, 134).
Subsequent work examined the pulmonary vascular input impedance modulus, which quantifies both the resistive (static) and compliant (dynamic) components of RV after-load. Impedance, like total volumetric compliance, provides a global measure of stiffening of the entire lungs; it derives from a simplified model of the vascular circuit known as a transmission line model (derived later in this article) which assumes pressure and flow waves propagate through the circuit without frequency dispersion or change in wave speed. Groups led by McDonald, Patel, and Milnor and Bergel appear to be the first to recognize its increased utility over PVR in studies that obtained impedance from animal models (10, 24, 119, 133).
Milnor et al. (119) used the measured harmonics of pressure and flow—necessary for the calculation of impedance—to determine the total, mean, and oscillatory RV power of anesthetized dogs. To do this, they first represented their measured pressure and flow waveforms in terms of a finite Fourier series, which for flow takes the form
| (1) |
in which Q0 is the mean flow, Qn and ϕn are the flow amplitude and phase at the nth harmonic, ω is the fundamental frequency of oscillation (i.e., the heart rate), and N is the maximum harmonic considered in the Fourier representation. With pressure expressed in the same form (with Pn and βn as the pressure amplitude and phase at the nth harmonic), the impedance magnitude (Zn ) and phase (θn ) are
| (2) |
The hydraulic power of the RV, as pressure energy, are then given by
| (3) |
where the total hydraulic power Ẇt is the sum of the mean (Ẇm ) and oscillatory (Ẇo) components. Each component in turn is defined as
| (4) |
and
| (5) |
This analysis established that the pulsatile components of pressure and flow could substantially contribute to the total workload. Milnor et al. also determined the flow kinetic energy, but this was found to be a minor contributor to total RV power. Three years later, Milnor et al. (120) published the first impedance studies in man that further confirmed that the pulsatile components of hemodynamics, governed by vascular stiffness, were a significant contributor to the total workload presented to the RV. Additionally, patients with PH were found to display right-shifted impedance curves in addition to larger characteristic impedances and phase velocities, all indicative of stiffer vessels and significantly increased RV work. These increases were consistent with proximal, site-specific measures of pulmonary stiffness (60) that had attributed stiffening purely to a passive phenomenon known as strain stiffening, although these authors acknowledged the magnitude of the increase suggested other factors, such as vascular remodeling were also at work.
Characteristic impedance (Zc), which is input impedance in the absence of wave reflection, has been used as a measure of PVS, and is included here as a global measure due to its nonspecificity to a particular vascular site, as are the measurements below. However, it shows greatest dependence on arteries closest to the measurement site (118). Zc has been determined experimentally by averaging the harmonics of a measured impedance curve from the first minimum up to the 8th or 10th harmonic (10, 45, 107). Determination of Zc also allows for the theoretical separation of forward-traveling and backward-traveling waves, as will be described later. While Zc can offer insight into vascular mechanics, it also depends on vascular diameter; thus vascular geometry must also be considered when interpreting its values.
Local measurement
Site-specific measures of stiffness have also been examined, and may be more relevant to proximal vascular remodeling. Patel (134) and Peterson (136) were the first to obtain local in vivo stiffness of dog main PAs with the invasive acquisition of local pressure and diameter though catheterization and application of a vascular cuff, respectively. The latter group further examined the artery distension from an engineering standpoint: with the small-strain approximation, their measures of pressure and diameter were converted into engineering stress and strain, respectively; these expressions are
| (6) |
in which σ is the circumferential (or “tangential”) stress, P is the transmural pressure, d and h are the artery diameter and thickness, respectively, ε is the circumferential strain, and d0 is the undeformed diameter (these quantities are derived in more detail below). This analysis yielded both the pressure-strain modulus (also known as the Peterson modulus) and the circumferential stress-strain modulus [equal to the slope of σ — vs. — ε in Eq. (6) above], both of which quantify the local stiffness of an artery. Bergel (8, 9) complemented this work by not only finding these moduli, but also noting their increase as a function of pressure, that is, he quantified the strain-stiffening nature of the dog PA, and analyzed their dynamic elastic, or viscoelastic, properties.
The invasive method to obtain diameter used in the animal studies above seem incompatible with human research; however, Greenfield and Griggs (60) utilized the same methods in determining in vivo local PA stiffness in patients undergoing open-heart surgery. They found the normal PA to be five times as distensible as the aorta. Finally, the PA area increased an average of 22.9% during systole, compared to other studies that found only an 11% increase in the aorta, which suggested that the pulmonary system has larger capacitance. Additionally, three patients with PH were found to have significantly higher values of main PA pressure-strain modulus.
Clinical measurement
Routine determination of PVS through measurement of impedance (measured using custom catheters equipped with both a pressure transducer and electromagnetic velocity sensor) or proximal vascular stiffness [using either intravascular ultrasound (IVUS) or catheterization coupled with noninvasive imaging] were attempted in the past. As noted above, these methods were largely neglected until recently in that there was no driving rationale for stiffness measurement. Additional methods for stiffness measurement (mostly applied in the systemic circulation) have been reviewed recently (23, 66, 131).
Impedance
Pulmonary vascular input impedance has always been challenging to measure given its requirement of complete pressure and flow waveforms. Haneda et al. (67) obtained these waveforms in 1983 using a catheter with a distal electromagnetic velocity sensor and pressure transducer at the tip, a measurement clearly less invasive than use of a cuff-type flowmeter (120). The velocity sensor provided only a relative measure of blood flow, and so required calibration to true flow with an additional measurement of cardiac output (i.e., mean flow). With this equipment, they demonstrated that high O2 and norepinephrine both significantly reduced characteristic impedance and PVR in a group of 10 patients with PAH but did not change these parameters in 8 patients with pulmonary venous hypertension and in 6 healthy control patients. Six years later, Tagawa (173) published a major clinical study in which characteristic impedance, measured in 52 patients with chronic pulmonary disease (CPD), increased significantly with mPAP. This, together with a rightward shift of the first minimum of the patients’ impedance curves, indicated increasing proximal stiffness in these patients. Tobise et al. (181) evaluated 11 patients with atrial septal defects undergoing repair to determine if vascular stiffness was chronically impaired because of shunting. After repair, patients displayed impedance spectra and characteristic impedance that were not significantly different from 10 control subjects. In each study, PH was found to significantly influence the impedance; however, no attempt was made to determine if impedance was a better diagnostic. Finally, in 1992 in patients with mitral stenosis or congestive heart failure, Kussmaul et al. demonstrated nitroprusside-induced decreases in the first harmonic of impedance were associated with increases in flow, occurred independently of decreases in PVR, and were an important mechanism in reducing RV work (96, 97). This work also suggested the use of lower harmonics, such as Z1, as indicators of vascular function, as opposed to the characteristic impedance.
In 2004, groups led by Vachiéry (77) and Shandas (195) independently reintroduced the clinical measurement of impedance in 22 adults and 12 children, respectively, both using a new and novel method to measure flow. In place of a specialized velocity sensor on the catheter, spectral Doppler echocardiography (echo) was used to measure blood velocity at the centerline of the main PA. True flow was estimated through the use of an “area correction factor” such that
| (7) |
where Acorr is the area correction factor, CO is the cardiac output, and v(t) is the spectral Doppler measured velocity signal. Transient flow was then computed as Acorrv(t). It was shown that use of this flow estimation method in impedance calculation yielded only small differences between impedance computed with flow measured from a traditional cuff-type ultrasonic flow meter (195), thus validating the method. Further, the first harmonic of impedance, Z1, was suggested as a new measure of vascular stiffness, in contrast to the characteristic impedance (195); Zc was avoided because of the potential for significant measurement error in computation of impedance higher harmonics. Reactivity in this harmonic was advocated as a new diagnostic tool in PH, indicative of vascular stiffness reactivity. Echo was noted to have two advantages: first, it was a more readily available modality to measure velocity than specialized catheters, which made routine measurements feasible (77); second, its use enabled the exploration and development of new, purely noninvasive modalities for diagnosis (195).
Local stiffness
Routine assessment of proximal vascular stiffness also required a less invasive method of measuring PA diameter. In 1988, Moore et al. (123) used computed tomography (CT) to obtain main PA dilation over the cardiac cycle in 17 patients with PH or pulmonary vascular disease and found it well correlated with PVR and reductions in CO but not with mPAP. Contrarily, no correlations were observed in a separate group of six patients with chronic lung disease. This was presented as a wholly noninvasive means of diagnosis, but it was noted that some knowledge of the underlying disease must be known a priori. The following year, Herve et al. (70) used cineangiography to obtain closed-chest PD curves of the right PA in nine patients with chronic obstructive pulmonary disease (COPD). Administration of almitrine caused the PA to dilate and significantly stiffen, resulting in decreased distensibility, a result which was attributed to increased motor tone, but has the hallmark of collagen engagement resulting from excessive dilation.
Intravascular ultrasound was also explored as a means to routinely obtain PD curves, in addition to other measures of proximal vascular stiffening. Borges et al. (17) used IVUS to detect increases in proximal vessel thickness or specific intimal thickening in a group of 39 patients with PH compared to 12 controls. Bressollette et al. (19) also focused on structural changes in the PAs—minimum diameter, wall thickness, lumen area—at both end-systole and end-diastole. They found that IVUS-measured vessel thickness in the smaller compliant vessels (1.7–6.5 mm) moderately correlated with mPAP but well-correlated with plasma endothelin-1 (ET-1) levels, which supported the idea of ET-1 as an indicator of vascular abnormality. Berger et al. (11) were the first to specifically pursue vascular mechanical properties through IVUS with simultaneous measures of vascular pulsatility (defined as the difference between systolic and diastolic vascular area divided by the diastolic area) and pulse pressure; these were combined to yield distensibility, or compliance. The former measure is similar in form to engineering strain, while the latter is simply an inverse of stiffness. In 43 children with PVD, distensibility decreased with disease severity and was independently correlated with mPAP-to-systemic mean pressure ratio, PVR/systemic resistance, and hemoglobin concentration; the two ratios were intended to denote derangement of pulmonary hemodynamics, while the latter (hemoglobin) was indicative of blood viscosity and thus potential changes in shear stress. That all three of these correlations were shown to be independent of mPAP indicated that the wall mechanics could be a valuable, independent measure of disease progression and thus disease prognosis. Finally, Rodés-Cabau et al. (147) measured distensibility in 20 patients with IPAH; this study will be discussed in the next subsection below.
In what may be the first completely noninvasive estimate of vascular stiffness, Dyer et al. (41) coupled right PA diameter assessed with color M-mode tissue Doppler imaging (CMM-TDI) with pressure estimated from color flow imaging measurement of tricuspid regurgitant velocity. The CMM-TDI measurement compared favorably to IVUS measured diameter, validating this component. Twenty-seven hypertensive children displayed significantly lower dynamic compliance of the right PA compared to 10 normal children; the compliance also showed an exponential decay with pressure, corresponding to the nonlinear behavior of artery mechanics.
Clinical relevance
PVR has long been used as a disease diagnostic and a predictor of patient outcomes in PH (4, 5, 6). By 2003, vascular stiffness measurement methods had been established, but reasons for their use were less established. The study of 20 patients with IPAH by Rodés-Cabau et al. (147) began to address the utility of such measurement in prognosis. They obtained the same two quantities as Berger et al. (11)—pulsatility and distensibility—but inverted the latter such that it was a stiffness measure, and established they were independent of standard hemodynamic measures. They however took the further step of demonstrating that reduced pulsatility was associated with high mortality at follow-up. Independence of the stiffness measures from other hemodynamic values indicated that vascular mechanics—specifically changes due to remodeling—were more responsible for the functional alterations found, while decreased area pulsatility—indicative of vascular stiffening—would indicate higher RV afterload and thus greater mortality.
Subsequently, three additional groups have shown stiffness measures to be more prognostic than PVR. Mahapatra et al. (109, 110) measured standard hemodynamic parameters and used them to estimate “global compliance” as stroke volume over pulse pressure (SV/PP). In a group of 104 patients with IPAH, SV/PP was the strongest Cox univariate predictor of all-cause mortality, with a hazard ratio of 17; this could be compared to PVR or cardiac index with hazard ratios of 1.05 and 3.2, respectively. Receiver-operator characteristic (ROC) curves further demonstrated that SV/PP was a better discriminator compared to these other two measures. Purely noninvasive prognostication was shown by Gan et al. (55), who measured vascular pulsatility (referred to as “relative area change” or RAC in the paper) with magnetic resonance imaging (MRI) in 70 adults with PAH. Cox univariate regression and ROC analysis again showed this stiffness proxy measure to be a superior prognostic compared to PVR. Finally, Hunter et al. (84) measured pulmonary vascular input impedance in 25 children with PAH. They computed the sum of the first three harmonics of impedance modulus (i.e., Z0 + Z1 + Z2), indexed to body surface area, as a new measure of total afterload and used it in a logistic regression model to predict three 1-year soft outcomes categories based on change in World Health Organization (WHO) functional status; here too, the stiffness-based measure (in this case incorporated into the two higher harmonics of impedance) outperformed PVR.
Models of Pulmonary Vascular Stiffness
Engineering models of mechanical systems have traditionally been used to predict or investigate system behavior when direct measurement was impractical or impossible. Applied to the pulmonary vasculature, models may predict blood flow and pressure, wall distension, normal and shear stresses, and/or energy requirements. These simulated endpoints may be useful in the diagnosis and treatment of disease, the design of vascular implants or surgical anastomoses, or simply for hypotheses testing. Generally, simple analytic models can provide insight into global or system-wide behavior, and their parameters have been proposed for use as diagnostics, while more complex models attempt to provide hemodynamic or structural details that cannot currently be measured. Below, we review the treatment of PVS in both local and global models of the pulmonary circulation.
Simple global structure/flow models
The earliest and simplest models of the vasculature represent blood pressure and flow as electrical voltage and current, respectively, and are usually displayed as electrical circuit diagrams. In these models the arterial concepts of resistance, compliance (stiffness), and inertance are analogous to the electrical components of resistance, capacitance, and inductance, respectively. This modeling analogy is most important in how it represents the total load imposed on the electrical driver (power source) due to the connected electrical circuit, known as the input impedance. Input impedance is a single parameter that quantitatively describes the entire circuit and may be used to determine the power or work requirements to move current through a given electrical circuit composed of resistive, inductive, and capacitive elements. This idea applied to the vasculature yields the vascular input impedance, or the measure of a peripheral vascular bed’s total opposition to blood flow.
Two general classes of these models exist: those which represent the entire circulation with a few conceptual components—known as lumped parameter or “Windkessel” models—and those that contain unique component groups for each branch or segment of the circulation, known as distributed or “transmission line” models. Each may be employed to compute the vascular input impedance and explore how changes in modeled vascular properties—either global or local—affect impedance.
Lumped parameter models
Frank (51) is universally credited with the first use of a lumped parameter model, which represents the entire pulmonary circulation using a single set of circuit components. His work explored the shape of the pressure wave during systole and diastole, indicated that diastolic pressure displayed an exponential decay proportional to the resistance and compliance of the system, and provided ordinary differential equations (similar to modern electrical circuit equations) that could be used to obtain the pressure given a constant or sinusoidal inflow rate.
Three general circuit configurations are primarily used today: the two-element model (51) (Fig. 3A), which contains only elements of total peripheral resistance and total arterial compliance; the three-element model (197) Figure 3B), which adds an additional resistor in series to the two-element model that represents the arterial characteristic impedance; and the four-element model (22) (Fig. 3C), which adds an inertial component in series with the characteristic impedance resistor of the three-element model. The latter two may be seen as incremental improvements to the two-element model, in that the addition of the characteristic impedance allows the three-element model to better represent the high-frequency components of vascular input impedance while the inclusion of an inertial component in the four-element model reduces low-frequency errors in the modeling of the impedance first minimum (198).
Figure 3.
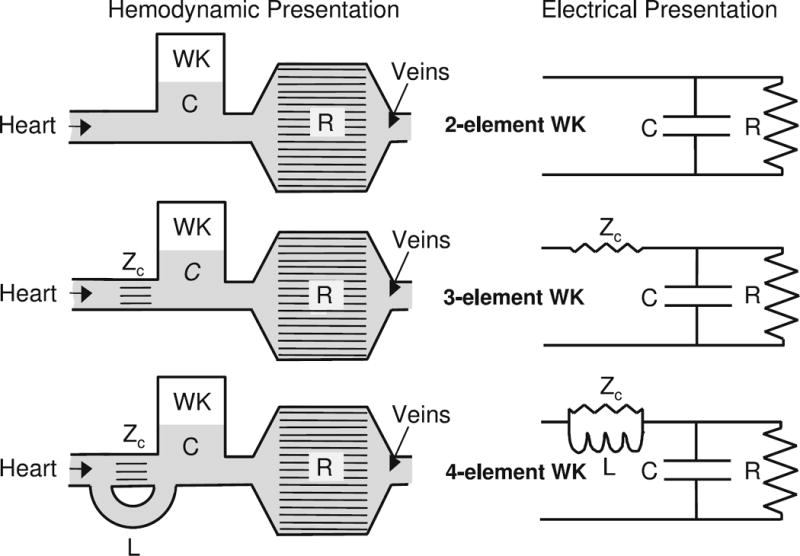
Idealized representations of the pulmonary circuit and their corresponding Windkessel models. C = (pulmonary artery) compliance, PAC; R = (total pulmonary) resistance, TPR; Zc = characteristic impedance; L = inertance. [From (198); used with permission]
The components of the lumped parameter models are determined using clinically measured data, either directly or through iterative numerical methods. In the simplest case, the total pulmonary (peripheral) resistance (TPR) is simply equal to mPAP divided by flow whereas the total pulmonary arterial compliance (PAC) is obtained either from assuming the pulse decays during diastole as exp(−t/RpC) where Rp and C are TPR and PAC, respectively (51, 197), or more recently from a host of procedures that utilize PAC as a fitting parameter that optimizes the model’s numerical prediction of either the pressure or flow wave (168). As seen below, this second, iterative method to obtain PAC has been most widely used to obtain clinical diagnostic information from the Windkessel model. For the three-element model, clinically measured pressure and flow waveforms are used to compute the average of the higher harmonics of pulmonary vascular input impedance; this average is, by various definitions, the characteristic impedance (10, 45, 107, 114, 151, 195). The inertance component of four-element models has been estimated from distributed models of the systemic circulation (169), although this methodology apparently has not been applied in the pulmonary circulation.
Lack of both wave propagation effects and the ability to model local vascular properties are the primary criticisms of Windkessel models, which lack any reference to true vascular geometry; in comparison, the circulation has location-dependent mechanical and geometric properties, hemodynamic quantities propagate with finite speed, reflections occur, and the relationship between pressure and flow varies by location. These limitations can result in misinterpretation of measured distal waveforms in the systemic circulation (131) and are likely sources of error in the estimation of PAC (188). Indeed, the initial development of distributed models in the 1960s was likely precipitated by this weakness (131).
Recent work
Van den Bos et al. (188) found that altering hemodynamics by serotonin infusion resulted in large changes in pulmonary hemodynamics but noted that these changes were reflected primarily in the lowest harmonics of the measured impedance curves. A Windkessel model of their data similarly predicted significant changes in modeled PAC and less definitive changes in characteristic impedance, which suggests that the latter may be a less sensitive measure of vasoreactivity. This work also found, based on computed characteristic impedance, a lower reflection index in the pulmonary circulation; this was given as an explanation for the similarity between pulmonary flow and pressure waveforms. Slife et al. (156) measured pulmonary flow and pressure in eight humans in both resting and exercise conditions. They compared TPR and characteristic impedance obtained using a three-element Windkessel model that optimally predicted flow from a measured pressure waveform to equivalent values extracted from measured impedance curves and found them to be similar in both conditions. However, their modeled compliance was significantly larger (2.5 times larger at rest, 3.4 times larger with exercise) than its simple hemodynamic equivalent, stroke volume over pulse pressure; this discrepancy was said to be expected because of the crudeness of SV/PP as an estimate of PAC. The increase in global compliance found could be reflective of increased pulmonary capillary recruitment that occurs during exercise (211). A Windkessel model was used to assess the independent effects of vascular resistance and compliance on ventricular-vascular coupling by Fourie et al. (50). Their analysis of data from an acute embolic model of PH in Landras white pigs suggests that increases in PAC are a significant factor in decoupling the ventricular-vascular system and results in decreased stroke volume. Segers et al. (150) compared three common methods for the estimation of PAC. By comparing their current results to previous PAC estimates they found that a two-element model that optimally predicted pulse pressure alone from measured flow provided the most consistent PAC predictions in both the pig and the dog under normal and pulmonary embolism conditions.
One common weakness of the Windkessel studies above is lack of a definitive validation measure for PAC. Regardless, as a derived measure of vascular compliance, PAC may have PH diagnostic potential; this question was explored by Lankhaar et at. (100), who used a three-element Windkessel and several parameter estimation techniques (22, 167, 168) to obtain PAC in normotensive, IPAH, and chronic thromboembolic PH (CTEPH) patient groups. While TPR was clearly higher in both PH groups, they also found larger characteristic impedance in the IPAH group than in the CTEPH group, suggesting additional proximal stiffening in this group. Both hypertensive groups displayed reduced, but not significantly different, PAC. Finally, the product of computed TPR and PAC in all three groups was found to display an inverse exponential relationship that suggested a pressure-dependence of PAC. The clinical utility of PAC (and indeed, Windkessel modeling) remains unknown, as no studies have yet indicated that PAC or characteristic impedance are useful diagnostics or prognostics for PH or other pulmonary diseases. As will be seen below, the simplicity of these models and their reasonable representation of both resistive and compliant afterload have made them useful in bounding the domains of more advanced computational models.
Distributed parameter models
A branching network of connected three- or four-element Windkessel-like circuit assemblies, wherein each individual assembly represents a specific vascular branch, is the physical form of a distributed parameter, or transmission line model. The model theory in simplified form was originally developed by Oliver Heaviside in the 19th century for the purpose of analyzing electrical power distribution. Womersley (208, 209, 210, 211) demonstrated that simplified versions of the one-dimensional (1D) fluid dynamics equations took the form of a simple transmission line model [useful additional derivation in (199, 201)]. Taylor (178, 179, 180) first modeled vascular structures with the method and thereby provided some theoretical underpinnings for the use of impedance in the vasculature; this initial work contrasted the input impedance of a single tube to that of a branching tube structure, and revealed that structural viscoelasticity tends to reduce oscillations in the higher harmonics of the impedance modulus. Westerhof et al. (199), building on this work and others (10, 114, 196), introduced the idea of forward and backward traveling waves in the vasculature, the means to separate them, and the first evidence in the systemic circulation of dogs that increased peripheral resistance coincided with increased wave reflection from the periphery. Together, these works brought to the forefront the concepts of vascular input impedance, characteristic impedance, and pulse-wave reflection from bifurcations and peripheral vascular beds.
The individual components of the circuit assemblies—resistor, capacitor (compliance) and inductor (inertance)—have properties that can typically be written as functions of the local wall diameter and thickness, the local elastic modulus, and the global blood viscosity. These functions allow for global or segmentally specific vessel compliance, and Womersley theory is frequently employed to represent the resistance (20, 56, 76, 128), although other, simpler theories exist (157). The branching geometry—which entails each segmental diameter and length, and branch connectivity—may be specified based on direct measurement (159) or is provided by fractals (20), fractal networks (127, 128), or random functions (179, 180). At the terminal (i.e., unconnected) end of a branch, an approximation of the opposition to flow leaving the end of the tube (i.e., the impedance) is required; this may be merely the resistance imparted by capillaries for pulmonary models, but can also represent complex impedance due to organs for systemic models (130). Once the geometry is established, the terminal impedances of the branches are then defined, and through the use of the transmission line equations the input impedance, terminal impedance, reflection coefficient, and propagation coefficient for each branch is found. These components respectively describe the opposition to flow at the inlet and exit of the branch, how much energy is transmitted through rather than reflected back at the branch, and how the flow attenuates and changes in phase as it is transmitted through the tree. When these quantities are summed for the entire tree, the input impedance for the entire structure is obtained.
Given the before mentioned model parameter requirements, it is clear that is it far more difficult to construct a distributed parameter model compared to a single Windkessel model. Additionally, input impedance is readily computed without the model—indeed it is far easier to obtain from pressure and flow measured at a single position. For these reasons, distributed parameter models have primarily been used in the systemic circulation to investigate wave transmission phenomenon such as transmission and reflection (71, 129), and to explore relationships between impedance and the components that define it (3): vascular geometry including vessel diameters and lengths, variable stiffness, and the connectivity of a vessel tree. Pulse-wave reflection has interesting diagnostic consequences, in that a double-humped velocity trace is usually seen in pulse-wave Doppler ultrasound obtained in the main PA patients with severe PH, known as a “Flying W” pattern. This theory has been used to show in multiple animal models that this early reflection yields additional right ventricular loading during ejection (64, 216). Additionally, one promising recent use of these models on the pulmonary side has been to place bounds on more advanced models of the pulmonary circulation (159); this usage will be described briefly below.
Material models: local vascular mechanics
In mechanics, constitutive models define the relationship between applied loads and the resultant deformation of materials. In the study of artery mechanics, such models allow for a quantitative examination of blood vessel material properties. Further, through the development and use of constitutive models, engineers are able to discover how the major structural elements of arterial tissue influence the mechanical behavior of that tissue in response to different operating conditions, disease or physical damage. Constitutive models rely on in vivo measurements of pressure and diameter or similar in vitro bench top mechanical measurements for their validation. A material’s mechanical “resistance” to deformation is given as the slope of such force-displacement curves. This “resistance” may be quantified intrinsically (i.e., only as a function of the continuum), in which case it is the slope of stress (normalized force)—vs—strain or stretch (normalized deformation) and is referred to as the material’s elastic modulus or Young’s modulus, or it may be quantified extrinsically, in which case it is the slope of an absolute force—vs—an absolute displacement and is referred to as its stiffness. Some applications of material models include the estimation of arterial mechanical properties in vivo, more quantitative assessment of how these properties change in disease states, and use in more advanced computational models to evalute how hemodynamics are affected by constitutive properties.
Given that the arterial wall is a layered structure, consisting of the intima, media, and adventitia and that passive artery mechanics are largely governed by the physiologic structure, quantity and intrinsic material properties (i.e., modulus) of the structural proteins collagen and elastin and by the active and passive mechanics of SMCs, artery constitutive modeling becomes a rather complicated engineering problem. In fact, the study of artery mechanics is over a century old and the study of arterial tissue constitutive models has received considerable attention in the past couple decades. Arteries display displacement-, direction-, and time-dependent passive material properties; these complicating factors are known respectively as strain-stiffening (due to large deformation), anisotropy, and viscoelasticity. The wall may also be treated as compressible (e.g., capable of changing in volume) or incompressible. Changes in artery wall geometry that occur during in vitro processing indicate that the wall carries an internal stress even when unloaded; histology clearly indicates the wall is heterogeneous; and SMCs may affect both wall geometry and stiffness. We believe separate examinations of passive properties (which may depend on time, direction, or displacement, as noted above) and active properties (which depend on the biological state of the SMC), that represent a component-wise or deconstructionist approach, may provide a useful means of separating active versus passive components within arterial mechanics.
For each constitutive model intricacy, simplifications exist that are useful for both approximate clinical assessment of vascular stiffness and for modeling vascular displacements over small (i.e., linear) ranges. Below we examine those models that incorporate simplifications of the above characteristics that are useful in the clinic, and more complex models currently used primarily in basic science or engineering treatments of arterial mechanics. Throughout, we primarily are concerned with passive properties, not because myogenic tone may not be important, but because a majority of current engineering work revolves around passive mechanics. More detailed theoretical treatments of arterial mechanics may be found in two recent reviews (57, 191).
Analytic/linear elastic models
Thin-walled and thick-walled theories are often applied to two-dimensional (2D) tube geometries to obtain a simple relationship between stress and strain, and thereby extract material properties. Here we will introduce these theories along with the basic concepts of stress and strain applied to a tube system, and discuss several simple engineering elasticity indices that have appeared in the literature (36, 59). Additional clinic-specific elasticity (stiffness) indices are reviewed elsewhere (131).
Thin-walled theory
Strain (ε) is a normalized measure of material deformation and is given for the circumferential direction by
| (8) |
where d0 and d are the initial (reference) and final diameters. For strains to be considered linear the deformation must be small relative to the total diameter, which implies that ε < 0.1. Stretch (λ) is also used as a measure of deformation and is merely
| (9) |
Both these quantities are dimensionless, and strain is occasionally presented as a percentage deformation. Clearly these equations are fairly simple, but it remains an open question as to which “initial” diameter is most appropriate for use (36): for example, the artery in vivo is always under some transmural load and may be additionally deformed because of SMC activity. In contrast, in vitro studies often examine vessel groups under the effects of vasoconstrictive and vasodilator drugs and thus technically have different “initial” diameters and therefore different initial reference configurations for each study group. This situation may be remedied through the use of stretch rather than strain, as it may be directly multiplied by reference diameter ratios to obtain different reference configurations. For example, if it was determined that a diameter d1 was a more appropriate reference compared to d0, the new, more appropriate stretch is simply
| (10) |
where λ1 is the stretch referenced to d1. Regardless, these measures quantify a typical material deformation, but consideration of reference diameter or length can become very important when considering coupled changes in artery dimensions and mechanics due to growth, remodeling, disease, or ex vivo tissue processing.
Stress is a normalized measure of force acting on a body and has dimensions of force per unit area. To obtain a simple estimate of the stress at work in a thin-walled tube, we examine the free-body diagram of a tube of internal radius ri and thickness h shown in Figure 4. In the figure the transmural pressure Pt = Pi − Po acts downward on the cutting plane of area 2 × ri × l, while the half-tube remains in place due to internal forces (tensions) T acting upward. The stress (σ) that corresponds to these forces is their combined value normalized by area of two tube sections (2 × h × l) over which they act,
Figure 4.

Tube geometry for the derivation of simple mechanics equations. Pi, Po = pressures acting on inner and outer walls; T, tension within the artery; ri, ro = inner, outer arterial radii; h = wall thickness, I = tube length.
| (11) |
or T = σ hl. Implicit in this equation is the idea that the stress is uniform across the area, which is true only for thin tubes. If the tube is in static equilibrium (i.e., is motionless), then
| (12) |
so we may solve for the stress as
| (13) |
Equation 13 has been referred to as Laplace’s equation due to its similarity to the Young-Laplace equation, which describes the relationship between pressure and surface curvature at a fluid-fluid interface due to surface tension. Also similar to the Young-Laplace relationship, an immediate takeaway from this equation is that wall stress increases not only with increasing pressure but with increasing internal radius; this idea has been used previously to explain the similar stresses in the left and right ventricles, despite their highly disparate operating pressures.
To find the linear elastic modulus, or Young’s modulus, of this example thin-walled tube, we consider changes in stress due to small strains. For an artery, whose modulus actually has a strong dependence on strain, we would call this approximation the incremental elastic modulus. This modulus is given as the instantaneous (tangent) or secant slope of the stress [Eq. (13)] plotted as a function of the strain [Eq. (8)]. This simple, linear approach was used in several early studies of local vascular mechanics (60, 136) and in modern studies either for the sake of simplicity or because thin-walled theory is applicable (95, 217). Generally, the theory remains useful both as an introduction to the mechanics of tubes and as a check on more advanced models.
Thick-walled theory
As the wall thickness increases, the stress is no longer uniform in the radial direction; this effect is neglected by the thin-walled theory. A simple explanation for this radial dependence was provided by Gow (59) and will be repeated here. Looking again at Figure 4, we may consider three separate forces in our equilibrium equation: (i) the dilating or expanding force 2riPil which expands the tube; (ii) the constricting force 2roPol, and the force due to the stress induced in the wall 2σ hl, which is equal to 2σ (ro − ri)l. For Pi > Po, we have the force balance
| (14) |
so that wall stress for a thick-walled cylinder becomes
| (15) |
Equation 15 is only one of several expressions for the stress in a thick-walled tube. Similarly, expressions to obtain elastic modulus from thick-walled theory are also numerous (9, 149, 185).
Clinical equations for stiffness
Quite recently, PD measurements were used to obtain elastic modulus approximations using both thin- and thick-walled theories, and compared to in vitro determined elastic modulus (82). It was shown (perhaps unsurprisingly) that elastic moduli computed from the thick-walled theory were superior estimates of ex vivo mechanics compared to those estimated with the thin-walled theory; these expressions are
| (16) |
for the thick-walled theory (TWPS, “Thick Walled Plane Strain”) and
| (17) |
for the thin-walled theory (PS, Pressure Strain). In these equations D is the vascular diameter, with subscripts ID, IS, and OD referring to this measurement’s time and location: inner diastole, inner systole, and outer diastole, respectively. Similarly P is the transmural pressure at diastole (subscript D) and systole (subscript S). Finally, in Eq. (16) ν is the assumed Poisson’s ratio and m is the regressed slope of the measured PD curve during systole; in Eq. (17), h is an approximate thickness computed from measured diameter and a thickness-to-diameter ratio assumption.
It is important to note that use of these simple approximations typically neglect a host of factors, including material nonlinearity and finite deformation effects; however, the measurements appear to offer a reasonable first estimate of in vivo stiffness, and have demonstrated initial diagnostic potential (83). Thus, more advanced finite deformation models have been developed for use in simulations, and potentially to analyze experimental data.
Strain energy function models of arterial tissue mechanics
Because of the breadth and depth of the field, this writing will not attempt to summarize the work devoted to the study of artery mechanics over the past several decades. In fact, there are several very good, and recent, review articles devoted to that particular subject (80, 81, 191). Nor is this writing a rigorous study in finite strain theory or nonlinear mechanics, both of which have been covered in great detail by others (16, 74). Rather, in this section, we will attempt to describe a particular type of constitutive model used to define arterial tissue as a pseudoelastic material through the use of strain energy functions (SEFs). This type of constitutive model is the most widely researched, directly correlates artery microstructure with specific material properties and forms the backbone of several other model types such as randomly elastic, poroelastic and viscoelastic arterial tissue models. It is hoped that by gaining a better understanding of the fundamental principles used in the derivation, application, and development of this specific model type, the reader will be better able to understand the underlying rationale for the study of arterial constitutive models and will be better able to read and understand other published materials related to this important field of study.
Early attempts at modeling arterial tissue mechanics
Arteries do not meet the definition of a purely elastic body insomuch as the relationship between stresses and strains are not single-valued. Arteries exhibit hysteresis in that the loading and unloading curves of these tissues do not directly correspond. Arterial tissues are anisotropic and therefore have different material properties in the longitudinal, circumferential, and radial directions. Finally, arteries demonstrate a highly nonlinear stress-strain response which is also a function of the maximum applied strain and preconditioning of the tissue being tested. All of these factors complicate significantly the formulation of a comprehensive mathematical model of the constitutive relationship between stress and strain in arterial tissues subjected to finite deformations. Because of these complications, SEFs are typically used to describe the complex material property relationships associated with the finite deformation of soft tissues.
Exponential and polynomial strain energy functions
The first attempts at developing SEFs representing the mechanical behavior of arterial tissues were purely phenomenological and simply represented the shape of the load-deformation curve through the use of an appropriate mathematical function (54, 174, 187). The best known of these early SEFs was the pseudoelastic SEF developed by Fung et al. in 1979 (54), which was an extension of the polynomial SEF developed by Vaishnav et al. in 1972 (187). In this article, Fung proposed pseudoelasticity as a convenient description of the stress-strain relationship of arterial tissues in which the complication of inelasticity was avoided by describing the cyclic loading and unloading curves as independent material curves. Following this approach, the method of the theory of elasticity may be applied to each of the loading and unloading regions separately. Fung then went on to define a mathematical representation of this pseudoelastic stress-strain relationship through the use of a SEF. By definition, the SEF (Ψ)
| (18) |
relates the second Piola-Kirchhoff stress (Sij) to the Green-Lagrange strain (Eij). Therefore, by defining the constitutive relationship in this manner for any given strain state, the material stress can be calculated directly. Given the ubiquity of the use of Green’s strain (E) and second Piola-Kirchhoff stress in artery mechanics SEFs it seems logical to describe these variables in more detail. Green’s strain is used in place of the more conceptually understandable Cauchy strain (ε):
| (19) |
due to the fact that Cauchy strain is not a valid measure of strain for finite large deformations. Here, l is the deformed length, L is the original length and λ is defined as the stretch ratio. Finite strain theory applies for any material that is deformed to a large enough degree such that the assumptions inherent in infinitesimal strain theory fail. In practice, however, finite deformations generally refer to changes in material dimension, in any direction, greater than approximately 2%. For these large strains, the assumptions used to derive ε fail and the strain cannot be thought of as a simple ratio of the change in length due to applied load by the original length of the material.
Cauchy stress (σ ) and Piola stress (T) are conceptually simple representations of stress, each referencing the deformed and original cross-sectional areas over which a given force is applied respectively. In the principal directions, these stress values can be represented as:
| (20) |
and are shown in Figure 5. However, neither Cauchy nor Piola stress tensors constitute a work conjugate relationship between stress and Green’s strain. Simply put, a stress-strain work conjugate ensures that for any physically realizable strain state, that the energy calculated from the stress-strain relation must be of a positive definite value. Fung therefore used the second Piola-Kirchhoff stress (Sij), which is the work conjugate of the Green’s strain, in the relation given in Eq. 18:
| (21) |
where ρ0 and ρ are the initial and final mass densities, respectively. Also, λ1 and λ2 are the principal stretch ratios and E1 and E2 are the principal Green’s strains defined as:
| (22) |
Figure 5.
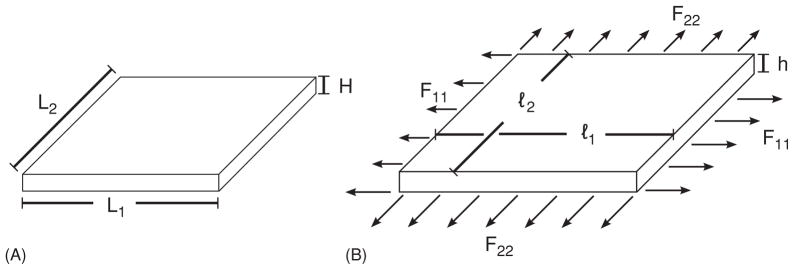
Deformation of a rectangular plate. (A) Un-deformed rectangular plate. (B) Deformed rectangular plate. Loads (F) are applied along the edges of the plate in the principle directions [circumferential (1) and longitudinal (2)].
The theory of elasticity states that if there is a one-to-one relationship between stresses and strains, then there exists a SEF that allows stresses to be calculated from the SEF by direct differentiation by the strain, from which Eq. (18) is derived.
While Eq. (18) is a general definition of an arterial SEF, in practice, the artery is often represented as a 2D body subjected to stresses and strains in the principal directions S11 S22 E11, and E22 only and the SEF can be specifically rewritten as Ψ(2) to indicate this two-dimensionality. This simplification of the SEF to two dimensions is the result of several assumptions, including the thin-wall assumption stating that the normal stress in the radial direction σrr is negligible in comparison to the circumferential normal stress σθθ and that the circumferential σθθ and longitudinal σzz stresses are approximately uniform throughout the vessel thickness. After further algebraic manipulations and several more simplifying assumptions, the circumferential and longitudinal stresses can be defined as (174):
| (23) |
| (24) |
| (25) |
| (26) |
Fung then used these relationships to calculate the experimental stress and strain based on measureable properties, and used these values to determine the four independent variables, C, a1, a2, and a4, in his proposed SEF (Ψ(2)expo).
| (27) |
Once the independent variables have been determined, the SEF can then be applied to Eq. (21), such that the stress (Sij) is univariate and fully defined for any strain state (Eij).
Fung compared the results obtained using his proposed exponential SEF to those found using a different, polynomial-based, SEF and found that the constants of the exponential SEF showed significantly less variability than the constants of the polynomial SEF. This reduction in constant variability held for both multiple fittings of the same artery as well as between arteries and therefore points to the conclusion that the exponential SEF is an inherently more stable model of artery mechanics than is the polynomial SEF. However, although the constant variability was different, the correlation between the theoretical and experimental data was the same for both SEFs. Fung’s conclusion was that if the goal is to simply fit the data with a function, then either SEF may be used, but if the goal is to look at variations in the values of the constants in response to age or disease, then the exponential SEF should be used. However, since neither the polynomial nor exponential SEF constants represent any direct physiologic or structural properties, no distinct answer can be drawn regarding how changes in these constants relate to changes artery material properties resulting from changes in tissue structure or physiology. To answer this question, more advanced structural SEF models have been developed.
Two-dimensional versus three-dimensional constitutive models and the effect of arterial ring-opening on residual strain of the zero-stress state
The SEF models reviewed thus far are derived using a thin-walled, plane-strain, assumption and therefore are 2D constitutive functions that do not account for any stresses or strains present in the radial direction. This 2D representation of the SEF is generally accepted as being accurate so long as its application is only used in solving problems consisting of tissue bending, inflation, and axial extension and avoiding any deformations due to torsion. A further limitation of the 2D formulation is that it does not allow for a detailed analysis of the change in the circumferential and longitudinal stresses (Sθθ, SZZ) and strains (Eθθ, EZZ) as a function of radial position within the tissue thickness. Modeling arterial tissues with a fully 3D SEF was attempted by Chuong and Fung in 1983 (27). The six-parameter, 3D SEF, which they used, showed a significant variation in circumferential stress across the tissue thickness (27). Further study indicated that this high variation in tissue stress as a function of radial position could be largely explained by changing the zero-stress state (ZSS) of the arteries from the unloaded configuration of an unpressurized cylindrical artery segment to the configuration of an artery ring that was axially bisected and allowed to open into an arterial arc segment (28). Since further sectioning of the arc segment did not result in appreciable changes in tissue dimension, this opened segment was taken to be the new ZSS. By incorporating the strain required to deform the open segment into the unloaded configuration, the variation in arterial stress as a function of radial position was significantly reduced, and use of the opened segment as the ZSS for artery modeling has now become standard.
Structural SEF models
While Fung’s exponential and Vaishnav’s polynomial SEFs are grounded in basic mechanics, they are purely phenomenological and therefore the model constants are not based on any particular anatomic or physiologic parameter that could be independently measured or verified. Further, while these functions accurately represent the material properties of arterial tissues within certain limitations, they fail to represent the constitutive relationship between load and deformation outside of their assumed valid parameters. Therefore, these models are typically valid only for limited pressure ranges and are often only capable of describing constitutive properties of the circumferential stress-strain relationship.
Collagen as fiber reinforcement for arterial tissue SEF modeling
In 2000, Holzapfel and Gasser made a significant advancement in arterial SEF constitutive models by incorporating the techniques used to describe the material behavior of fiber-reinforced composites into a two-layer model of arterial tissues (75), Figure 6B. The basic concept was to treat the medial and adventitial layers of elastic arteries separately and to model the mechanically relevant non-collagenous material as an isotropic, linearly elastic SEF (ψiso) which was primarily associated with the mechanics of elastin. Collagen mechanics were modeled as a pair of helical fibers with equal, but opposite, pitch (α, α′). By modeling collagen as a fiber weave, the anisotropy of the model was solely a function of the fiber helical pitch representing collagen physiology in arterial tissues. This anisotropic SEF was modeled separately as (ψaniso), and was summed with the isotropic portion to define the total SEF (ψHolz).
Figure 6.
Schematic diagram of artery wall in the zero-stress state used in the formulation of several arterial models. Arterial wall as modeled by (A) Fung (54), (B) Holzapfel (75), (C) Zulliger (218). Figures used with permission.
| (28) |
| (29) |
| (30) |
where the material parameters c and k1 are positive with dimensions of stress and k2 is dimensionless. The invariants (I1, I4, and I4′) are functions of the strain deformation tensor that represent physical quantities in a manner that is independent of the coordinate system used, a requirement that is essential in the formulation of any useful SEF. Since Holzapfel modeled the arterial wall as two layers, there were a total of six material parameters to solve for: cM, k1M, and k2M for the media and cA, k1A, and k2A for the adventitia. Geometric and experimental data from a single rabbit carotid artery [experiment 71 in (54)] were used for determining these parameters. Further, the ratio of medial to circumferential stiffness of the isotropic SEF was set to cM = 10 cA, further reducing the number of independent material parameters to five. This relationship between medial and adventitial stiffness was justified based on experimental results (192, 210, 212), which showed the media to be much stiffer than the adventitia. The medial thickness was assumed to be two-third of the arterial wall thickness, and the medial and adventitial wall thicknesses were assumed to remain constant regardless of axial deformation. The remaining model parameters were then found using the standard nonlinear Levenberg-Marquardt algorithm (75).
While Holzapfel’s SEF model showed somewhat improved fit quality between experimental and predicted data sets, the structural details incorporated within this new model were the real advancement over previous phenomenological constitutive models. This new SEF modeled the arterial wall as two separate layers, representing both the media and adventitia. Each layer used its own independent material parameters to define the 3D anisotropic behavior of arterial tissue. Noncollagenous tissue was modeled as an isotropic material, and tissue anisotropy was included by modeling collagen as a fiber weave of oppositely oriented helical collagen fibers. This structural approach resulted in Holzapfel’s model’s ability to at least partly correlate changes in arterial stiffness with changes in the underlying histological structure. While this model was a significant advancement over the phenomenological models of the time, it does have some limitations, mainly that the predicted collagen fiber angle is greater than what was expected on the basis of the physiologic structure of arterial collagen (35, 203). This elevated collagen fiber angle predicted by Holzapfel’s SEF may be at least partly due to the fact that in this model collagen is represented using straight fibers which have no tortuosity.
Inclusion of collagen engagement strain probability distribution in fiber-reinforced model
Zulliger made further refinements to the arterial constitutive model by developing a SEF very similar to that used by Holazpfel, but one which also incorporated elastin and collagen area fractions, elastin non-linearity and collagen tortuosity (218), Figure 6C. In arterial tissues, the structure of collagen has been described as long wavy bundles with mainly circumferential orientation (158) and appears as a coiled and wavy structure in the unloaded state (29, 35). Zulliger incorporated this wavy collagen structure by assuming that the point at which collagen is straightened and able to carry load is distributed across the deformation strain range in some statistical manner. This collagen engagement strain was then incorporated into the SEF model through a log-logistic probability distribution function (ρfiber). Like Holzapfel, Zulliger’s SEF (ΨZull) models arterial tissues using a noncollagenous isotropic component (Ψiso) and has anisotropy incorporated as a second SEF based on collagen morphology (Ψaniso).
| (31) |
| (32) |
| (33) |
| (34) |
where celast is the elastic constant and felast and fcoll are the area fractions of elastin and collagen, respectively. Ψfiber is the SEF of an individual collagen fiber, which is modeled as a linearly elastic hookean material of modulus ccoll for strains greater than zero. Equation 33 convolves the collagen fiber SEF with the probability distribution function for collagen engagement strain to define the collagen SEF (Ψcoll) as a function of strain. Ψaniso is then computed using the invariants (I4 and I4′) to account for collagen helical pitch in the same manner as used by Holzapfel (75). Defined in this way, Zulliger’s SEF has seven material parameters. felast and fcoll were experimentally measured and ccoll was set to 200 MPa and imposed on the model, leaving four independent material parameters to solve for simultaneously.
The SEF proposed by Zulliger advanced the idea of modeling arterial tissue as an isotropic ground matrix with anisotropic collagen fiber-reinforcement by including a collagen engagement probability distribution function. This distribution function more accurately represents the physiologic structure of arterial collagen and therefore results in a more structure-based constitutive model. By including measureable structural elements, this SEF has greater potential for being predictive of how changes in artery structure, resulting from age or disease, will affect vessel wall properties. However, as of this writing, the predictive capabilities of this model have not been tested.
In 2006, the research group who developed Holzapfel’s fiber-reinforced artery SEF, revised their original model to incorporate an additional scalar structural parameter that characterized how collagen orientation was dispersed within the vessel wall (57). Evidence suggests that while medial collagen is oriented nearly circumferentially adventitial collagen orientation is much more dispersed. By incorporating collagen dispersion, Holzapfel’s model was better able to represent arterial structure and without the large computational overhead of solving the convolution integral in Zulliger’s model, this new SEF is more easily adapted for use in finite element modeling of arterial tissues. However, like the Zulliger model, the predictive capabilities of this model and a detailed study of the physiologic validity of the structural material parameters have yet to be independently tested and verified.
Although the newest generation of pseudoelastic SEF arterial models incorporate a substantial degree of structural information, there is a lack of detailed microstructural information available for model development and validation. In particular, there is a need for more quantitative information on the distribution of collagen fiber orientation and tortuosity throughout the thickness of the vessel wall. Further more, detailed studies are needed to determine how applied loads and concomitant tissue deformation affects collagen fiber structure in vivo. Finally, it is important to validate that the changes in collagen, elastin, and SMC geometry and structure resulting from PH vascular remodeling correspond with changes in the model parameters that are supposed to reflect these structural properties. Only then will these models be truly predictive of the disease state and will offer valuable insight into how anatomical structure and physiological changes in arterial components directly influence bulk artery material properties and vascular compliance.
Computational mechanics modeling approaches
The detailed modeling of blood flow alone or coupled to arterial motion has been accomplished through the use of computational mechanics, primarily the finite element method (FEM). Such methods first require the discretization of fluid and/or solid systems of arbitrary and complex geometries into discrete parcels of material; this abstraction then allows for the computational solution of the equations governing the behavior of these systems. The application of computational mechanics to vascular dynamics is a relatively new—no mention of it appears in previous AJP Handbook of Physiology articles covering the vasculature. Below we briefly and broadly review computational approaches; more detail can be found elsewhere (163, 175, 176, 177).
The number of dimensions simulated by FEM can vary the detail of the solution. The simplest, one dimensional (1D) simulations track flow only along the longitudinal dimension of a vascular tree. They offer rapid runtime, require little data to create a mesh (model domain) or create input/outlet conditions, provide better accuracy compared to a transmission line model, and can consider wave propagation effects (47,48, 78, 127, 154, 193). It has been proposed that their accuracy may be sufficient for surgical planning (161), in that gross estimates of pressure and flow distributions can provide insight into outcomes. 1D simulations are too simple, however, to examine local hemodynamic variations; wall shear stress, for example, cannot be predicted. In comparison, three dimensional (3D) fluid models can provide far more flow and pressure detail than is available from any clinical measurement modality. They frequently utilize patient-specific meshes that are reconstructed from image data (177), use MRI-based 3D inlet or exit boundary conditions, have longer runtimes, and offer unparalleled hemodynamic detail. A preponderance of recent 3D modeling work on the pulmonary circuit has focused on optimizing vascular flow geometries associated with the Fontan surgical procedure (92, 200) or understanding the impact of respiration and exercise (111, 172) or ventricular assist devices (98, 135) on this vascular circuit. An ongoing problem with either type of simulation is a dearth of comparisons between simulations and real clinical measurements; without such validation, simulation results cannot be used with confidence in diagnosis or prognosis.
Both 1D and 3D simulations can also incorporate fluid-structure interaction (FSI), in which the vascular wall is additionally modeled and allowed to deform. Given the links between vascular stiffening and disease progression, such modeling capability seems vital for these advanced methods to be clinically relevant. Interestingly, even if wall motion does not strongly affect hemodynamics, it is becoming more appreciated that pathological response may be dependent on wall mechanics (e.g., vessel stress or strain). The finite deformation mechanics models described above provide support for FSI efforts; however, until patient-specific, or better, disease-specific material properties can be obtained for use in simulations, their results may not be worth the extra effort (163).
Computational models can currently only represent specific segments of the vasculature—thus the upstream and downstream sides of vascular models require special treatment. Such treatments are known as boundary conditions; early efforts (and indeed, some current work) utilized simple sinusoidal or clinically obtained waveforms for inlet flow, and constant pressure conditions at outlets. Clinically obtained inlet waveforms only represent a particular heart condition and thus are not useful in predictive modeling, while the outlet condition—constant pressure—is clearly quite different from true of distal vascular conditions as represented by impedance. Improvements to these approaches come in the form of coupling simple models—such as lumped models—to the more advanced primary computational model. Mathematical methods to couple such models of the heart and vasculature to more advanced models are not especially new (47, 138, 189, 190) but appear underused in the literature, likely due to their limited availability in commercial codes. Recently, several groups have applied these methods; on the pulmonary side, Spilker et al. (159) coupled a 1D proximal pulmonary vessel model to multiple distributed (i.e., transmission line) models of the distal lung circulation. Through iterating on the distal vessel diameters in the distal models, the proximal model results were brought into good correspondence with measurements of pressure and flow to the right and left lungs. The same computational techniques also allow for combining heart (152) and vascular models, thus simulating ventricular-vascular coupling, although this has currently only been performed in models of the systemic circulation (49, 94).
Concluding Thoughts
This article is meant to provide some perspectives on PVS, with particular focus on the emerging importance of stiffness as a disease diagnostic and methods to evaluate stiffness via clinical, experimental, and computational studies. Nearly 50 years ago, McDonald, Milnor, Patel, and others determined that proximal vascular stiffening was innate to the PH disease process, but this knowledge was not further explored because of difficulties in measurement and lack of any clear benefits that such measurements would provide. Earlier studies of the systemic circulation established connections between stiffening and outcomes, between flow pulsatility and cellular signaling derangement, and even between stiffening and inflammation (115); the first two of these have now also been demonstrated on the pulmonary side. New noninvasive and minimally invasive measurement modalities are emerging that will make routine stiffness measurement possible in the pulmonary circulation; increased appreciation of the importance of stiffening should encourage their use. Together with PVR, the traditional measure of PH severity, stiffness measurement will improve basic science and clinical studies of the disease, with better outcomes the result.
Acknowledgments
This work was supported in part by grants from the National Heart, Lung, and Blood Institute (T32-HL072738, K24-HL081506, K25-HL094749, and P50-HL084923) and from the American Heart Association (09SDG2260194).
References
- 1.Aaron BB, Gosline JM. Elastin as a random-network elastomer—a mechanical and optical analysis of single elastin fibers. Biopolymers. 1981;20(6):1247–1260. [Google Scholar]
- 2.Arribas SM, Hinek A, Gonzalez MC. Elastic fibres and vascular structure in hypertension. Pharmacol Ther. 2006;111(3):771–791. doi: 10.1016/j.pharmthera.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Avolio A. Input impedance of distributed arterial structures as used in investigations of underlying concepts in arterial haemodynamics. Med Biol Eng Comput. 2009;47(2):143–151. doi: 10.1007/s11517-008-0413-0. [DOI] [PubMed] [Google Scholar]
- 4.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, Torbicki A. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S55–S66. doi: 10.1016/j.jacc.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 5.Balzer DT, Kort HW, Day RW, Corneli HM, Kovalchin JP, Cannon BC, Kaine SF, Ivy DD, Webber SA, Rothman A, Ross RD, Aggarwal S, Takahashi M, Waldman JD. Inhaled nitric oxide as a preoperative test (INOP Test I) - The INOP Test Study Group. Circulation. 2002;106(13 Suppl S):I76–I81. [PubMed] [Google Scholar]
- 6.Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12):40S–47S. doi: 10.1016/j.jacc.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 7.Bayer IM, Adamson SL, Langille BL. Atrophic remodeling of the artery-cuffed artery. Arterioscler Thromb Vasc Biol. 1999;19(6):1499–1505. doi: 10.1161/01.atv.19.6.1499. [DOI] [PubMed] [Google Scholar]
- 8.Bergel DH. The dynamic elastic properties of the arterial wall. J Physiol. 1961a;156(3):458–469. doi: 10.1113/jphysiol.1961.sp006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergel DH. The static elastic properties of the arterial wall. J Physiol. 1961b;156(3):445–457. doi: 10.1113/jphysiol.1961.sp006686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergel DH, Milnor WR. Pulmonary vascular impedance in the dog. Circ Res. 1965;16(5):401–405. doi: 10.1161/01.res.16.5.401. [DOI] [PubMed] [Google Scholar]
- 11.Berger RMF, Cromme-Dijkhuis AH, Hop WCJ, Kruit MN, Hess J. Pulmonary arterial wall distensibility assessed by intravascular ultrasound in children with congenital heart disease. Chest. 2002;122(2):549–557. doi: 10.1378/chest.122.2.549. [DOI] [PubMed] [Google Scholar]
- 12.Bezie Y, Lacolley P, Laurent S, Gabella G. Connection of smooth muscle cells to elastic lamellae in aorta of spontaneously hypertensive rats. Hypertension. 1998;32(1):166–169. doi: 10.1161/01.hyp.32.1.166. [DOI] [PubMed] [Google Scholar]
- 13.Bezie Y, Lamaziere JMD, Laurent S, Challande P, Cunha RS, Bonnet J, Lacolley P. Fibronectin expression and aortic wall elastic modulus in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 1998;18(7):1027–1034. doi: 10.1161/01.atv.18.7.1027. [DOI] [PubMed] [Google Scholar]
- 14.Bia D, Armentano R, Craiem D, Grignola J, Gines F, Simon A, Levenson J. Smooth muscle role on pulmonary arterial function during acute pulmonary hypertension in sheep. Acta Physiol Scand. 2004;181(3):359–366. doi: 10.1111/j.1365-201X.2004.01294.x. [DOI] [PubMed] [Google Scholar]
- 15.Bia D, Armentano R, Grignola JC, Craiem D, Zocalo YA, Gines FF, Levenson J. The vascular smooth muscle of the great arteries: Local control site of arterial buffering function? Rev Esp Cardiol. 2003;56(12):1202–1209. doi: 10.1016/s0300-8932(03)77039-0. [DOI] [PubMed] [Google Scholar]
- 16.Boresi AP, Chong KP. Elasticity in Engineering Mechanics. Wiley-Interscience; New York, NY: 1999. [Google Scholar]
- 17.Borges AC, Wensel R, Opitz C, Bauer U, Baumann G, Kleber FX. Relationship between haemodynamics and morphology in pulmonary hypertension. A quantitative intravascular ultrasound study. Eur Heart J. 1997;18(12):1988–1994. doi: 10.1093/oxfordjournals.eurheartj.a015210. [DOI] [PubMed] [Google Scholar]
- 18.Boumaza S, Arribas SM, Osborne-Pellegrin M, McGrath JC, Laurent S, Lacolley P, Challande P. Fenestrations of the carotid internal elastic lamina and structural adaptation in stroke-prone spontaneously hypertensive rats. Hypertension. 2001;37(4):1101–1107. doi: 10.1161/01.hyp.37.4.1101. [DOI] [PubMed] [Google Scholar]
- 19.Bressollette E, Dupuis J, Bonan R, Doucet S, Cernacek P, Tardif JC. Intravascular ultrasound assessment of pulmonary vascular disease in patients with pulmonary hypertension. Chest. 2001;120(3):809–815. doi: 10.1378/chest.120.3.809. [DOI] [PubMed] [Google Scholar]
- 20.Brown DJ. Input impedance and reflection coefficient in fractal-like models of asymmetrically branching compliant tubes. IEEE Trans Biomed Eng. 1996;43(7):715–722. doi: 10.1109/10.503179. [DOI] [PubMed] [Google Scholar]
- 21.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2):159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 22.Burattini R, Gnudi G. Computer identification of models for the arterial tree input impedance: Comparison between two new simple models and first experimental results. Med Biol Eng Comput. 1982;20:134–144. doi: 10.1007/BF02441348. [DOI] [PubMed] [Google Scholar]
- 23.Cameron J. Estimation of arterial mechanics in clinical practice and as a research technique. Clin Exp Pharmacol Physiol. 1999;26(4):285–294. doi: 10.1046/j.1440-1681.1999.03032.x. [DOI] [PubMed] [Google Scholar]
- 24.Caro CG, McDonald DA. The relation of the pulsatile pressure and flow in the pulmonary vascular bed. J Physiol. 1961;157(3):426–453. doi: 10.1113/jphysiol.1961.sp006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chamiot Clerc P, Renaud JF, Blacher J, Legrand M, Samuel JL, Levy BI, Sassard J, Safar ME. Collagen I and III and mechanical properties of conduit arteries in rats with genetic hypertension. J Vasc Res. 1999;36(2):139–146. doi: 10.1159/000025637. [DOI] [PubMed] [Google Scholar]
- 26.Chesler NC, Thompson-Figueroa J, Millburne K. Measurements of mouse pulmonary artery biomechanics. J Biomech Eng. 2004;126(2):309–314. doi: 10.1115/1.1695578. [DOI] [PubMed] [Google Scholar]
- 27.Chuong CJ, Fung YC. 3-Dimensional stress-distribution in arteries. J Biomech Eng Trans. 1983;105(3):268–274. doi: 10.1115/1.3138417. [DOI] [PubMed] [Google Scholar]
- 28.Chuong CJ, Fung YC. On residual stresses in arteries. J Biomech Eng. 1986;108:189. doi: 10.1115/1.3138600. [DOI] [PubMed] [Google Scholar]
- 29.Clark JM, Glagov S. Transmural organization of the arterial media. The lamellar unit revisited. Arterioscler Thromb Vasc Biol. 1985;5(1):19. doi: 10.1161/01.atv.5.1.19. [DOI] [PubMed] [Google Scholar]
- 30.Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, Tuder RM. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155:411–419. doi: 10.1016/S0002-9440(10)65137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Godring RM, Groves BM, Kernis JT. Survival in patients with primary pulmonary hypertension: Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- 32.De Canniere D, Stefanidis C, Brimioulle S, Naeije R. Effects of a chronic aortopulmonary shunt on pulmonary hemodynamics in piglets. J Appl Physiol. 1994;77(4):1591–1596. doi: 10.1152/jappl.1994.77.4.1591. [DOI] [PubMed] [Google Scholar]
- 33.Díez J. Arterial stiffness and extracellular matrix. Adv Cardiol. 2007;44:76–95. doi: 10.1159/000096722. [DOI] [PubMed] [Google Scholar]
- 34.Dingemanns KP, Wagenvoort CA. Pulmonary arteries and veins in experimental hypoxia. Am J Pathol. 1978;93:353–368. [PMC free article] [PubMed] [Google Scholar]
- 35.Dingemans KP, Teeling P, Lagendijk JH, Becker AE. Extracellular matrix of the human aortic media: An ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec. 2000;258(1):1–14. doi: 10.1002/(SICI)1097-0185(20000101)258:1<1::AID-AR1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Dobrin PB. Handbook of Physiology. The Cardiovascular System. Peripheral Circulation and Organ Blood Flow. III. Bethesda, MD: American Physiological Society; 1983. Vascular mechanics; pp. 65–102. sect. 2. chapt. 3. [Google Scholar]
- 37.Domanski MJ, Mitchell GF, Norman JE, Exner DV, Pitt B, Pfeffer MA. Independent prognostic information provided sphygmomanometrically determined pulse pressure and mean arterial pressure in patients with left ventricular dysfunction. J Am Coll Cardiol. 1999;33(4):951–958. doi: 10.1016/s0735-1097(98)00679-2. [DOI] [PubMed] [Google Scholar]
- 38.Drexler E, Bischoff JE, Slifka AJ, McCowan CN, Quinn TP, Shandas R, Ivy DD, Stenmark K. Stiffening of the extrapulmonary arteries from rats in chronic hypoxic pulmonary hypertension. J Res Natl Inst Stand Technol. 2008;113:239–249. doi: 10.6028/jres.113.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drexler ES, McCowan C, Wright J, Slifka AJ, Ivy DD, Shandas R. Comparison of strength properties of normotensive and hypertensive rat pulmonary arteries. Biomed Sci Instrum. 2004;40:297–302. [PubMed] [Google Scholar]
- 40.Durmowicz AG, Orton EC, Stenmark KR. Progressive loss of vasodilator responsive component of pulmonary-hypertension in neonatal calves exposed to 4,570 M. Am J Physiol. 1993;265(6):H2175–H2183. doi: 10.1152/ajpheart.1993.265.6.H2175. [DOI] [PubMed] [Google Scholar]
- 41.Dyer K, Lanning CJ, Das B, Lee PF, Ivy DD, Valdes-Cruz L, Shandas R. Noninvasive Doppler tissue measurement of pulmonary artery compliance in children with pulmonary hypertension. J Am Soc Echocardiogr. 2006;19(4):403–412. doi: 10.1016/j.echo.2005.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engelberg J, DuBois AB. Mechanics of pulmonary circulation in isolated rabbit lungs. Am J Physiol. 1959;196(2):401–414. doi: 10.1152/ajplegacy.1959.196.2.401. [DOI] [PubMed] [Google Scholar]
- 43.Estrada KD, Chesler NC. Collagen-related gene and protein expression changes in the lung in response to chronic hypoxia. Biomech Model Mechanobiol. 2009;8(4):263–272. doi: 10.1007/s10237-008-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L656–L664. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 45.Finkelstein SM, Collins VR, Cohn JN. Arterial vascular compliance response to vasodilators by Fourier and pulse contour analysis. Hypertension. 1988;12:380–387. doi: 10.1161/01.hyp.12.4.380. [DOI] [PubMed] [Google Scholar]
- 46.Finlay HM, McCullough L, Canham PB. Three-dimensional collagen organization of human brain arteries at different transmural pressures. J Vasc Res. 1995;32(5):301–312. doi: 10.1159/000159104. [DOI] [PubMed] [Google Scholar]
- 47.Formaggia L, Gerbeau JR, Nobile F, Quarteroni A. On the coupling of 3D and 1D Navier-Stokes equations for flow problems in compliant vessels. Comp Methods Appl Mech Engrg. 2001;191(6–7):561–582. [Google Scholar]
- 48.Formaggia L, Lamponi D, Quarteroni A. One-dimensional models for blood flow in arteries. J Eng Math. 2003;47(3/4):251–276. [Google Scholar]
- 49.Formaggia L, Lamponi D, Tuveri M, Veneziani A. Numerical modeling of 1D arterial networks coupled with a lumped parameters description of the heart. Comput Methods Biomech Biomed Engin. 2006;9(5):273–288. doi: 10.1080/10255840600857767. [DOI] [PubMed] [Google Scholar]
- 50.Fourie PR, Coetzee AR, Bolliger CT. Pulmonary artery compliance: Its role in right ventricular-arterial coupling. Cardiovasc Res. 1992;26(9):839–844. doi: 10.1093/cvr/26.9.839. [DOI] [PubMed] [Google Scholar]
- 51.Frank O. Die grundform des arteriellen pulses. Erste Abhandlung. mathematische analyse. Z Biol. 1899;37:483–526. [Google Scholar]
- 52.Frasher WG, Sobin SS. Distensible behavior of pulmonary artery. Am J Physiol. 1960;199(3):472–480. doi: 10.1152/ajplegacy.1960.199.3.472. [DOI] [PubMed] [Google Scholar]
- 53.Fridez P, Makino A, Kakoi D, Miyazaki H, Meister JJ, Hayashi K, Stergiopulos N. Adaptation of conduit artery vascular smooth muscle tone to induced hypertension. Ann Biomed Eng. 2002;30(7):905–916. doi: 10.1114/1.1507326. [DOI] [PubMed] [Google Scholar]
- 54.Fung YC, Fronek K, Patitucci P. Pseudoelasticity of arteries and the choice of its mathematical expression. Am J Physiol. 1979;237(5):H620–H631. doi: 10.1152/ajpheart.1979.237.5.H620. [DOI] [PubMed] [Google Scholar]
- 55.Gan CT-J, Lankhaar JW, Weterhof N, Marcus JT, Becker A, Twisk JWR, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Noinvasively assessed pulmonary artery stiffness predicts mortality in pulmonary arterial hypertension. Chest. 2007;32(6):1906–1914. doi: 10.1378/chest.07-1246. [DOI] [PubMed] [Google Scholar]
- 56.Gan RZ, Yen RT. Vascular impedance analysis in dog lung with detailed morphometric and elasticity data. J Appl Physiol. 1994;77(2):706–717. doi: 10.1152/jappl.1994.77.2.706. [DOI] [PubMed] [Google Scholar]
- 57.Gasser TC, Ogden RW, Holzapfel GA. Hyperelastic modelling of arterial layers with distributed collagen fibre orientations. J R Soc Interface. 2006;3(6):15–35. doi: 10.1098/rsif.2005.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glasser SP, Arnett DK. Vascular stiffness and the “chicken-or-the-egg” question. Hypertension. 2008;51(2):177–178. doi: 10.1161/HYPERTENSIONAHA.107.098830. [DOI] [PubMed] [Google Scholar]
- 59.Gow BS. Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle. II. Bethesda, MD: American Physiological Society; 1980. Circulatory correlates: Vascular impedance, resistance, and capacity; pp. 353–408. sect. 2. chapt. 14. [Google Scholar]
- 60.Greenfield JC, Griggs DM. Relation between pressure and diameter in main pulmonary artery of man. J Appl Physiol. 1963;18(3):557–559. doi: 10.1152/jappl.1963.18.3.557. [DOI] [PubMed] [Google Scholar]
- 61.Grignola JC, Bia D, Ginés F, Armentano RL. Acute pulmonary hypertension: Protective role of vascular smooth muscle activation. Rev Esp Cardiol. 2003;56(11):1077–1084. doi: 10.1016/s0300-8932(03)77018-3. [DOI] [PubMed] [Google Scholar]
- 62.Grignola JC, Ginés F, Bia D, et al. Improved right ventricular-vascular coupling during active pulmonary hypertension. Int J Cardiol. 2007;115(2):171–182. doi: 10.1016/j.ijcard.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 63.Grover RF, Wagner WW, McMurtry IF, Reeves JT. Handbook of Physiology. The Cardiovascular System. Vascular Smooth Muscle. III. Bethesda, MD: American Physiological Society; 1980. Pulmonary circulation; pp. 103–136. sect. 2. chapt 4. [Google Scholar]
- 64.Ha B, Lucas CL, Henry GW, Frantz EG, Ferreiro JI, Wilcox BR. Effects of chronically elevated pulmonary arterial pressure and flow on right ventricular afterload. Am J Physiol. 1994;267(1 Pt 2):H155–H165. doi: 10.1152/ajpheart.1994.267.1.H155. [DOI] [PubMed] [Google Scholar]
- 65.Hall JB, Schmidt GA, Wood LDH, Taylor CD. Principles of Critical Care. chapt. 26 New York: McGraw-Hill; 2005. [Google Scholar]
- 66.Hamilton PK, Lockhart CJ, Quinn CE, McVeigh GE. Arterial stiffness: Clinical relevance, measurement and treatment. Clin Sci (Lond) 2007;113(4):157–170. doi: 10.1042/CS20070080. [DOI] [PubMed] [Google Scholar]
- 67.Haneda T, Nakajima T, Shirato K, Onodera S, Takishima T. Effects of oxygen breathing on pulmonary vascular input impedance in patients with pulmonary hypertension. Chest. 1983;83(3):520–527. doi: 10.1378/chest.83.3.520. [DOI] [PubMed] [Google Scholar]
- 68.Harris P, Heath D. Ultrastructure of plexogenic arteriopathy. In: Harris P, Heath D, editors. The Human Pulmonary Circulation. Its Form and Function in Health and Disease. London: Churchill Livingston; 1986. pp. 262–272. [Google Scholar]
- 69.Hay ED. Cell Biology of Extracellular Matrix. Springer; New York: 1981. [Google Scholar]
- 70.Herve P, Musset D, Simonneau G, Wagner W, Jr, Duroux P. Almitrine decreases the distensibility of the large pulmonary arteries in man. Chest. 1989;96(3):572–577. doi: 10.1378/chest.96.3.572. [DOI] [PubMed] [Google Scholar]
- 71.Hirata K, Kawakami M, O’Rourke MF. Pulse wave analysis and pulse wave velocity: A review of blood pressure interpretation 100 years after Korotkov. Circ J. 2006;70(10):1231–1239. doi: 10.1253/circj.70.1231. [DOI] [PubMed] [Google Scholar]
- 72.Hislop A, Reid L. Changes in the pulmonary arteries of the rat during recovery from hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1977;58:653–662. [PMC free article] [PubMed] [Google Scholar]
- 73.Hislop A, Reid L. New findings in pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. Br J Exp Pathol. 1976;57:542–554. [PMC free article] [PubMed] [Google Scholar]
- 74.Holzapfel GA. Nonlinear Solid Mechanics: A Continuum Approach for Engineering. Wiley; West Sussex, UK: 2000. [Google Scholar]
- 75.Holzapfel GA, Gasser TC, Ogden RW. A new constitutive framework for arterial wall mechanics and a comparative study of material models. Journal of Elasticity. 2000;61(1–3):1–48. [Google Scholar]
- 76.Huang W, Tian Y, Gao J, Yen RT. Comparison of theory and experiment in pulsatile flow in cat lung. Ann Biomed Eng. 1998;26(5):812–820. doi: 10.1114/1.107. [DOI] [PubMed] [Google Scholar]
- 77.Huez S, Brimioulle S, Naeije R, Vachiéry JL. Feasibility of routine pulmonary artery impedance measurements in pulmonary hypertension. Chest. 2004;125(6):2121–2128. doi: 10.1378/chest.125.6.2121. [DOI] [PubMed] [Google Scholar]
- 78.Hughes TJR, Lubliner J. On the one-dimensional theory of blood flow in the larger vessels. Math Biosci. 1973;18(1/2):161–170. [Google Scholar]
- 79.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12):13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 80.Humphrey JD. Mechanics of the arterial wall: Review and directions. Crit Rev Biomed Eng. 1995;23(1–2):1–162. [PubMed] [Google Scholar]
- 81.Humphrey JD. Review paper. Continuum biomechanics of soft biological tissues. Proc Math Phys Eng Sci. 2003;459(2029):3–46. [Google Scholar]
- 82.Hunter KS, Albietz JA, Lee PF, Lanning CJ, Lammers SR, Hofmeister SH, Kao PH, Qi H, Stenmark KR, Shandas R. In-vivo measurement of proximal pulmonary artery elastic modulus in the neonatal calf model of pulmonary hypertension: Development and ex-vivo validation. J Appl Physiol. 2010;108(4):968–975. doi: 10.1152/japplphysiol.01173.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hunter KS, Lanning CJ, Kirby KS, Ivy DD, Shandas R. In-vivo pulmonary vascular stiffness obtained from color M-mode tissue doppler imaging and pressure measurements predicts clinical outcomes better than indexed pulmonary vascular resistance in pediatric patients with pulmonary arterial hypertension [Abstract 4388] Circulation. 2008;118(18):S879. [Google Scholar]
- 84.Hunter KS, Lee PF, Lanning CJ, Ivy DD, Kirby KS, Claussen LR, Chan KC, Shandas R. Pulmonary vascular input impedance is a combined measure of pulmonary vascular resistance and stiffness and predicts clinical outcomes better than PVR alone in pediatric patients with pulmonary hypertension. Am Heart J. 2008;155(1):166–174. doi: 10.1016/j.ahj.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLough-lin P. Inhibition of rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res. 2005;97(2):185–191. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 86.Intengan HD, Schiffrin EL. Structure and mechanical properties of resistance arteries in hypertension—Role of adhesion molecules and extracellular matrix determinants. Hypertension. 2000;36(3):312–318. doi: 10.1161/01.hyp.36.3.312. [DOI] [PubMed] [Google Scholar]
- 87.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: Roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38(3 Pt 2):581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 88.Intengan HD, Thibault G, Li JS, Schiffrin EL. Resistance artery mechanics, structure, and extracellular components in spontaneously hypertensive rats—Effects of angiotensin receptor antagonism and converting enzyme inhibition. Circulation. 1999;100(22):2267–2275. doi: 10.1161/01.cir.100.22.2267. [DOI] [PubMed] [Google Scholar]
- 89.Jacob MP. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed Pharmacother. 2003;57(5–6):195–202. doi: 10.1016/s0753-3322(03)00065-9. [DOI] [PubMed] [Google Scholar]
- 90.Jacob MP, Badier-Commander C, Fontaine V, Benazzoug Y, Feldman L, Michel JB. Extracellular matrix remodeling in the vascular wall. Pathol Biol. 2001;49(4):326–332. doi: 10.1016/s0369-8114(01)00151-1. [DOI] [PubMed] [Google Scholar]
- 91.Jones R, Reid L. Vascular Remodeling in Clinical and Experimental Pulmonary Hypertensions. London: Portland Press; 1995. [Google Scholar]
- 92.Khunatorn Y, Mahalingam S, DeGroff CG, Shandas R. Influence of connection geometry and SVC-IVC flow rate ratio on flow structures within the total cavopulmonary connection: A numerical study. J Biomech Eng. 2002;124(4):364–377. doi: 10.1115/1.1487880. [DOI] [PubMed] [Google Scholar]
- 93.Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002;115(14):2817–2828. doi: 10.1242/jcs.115.14.2817. [DOI] [PubMed] [Google Scholar]
- 94.Kim HJ, Vignon-Clementel IE, Figueroa CA, LaDisa JF, Jansen KE, Feinstein JA, Taylor CA. On coupling a lumped parameter heart model and a three-dimensional finite element aorta model. Ann Biomed Eng. 2009;37(11):2153–2169. doi: 10.1007/s10439-009-9760-8. [DOI] [PubMed] [Google Scholar]
- 95.Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodeling in mouse pulmonary arteries with hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2005;288(3):H1209–H1217. doi: 10.1152/ajpheart.01129.2003. [DOI] [PubMed] [Google Scholar]
- 96.Kussmaul WG, Altschuler JA, Herrmann HC, Laskey WK. Effects of pacing tachycardia and balloon valvuloplasty on pulmonary artery impedance and hydraulic power in mitral stenosis. Circulation. 1992;86(6):1770–1779. doi: 10.1161/01.cir.86.6.1770. [DOI] [PubMed] [Google Scholar]
- 97.Kussmaul WG, Altschuler JA, Matthai WH, Laskey WK. Right ventricular-vascular interaction in congestive heart failure/importance of low-frequency impedance. Circulation. 1993;88(3):1010–1015. doi: 10.1161/01.cir.88.3.1010. [DOI] [PubMed] [Google Scholar]
- 98.Lacour-Gayet FG, Lanning CJ, Stoica S, Wang R, Rech BA, Goldberg S, Shandas R. An artificial right ventricle for failing fontan: In vitro and computational study. Ann Thorac Surg. 2009;88(1):170–176. doi: 10.1016/j.athoracsur.2009.03.091. [DOI] [PubMed] [Google Scholar]
- 99.Lammers SR, Kao P, Qi HJ, Hunter KS, Lanning CJ, Albietz JA, Hofmeister SE, Mecham R, Stenmark KR, Shandas R. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol. 2008;295(4):H1451–H1459. doi: 10.1152/ajpheart.00127.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lankhaar JW, Westerhof N, Faes TJ, Marques KM, Marcus JT, Postmus PE, Vonk-Noordegraaf A. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2006;291(4):H1731–H1737. doi: 10.1152/ajpheart.00336.2006. [DOI] [PubMed] [Google Scholar]
- 101.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37(5):1236–1241. doi: 10.1161/01.hyp.37.5.1236. [DOI] [PubMed] [Google Scholar]
- 102.Lee HW, Karam J, Hussain B, Winer N. Vascular compliance in hypertension: Therapeutic implications. Curr Diab Rep. 2008;8(3):208–213. doi: 10.1007/s11892-008-0036-8. [DOI] [PubMed] [Google Scholar]
- 103.Leibovitch ER. Hypertension 2008, refining our treatment. Geriatrics. 2008;63(10):14–15. 17–20. [PubMed] [Google Scholar]
- 104.Li M, Scott DE, Shandas R, Stenmark KR, Tan W. High pulsatility flow induces adhesion molecule and cytokine mRNA expression in distal pulmonary artery endothelial cells. Ann Biomed Eng. 2009;37(6):1082–1092. doi: 10.1007/s10439-009-9684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li M, Stenmark KR, Shandas R, Tan W. Effects of pathological flow on pulmonary artery endothelial production of vasoactive mediators and growth factors. J Vasc Res. 2009;46(6):561–571. doi: 10.1159/000226224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liao D, Arnett DK, Tyroler HA, Riley WA, Chambless LE, Szklo M, Heiss G. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension. 1999;34(2):201–206. doi: 10.1161/01.hyp.34.2.201. [DOI] [PubMed] [Google Scholar]
- 107.Lucas CL, Wilcox BR, Ha B, Henry GW. Comparison of time domain algorithms for estimating aortic characteristic impedance in humans. IEEE Trans Biomed Eng. 1988;35:62–68. doi: 10.1109/10.1337. [DOI] [PubMed] [Google Scholar]
- 108.Madden JA, Keller PA, Effros RM, Seavitte C, Choy JS, Hacker AD. Responses to pressure and vasoactive agents by isolated pulmonary arteries from monocrotaline-treated rats. J Appl Physiol. 1994;76(4):1589–1593. doi: 10.1152/jappl.1994.76.4.1589. [DOI] [PubMed] [Google Scholar]
- 109.Mahapatra S, Nishimura RA, Oh JK, McGoon MD. The prognostic value of pulmonary vascular capacitance determined by Doppler echocardiography in patients with pulmonary arterial hypertension. J Am Soc Echocardiogr. 2006;19(8):1045–1050. doi: 10.1016/j.echo.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 110.Mahapatra S, Nishimura RA, Sorajja P, Cha S, McGoon MD. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Coll Cardiol. 2006;47(4):799–803. doi: 10.1016/j.jacc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 111.Marsden AL, Vignon-Clementel IE, Chan FP, Feinstein JA, Taylor CA. Effects of exercise and respiration on hemodynamic efficiency in CFD simulations of the total cavopulmonary connection. Ann Biomed Eng. 2007;35(2):250–263. doi: 10.1007/s10439-006-9224-3. [DOI] [PubMed] [Google Scholar]
- 112.Maruyama K, Ye CL, Woo M, Venkatacharya H, Lines LD, Silver MM, Rabinovitch M. Chronic hypoxic pulmonary-hypertension in rats and increased elastolytic activity. Am J Physiol. 1991;261(6):H1716–H1726. doi: 10.1152/ajpheart.1991.261.6.H1716. [DOI] [PubMed] [Google Scholar]
- 113.Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: Role of ion channels. J Appl Physiol. 2005;98:415–420. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- 114.McDonald DA. Blood Flow in Arteries. London: Edward Arnold; 1974. [Google Scholar]
- 115.McEniery CM, Wilkinson IB. Large artery stiffness and inflammation. J Hum Hypertens. 2005;19(7):507–509. doi: 10.1038/sj.jhh.1001814. [DOI] [PubMed] [Google Scholar]
- 116.Meyrick B, Reid L. Hypoxia-induced structural changes in the media and adventitia of the rat hilar pulmonary artery and their regression. Am J Pathol. 1980;100:151–178. [PMC free article] [PubMed] [Google Scholar]
- 117.Meyrick B, Reid L. Pulmonary hypertension. Anatomic and physiologic correlates. Clin Chest Med. 1983;4:199–217. [PubMed] [Google Scholar]
- 118.Milnor WR. Hemodynamics. Williams & Wilkins; Baltimore, MD: 1989. [Google Scholar]
- 119.Milnor WR, Bergel DH, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res. 1966;19(3):467–480. doi: 10.1161/01.res.19.3.467. [DOI] [PubMed] [Google Scholar]
- 120.Milnor WR, Conti CR, Lewis KB, et al. Pulmonary arterial pulse wave velocity and impedance in man. Circ Res. 1969;25(6):637–649. doi: 10.1161/01.res.25.6.637. [DOI] [PubMed] [Google Scholar]
- 121.Milnor WR, Jose AD, McGaff CJ. Pulmonary vascular volume, resistance, and compliance in man. Circulation. 1960;22:130–137. doi: 10.1161/01.cir.22.1.130. [DOI] [PubMed] [Google Scholar]
- 122.Mitchell GF, Moyé LA, Braunwald E, Rouleau JL, Bernstein V, Geltman EM, Flaker GC, Pfeffer MA. Sphygmomanometrically determined pulse pressure is a powerful independent predictor of recurrent events after myocardial infarction in patients with impaired left ventricular function. Circulation. 1997;96(12):4254–4260. doi: 10.1161/01.cir.96.12.4254. [DOI] [PubMed] [Google Scholar]
- 123.Moore NR, Scott JP, Flower CD, Higenbottam TW. The relationship between pulmonary artery pressure and pulmonary artery diameter in pulmonary hypertension. Clin Radiol. 1988;39(5):486–489. doi: 10.1016/s0009-9260(88)80205-8. [DOI] [PubMed] [Google Scholar]
- 124.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98:390–403. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- 125.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry IF, Oka M. Inhaled rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171(5):494–499. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 126.Olsen MH, Christensen MK, Wachtell K, Tuxen C, Fossum E, Bang LE, Wiinberg N, Devereux RB, Kjeldsen SE, Hildebrandt P, Rokkedal J, Ibsen H. Markers of collagen synthesis is related to blood pressure and vascular hypertrophy: A life substudy. J Hum Hypertens. 2005;19(4):301–307. doi: 10.1038/sj.jhh.1001819. [DOI] [PubMed] [Google Scholar]
- 127.Olufsen MS, Peskin CS, Kim WY, Pedersen EM, Nadim A, Larsen J. Numerical simulation and experimental validation of blood flow in arteries with structured-tree outflow conditions. Ann Biomed Eng. 2000;28(11):1281–1299. doi: 10.1114/1.1326031. [DOI] [PubMed] [Google Scholar]
- 128.Olufsen MS. Structured tree outflow condition for blood flow in larger systemic arteries. Am J Physiol. 1999;276(1 Pt 2):H257–H268. doi: 10.1152/ajpheart.1999.276.1.H257. [DOI] [PubMed] [Google Scholar]
- 129.O’Rourke MF, Avolio AP. Arterial transfer functions: Background, applications and reservations. J Hypertens. 2008;26(1):8–10. doi: 10.1097/HJH.0b013e3282f0ca1a. [DOI] [PubMed] [Google Scholar]
- 130.O’Rourke MF, Avolio AP. Pulsatile flow and pressure in human systemic arteries. Studies in man and in a multibranched model of the human systemic arterial tree. Circ Res. 1980;46(3):363–372. doi: 10.1161/01.res.46.3.363. [DOI] [PubMed] [Google Scholar]
- 131.O’Rourke MF. Vascular mechanics in the clinic. J Biomech. 2003;36(5):623–630. doi: 10.1016/s0021-9290(02)00440-2. [DOI] [PubMed] [Google Scholar]
- 132.Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, Lemarchand P, Eddahibi S. Adenovirus-mediated lung vascular endothelial endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23:762–771. doi: 10.1165/ajrcmb.23.6.4106. [DOI] [PubMed] [Google Scholar]
- 133.Patel DJ, deFreitas FM, Fry DL. Hydraulic input impedance to aorta and pulmonary artery in dogs. J Appl Physiol. 1963;18(1):134–140. doi: 10.1152/jappl.1963.18.1.134. [DOI] [PubMed] [Google Scholar]
- 134.Patel DJ, Schilder DP, Mallos AJ. Mechanical properties and dimensions of the major pulmonary arteries. J Appl Physiol. 1960;15(1):92–96. doi: 10.1152/jappl.1960.15.1.92. [DOI] [PubMed] [Google Scholar]
- 135.Pekkan K, Frakes D, De Zelicourt D, Lucas CW, Parks WJ, Yoganathan AP. Coupling pediatric ventricle assist devices to the Fontan circulation: Simulations with a lumped-parameter model. ASAIO J. 2005;51(5):618–628. doi: 10.1097/01.mat.0000176169.73987.0d. [DOI] [PubMed] [Google Scholar]
- 136.Peterson LH, Jensen RE, Parnell J. Mechanical properties of arteries in vivo. Circ Res. 1960;8(3):622–639. [Google Scholar]
- 137.Pietri P, Vyssoulis G, Vlachopoulos C, Zervoudaki A, Gialernios T, Aznaouridis K, Stefanadis C. Relationship between low-grade inflammation and arterial stiffness in patients with essential hypertension. J Hypertens. 2006;24(11):2231–2238. doi: 10.1097/01.hjh.0000249701.49854.21. [DOI] [PubMed] [Google Scholar]
- 138.Quarteroni A, Ragni S, Veneziani A. Coupling between lumped and distributed models for blood flow problems. Comp Visual Sci. 2001;4(2):111–124. [Google Scholar]
- 139.Reeves JT, Groves BM, Turkevich D. The case for treatment of selected patients with primary pulmonary hypertension. Am Rev Respir Dis. 1986;134(2):342–346. doi: 10.1164/arrd.1986.134.2.342. [DOI] [PubMed] [Google Scholar]
- 140.Reid L, Davies P. Control of Cell Proliferation in Pulmonary Hypertension. Vol. 38. New York: Marcel Dekker Inc; 1989. [Google Scholar]
- 141.Remillard CV, Yuan JX. High altitude pulmonary hypertension: Role of K and Ca2 channels. High Alt Med Biol. 2005;6:133–146. doi: 10.1089/ham.2005.6.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK, Levy PC, Reid LM, Vreim CE, Williams GW. Primary pulmonary hypertension: A national prospective study. Ann Intern Med. 1987;107(2):216–223. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- 143.Rigolin VH, Robiolio PA, Wilson JS, Harrison JK, Bashore TM. The forgotten chamber: The importance of the right ventricle. Cathet Cardiovasc Diagn. 1995;35(1):18–28. doi: 10.1002/ccd.1810350105. [DOI] [PubMed] [Google Scholar]
- 144.Roach MR, Burton AC. The reason for the shape of the distensibility curves of arteries. Can J Biochem Physiol. 1957;35(8):681–690. [PubMed] [Google Scholar]
- 145.Robert L, Hornebeck W. Elastin and Elastases. I. CRC Press; Boca Raton, FL: 1989a. [Google Scholar]
- 146.Robert L, Hornebeck W. Elastin and Elastases. II. CRC Press; Boca Raton, FL: 1989b. [Google Scholar]
- 147.Rodés-Cabau J, Domingo E, Román A, Majó J, Lara B, Padilla F, Anívarro I, Angel J, Tardif JC, Soler-Soler J. Intravascular ultrasound of the elastic pulmonary arteries: A new approach for the evaluation of primary pulmonary hypertension. Heart. 2003;89(3):311–315. doi: 10.1136/heart.89.3.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rondelet B, Van Beneden R, Kerbaul F, Motte S, Fesler P, McEntee K, Brimioulle S, Ketelslegers JM, Naeije R. Expression of the serotin 1b receptor in experimental pulmonary hypertension. Eur Respir J. 2003;22(3):408–412. doi: 10.1183/09031936.03.00036203. [DOI] [PubMed] [Google Scholar]
- 149.Santana DB, Barra JG, Grignola JC, Ginés FF, Armentano RL. Pulmonary artery smooth muscle activation attenuates arterial dysfunction during acute pulmonary hypertension. J Appl Physiol. 2005;98(2):605–613. doi: 10.1152/japplphysiol.00361.2004. [DOI] [PubMed] [Google Scholar]
- 150.Segers P, Brimioulle S, Stergiopulos N, Westerhof N, Naeije R, Maggiorini M, Verdonck P. Pulmonary arterial compliance in dogs and pigs: The three-element windkessel model revisited. Am J Physiol. 1999;277(2 Pt 2):H725–H731. doi: 10.1152/ajpheart.1999.277.2.H725. [DOI] [PubMed] [Google Scholar]
- 151.Segers P, Rietzschel ER, De Buyzere ML, Vermeersch SJ, De Bacquer D, Van Bortel LM, De Backer G, Gillebert TC, Verdonck PR. Non-invasive (input) impedance, pulse wave velocity, and wave reflection in healthy middle-aged men and woman. Hypertension. 2007;49:1248–1255. doi: 10.1161/HYPERTENSIONAHA.106.085480. [DOI] [PubMed] [Google Scholar]
- 152.Segers P, Stergiopulos N, Westerhof N, Wouters P, Kolh P, Verdonck P. Systemic and pulmonary hemodynamics assessed with a lumped-parameter heart-arterial interaction model. J Eng Math. 2003;47(3):185–199. [Google Scholar]
- 153.Shadwick RE. Mechanical design in arteries. J Exp Biol. 1999;202(23):3305–3313. doi: 10.1242/jeb.202.23.3305. [DOI] [PubMed] [Google Scholar]
- 154.Sherwin SJ, Franke V, Peiro J, Parker K. One-dimensional modeling of a vascular network in space-time variables. J Eng Math. 2003;47(3/4):217–250. [Google Scholar]
- 155.Shewry PR, Tatham AS, Bailey JB. Elastomeric Proteins: Structures, Biomechanical Properties and Biological Roles. Cambridge University Press; Cambridge, UK: 2003. [Google Scholar]
- 156.Slife DM, Latham RD, Sipkema P, Westerhof N. Pulmonary arterial compliance at rest and exercise in normal humans. Am J Physiol Heart Circ Physiol. 1990;258:H1823–H1828. doi: 10.1152/ajpheart.1990.258.6.H1823. [DOI] [PubMed] [Google Scholar]
- 157.Snyder MF, Rideout VC, Hillestad RJ. Computer modeling of the human systemic arterial tree. J Biomech. 1968;1(4):341–353. doi: 10.1016/0021-9290(68)90029-8. [DOI] [PubMed] [Google Scholar]
- 158.Sokolis DP, Kefaloyannis EM, Kouloukoussa M, Marinos E, Boudoulas H, Karayannacos PE. A structural basis for the aortic stress-strain relation in uniaxial tension. J Biomech. 2006;39(9):1651–1662. doi: 10.1016/j.jbiomech.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 159.Spilker RL, Feinstein JA, Parker DW, Reddy VM, Taylor CA. Morphometry-based impedance boundary conditions for patient-specific modeling of blood flow in pulmonary arteries. Ann Biomed Eng. 2007;35(4):546–559. doi: 10.1007/s10439-006-9240-3. [DOI] [PubMed] [Google Scholar]
- 160.St John Sutton M. Aortic stiffness: A predictor of acute coronary events? Eur Heart J. 2000;21:342–344. doi: 10.1053/euhj.1999.1959. [DOI] [PubMed] [Google Scholar]
- 161.Steele BN, Wan J, Ku JP, Hughes TJ, Taylor CA. In vivo validation of a one-dimensional finite-element method for predicting blood flow in cardiovascular bypass grafts. IEEE Trans Biomed Eng. 2003;50(6):649–656. doi: 10.1109/TBME.2003.812201. [DOI] [PubMed] [Google Scholar]
- 162.Stefanadis C, Dernellis J, Tsiamis E, Stratos C, Diamantopoulos L, Michaelides A, Toutouzas P. Aortic stiffness as a risk factor for recurrent acute coronary events in patients with ischaemic heart disease. Eur Heart J. 2000;21:390–396. doi: 10.1053/euhj.1999.1756. [DOI] [PubMed] [Google Scholar]
- 163.Steinman DA, Taylor CA. Flow imaging and computing: Large artery hemodynamics. Ann Biomed Eng. 2005;33(12):1704–1709. doi: 10.1007/s10439-005-8772-2. [DOI] [PubMed] [Google Scholar]
- 164.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006;21:134–145. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 165.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling—Cellular and molecular mechanisms. Circ Res. 2006;99(7):675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 166.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension—A time for reappraisal? Circ Res. 2005;97(2):95–98. doi: 10.1161/01.RES.00000175934.68087.29. [DOI] [PubMed] [Google Scholar]
- 167.Stergiopulos N, Meister JJ, Westerhof N. Simple and accurate way for estimating total and segmental arterial compliance: The pulse pressure method. Ann Biomed Eng. 1994;22:392–397. doi: 10.1007/BF02368245. [DOI] [PubMed] [Google Scholar]
- 168.Stergiopulos N, Meister JJ, Westerhof N. Evaluation of methods for estimation of total arterial compliance. Am J Physiol. 1995;268(4 Pt 2):H1540–H1548. doi: 10.1152/ajpheart.1995.268.4.H1540. [DOI] [PubMed] [Google Scholar]
- 169.Stergiopulos N, Westerhof BE, Westerhof N. Total arterial inertance as the fourth element of the windkessel model. 1999;276(1):H81–H88. doi: 10.1152/ajpheart.1999.276.1.H81. [DOI] [PubMed] [Google Scholar]
- 170.Stewart S. Pathology of adult pulmonary hypertension. In: Morice AH, editor. Clinical Pulmonary Hypertension. London: Portland Press; 1995. pp. 43–79. [Google Scholar]
- 171.Sugawa K, Maughan L, Suga H, Sunagawa K. Cardiac Contraction and the Pressure-Volume Relationship. Oxford, UK: Oxford University Press; 1988. [Google Scholar]
- 172.Sundareswaran KS, Pekkan K, Dasi LP, Whitehead K, Sharma S, Kanter KR, Fogel MA, Yoganathan AP. The total cavopulmonary connection resistance: A significant impact on single ventricle hemodynamics at rest and exercise. Am J Physiol Heart Circ Physiol. 2008;295(6):H2427–H2435. doi: 10.1152/ajpheart.00628.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tagawa H. Pulmonary arterial input impedance in patients with chronic pulmonary diseases. Nihon Kyobu Shikkan Gakkai Zasshi. 1989;27(9):1031–1039. [PubMed] [Google Scholar]
- 174.Takamizawa K, Hayashi K. Strain energy density function and uniform strain hypothesis for arterial mechanics. J Biomech. 1987;20(1):7. doi: 10.1016/0021-9290(87)90262-4. [DOI] [PubMed] [Google Scholar]
- 175.Tawhai MH, Burrowes KS. Modelling pulmonary blood flow. Respir Physiol Neurobiol. 2008;163(1–3):150–157. doi: 10.1016/j.resp.2008.02.016. Epub 2008 Mar 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Taylor CA, Draney MT. Experimental and computational methods in cardiovascular fluid mechanics. Annu Rev Fluid Mech. 2004;36:197–231. [Google Scholar]
- 177.Taylor CA, Figueroa CA. Patient-specific modeling of cardiovascular mechanics. Annu Rev Biomed Eng. 2009;11:109–134. doi: 10.1146/annurev.bioeng.10.061807.160521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Taylor MG. An experimental determination of the propagation of fluid oscillations in a tube with a visco-elastic wall; together with an analysis of the characteristics required in an electrical analogue. Phys Med Biol. 1959;4:63–82. doi: 10.1088/0031-9155/4/1/308. [DOI] [PubMed] [Google Scholar]
- 179.Taylor MG. The input impedance of an assembly of randomly branching elastic tubes. Biophys J. 1966a;6(1):29–51. doi: 10.1016/S0006-3495(66)86638-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Taylor MG. Wave transmission through an assembly of randomly branching elastic tubes. Biophys J. 1966b;6(6):697–716. doi: 10.1016/S0006-3495(66)86689-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tobise K, Haneda T, Onodera S. Changes in the pulmonary vascular input impedance in patients with atrial septal defect after surgical correction. Jpn Circ J. 1990;54(2):175–182. doi: 10.1253/jcj.54.175. [DOI] [PubMed] [Google Scholar]
- 182.Tozzi CA, Christiansen DL, Poiani GJ, Riley DJ. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am J Respir Crit Care Med. 1994;149(5):1317–1326. doi: 10.1164/ajrccm.149.5.8173773. [DOI] [PubMed] [Google Scholar]
- 183.Tuder RM, Abman SH, Braun T, Capron F, Stevens T, Thistlethwaite PA, Haworth SG. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S3–S9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 184.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28(1):23–42. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ugural AC, Fenster SK. Advanced Strength and Applied Elasticity. 4. Upper Saddle River, NJ: Prentice Hall; 2003. pp. 315–320. [Google Scholar]
- 186.Uren NG, Oakley CM. The treatment of primary pulmonary hypertension. Br Heart J. 1991;66(2):119–121. doi: 10.1136/hrt.66.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vaishnav RN, Young JT, Janicki JS, Patel DJ. Nonlinear anisotropic elastic properties of the canine aorta. Biophys J. 1972;12(8):1008–1027. doi: 10.1016/S0006-3495(72)86140-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Van Den Bos GC, Westerhof N, Randall OS. Pulse wave reflection: Can it explain the differences between systemic and pulmonary pressure and flow waves? A study in dogs. Circ Res. 1982;51:479–485. doi: 10.1161/01.res.51.4.479. [DOI] [PubMed] [Google Scholar]
- 189.Vignon IE, Taylor CA. Outflow boundary conditions for one-dimensional finite element modeling of blood flow and pressure waves in arteries. Wave Motion. 2004;39(4):361–374. [Google Scholar]
- 190.Vignon-Clementel IE, Figueroa CA, Jansen KE, Taylor CA. Outflow boundary conditions for three-dimensional finite element modeling of blood flow and pressure in arteries. Comput Methods Appl Mech Engrg. 2006;195(29):3776–3796. [Google Scholar]
- 191.Vito RP, Dixon SA. Blood vessel constitutive models-1995–2002. Annu Rev Biomed Eng. 2003;5:413–439. doi: 10.1146/annurev.bioeng.5.011303.120719. [DOI] [PubMed] [Google Scholar]
- 192.Von Maltzahn WW, Warriyar RG, Keitzer WF. Experimental measurements of elastic properties of media and adventitia of bovine carotid arteries. J Biomech. 1984;17(11):839. doi: 10.1016/0021-9290(84)90142-8. [DOI] [PubMed] [Google Scholar]
- 193.Wan J, Steele B, Spicer SA, Strohband S, Feijóo GR, Hughes TJ, Taylor CA. A one-dimensional finite element method for simulation-based medical planning for cardiovascular disease. Comput Methods Biomech Biomed Engin. 2002;5(3):195–206. doi: 10.1080/10255840290010670. [DOI] [PubMed] [Google Scholar]
- 194.Wauthy P, Abdel Kafi S, Mooi WJ, Naeije R, Brimioulle S. Inhaled nitric oxide versus prostacyclin in chronic shunt-induced pulmonary hypertension. J Thorac Cardiovasc Surg. 2003;126(5):1434–1441. doi: 10.1016/s0022-5223(03)01183-8. [DOI] [PubMed] [Google Scholar]
- 195.Weinberg C, Hertzberg J, Valdes-Cruz LM, Shandas R. Extraction of pulmonary vascular compliance, PVR and RV work from single-pressure and Doppler flow measurements in children with pulmonary hypertension—a new method for evaluating reactivity: In vitro and clinical studies. Circulation. 2004;110(7):2609–2617. doi: 10.1161/01.CIR.0000146818.60588.40. [DOI] [PubMed] [Google Scholar]
- 196.Westerhof N, Bosman F, de Vries CJ, Noordergraaf A. Analog studies of the human systemic arterial tree. J Biomech. 1969;2:121–143. doi: 10.1016/0021-9290(69)90024-4. [DOI] [PubMed] [Google Scholar]
- 197.Westerhof N, Elzinga G, Sipkema P. An artificial arterial system for pumping hearts. J Appl Physiol. 1971;31(5):776–781. doi: 10.1152/jappl.1971.31.5.776. [DOI] [PubMed] [Google Scholar]
- 198.Westerhof N, Lankhaar JW, Westerhof BE. The arterial Windkessel. Med Biol Eng Comput. 2009;47(2):131–141. doi: 10.1007/s11517-008-0359-2. [DOI] [PubMed] [Google Scholar]
- 199.Westerhof N, Sipkema P, Van Den Bos GC, Elzinga G. Forward and backward waves in the arterial system. Cardiovasc Res. 1972;6(6):648–656. doi: 10.1093/cvr/6.6.648. [DOI] [PubMed] [Google Scholar]
- 200.Whitehead KK, Pekkan K, Kitajima HD, Paridon SM, Yoganathan AP, Fogel MA. Nonlinear power loss during exercise in single-ventricle patients after the Fontan: Insights from computational fluid dynamics. Circulation. 2007;116(11 Suppl):I165–I171. doi: 10.1161/CIRCULATIONAHA.106.680827. [DOI] [PubMed] [Google Scholar]
- 201.Wiener F, Morkin E, Skalak R, Fishman AP. Wave propagation in the pulmonary circulation. Circ Res. 1966;19:834–850. doi: 10.1161/01.res.19.4.834. [DOI] [PubMed] [Google Scholar]
- 202.Wohrley JD, Frid MG, Moiseeva EP, Orton EC, Belknap JK, Stenmark KR. Hypoxia selectively induces proliferation in a specific subpopulation of smooth-muscle cells in the bovine neonatal pulmonary arterial media. J Clin Invest. 1995;96(1):273–281. doi: 10.1172/JCI118031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wolinsky H, Glagov S. Structural basis for the static mechanical properties of the aortic media. Circ Res. 1964;14(5):400. doi: 10.1161/01.res.14.5.400. [DOI] [PubMed] [Google Scholar]
- 204.Womersley JR. An elastic theory of pulse transmission and oscillatory flow in mammalian arteries, TR 56–614. Wright Air Development Center; Dayton, Ohio, U.S.A: 1957. [Google Scholar]
- 205.Womersley JR. Mathematical theory of oscillating flow in an elastic tube. J Physiol. 1955a;127(2):37–38P. [PubMed] [Google Scholar]
- 206.Womersley JR. Method for the calculation of velocity, rate of flow and viscous drag in arteries when the pressure gradient is known. J Physiol. 1955b;127(3):553–563. doi: 10.1113/jphysiol.1955.sp005276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Womersley JR. Oscillatory flow in arteries: The constrained elastic tube as a model of arterial flow and pulse transmission. Phys Med Biol. 1957;2(2):178–187. doi: 10.1088/0031-9155/2/2/305. [DOI] [PubMed] [Google Scholar]
- 208.Wright JE, Drexler ES, Slifka AJ, McCowan CN, Ivy DD, Shandas R. Stress and strain in rat pulmonary artery material during a biaxial bubble test. Biomed Sci Instrum. 2004;40:303–308. [PubMed] [Google Scholar]
- 209.Wuyts FL, Vanhuyse VJ, Langewouters GJ, Decraemer WF, Raman ER, Buyle S. Elastic properties of human aortas in relation to age and atherosclerosis: A structural model. 1995;40:1577–1597. doi: 10.1088/0031-9155/40/10/002. [DOI] [PubMed] [Google Scholar]
- 210.Xie J, Zhou J, Fung YC. Bending of blood vessel wall: Stress-strain laws of the intima-media and adventitial layers. J Biomech Eng. 1995;117:136. doi: 10.1115/1.2792261. [DOI] [PubMed] [Google Scholar]
- 211.Yu PN, Murphy GW, Schreiner BF, James DH. Distensibility characteristics of the human pulmonary vascular bed. Circulation. 1967;35:710–723. doi: 10.1161/01.cir.35.4.710. [DOI] [PubMed] [Google Scholar]
- 212.Yu Q, Zhou J, Fung YC. Neutral axis location in bending and Young’s modulus of different layers of arterial wall. Am J Physiol Heart Circ Physiol. 1993;265(1):H52. doi: 10.1152/ajpheart.1993.265.1.H52. [DOI] [PubMed] [Google Scholar]
- 213.Zaidi SHE, You XM, Ciura S, Husain M, Rabinovitch M. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105(4):516–521. doi: 10.1161/hc0402.102866. [DOI] [PubMed] [Google Scholar]
- 214.Zhang YH, Dunn ML, Drexler ES, McCowan CN, Slifka AJ, Ivy DD, Shandas R. A microstructural hyperelastic model of pulmonary arteries under normo- and hypertensive conditions. Ann Biomed Eng. 2005;33(8):1042–1052. doi: 10.1007/s10439-005-5771-2. [DOI] [PubMed] [Google Scholar]
- 215.Zhang YH, Dunn ML, Hunter KS, Lanning C, Ivy DD, Claussen L, Chen SJ, Shandas R. Application of a microstructural constitutive model of the pulmonary artery to patient-specific studies: Validation and effect of orthotropy. J Biomech Eng. 2007;129(2):193–201. doi: 10.1115/1.2485780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zuckerman BD, Orton EC, Latham LP, Barbiere CC, Stenmark KR, Reeves JT. Pulmonary vascular impedance and wave reflections in the hypoxic calf. J Appl Physiol. 1992;72(6):2118–2127. doi: 10.1152/jappl.1992.72.6.2118. [DOI] [PubMed] [Google Scholar]
- 217.Zuckerman BD, Orton EC, Stenmark KR, Trapp JA, Murphy JR, Coffeen PR, Reeves JT. Alteration of the pulsatile load in the high-altitude calf model of pulmonary hypertension. J Appl Physiol. 1992;70(2):859–868. doi: 10.1152/jappl.1991.70.2.859. [DOI] [PubMed] [Google Scholar]
- 218.Zulliger MA, Fridez P, Hayashi K, Stergiopulos N. A strain energy function for arteries accounting for wall composition and structure. J Biomech. 2004;37(7):989–1000. doi: 10.1016/j.jbiomech.2003.11.026. [DOI] [PubMed] [Google Scholar]