

Abstract
Pseudomonas aeruginosa can grow to very high-cell-density (HCD) during infection of the cystic fibrosis (CF) lung. Phosphatidylcholine (PC), the major component of lung surfactant, has been hypothesized to support HCD growth of P. aeruginosa in vivo. The phosphorylcholine headgroup, a glycerol molecule, and two long-chain fatty acids (FAs) are released by enzymatic cleavage of PC by bacterial phospholipase C and lipases. Three different bacterial pathways, the choline, glycerol, and fatty acid degradation pathways, are then involved in the degradation of these PC components. Here, we identified five potential FA degradation (Fad) related fadBA-operons (fadBA1-5, each encoding 3-hydroxyacyl-CoA dehydrogenase and acyl-CoA thiolase). Through mutagenesis and growth analyses, we showed that three (fadBA145) of the five fadBA-operons are dominant in medium-chain and long-chain Fad. The triple fadBA145 mutant also showed reduced ability to degrade PC in vitro. We have previously shown that by partially blocking Fad, via mutagenesis of fadBA5 and fadDs, we could significantly reduce the ability of P. aeruginosa to replicate on FA and PC in vitro, as well as in the mouse lung. However, no studies have assessed the ability of mutants, defective in choline and/or glycerol degradation in conjunction with Fad, to grow on PC or in vivo. Hence, we constructed additional mutants (ΔfadBA145ΔglpD, ΔfadBA145ΔbetAB, and ΔfadBA145ΔbetABΔglpD) significantly defective in the ability to degrade FA, choline, and glycerol and, therefore, PC. The analysis of these mutants in the BALB/c mouse lung infection model showed significant inability to utilize PC in vitro, resulted in decreased replication fitness and competitiveness in vivo compared to the complement strain, although there was little to no variation in typical virulence factor production (e.g., hemolysin, lipase, and protease levels). This further supports the hypothesis that lung surfactant PC serves as an important nutrient for P. aeruginosa during CF lung infection.
Introduction
Pseudomonas aeruginosa is widespread in nature, inhabiting soil, water, plants and animals. In hospitals, it can be found in sinks, respirators, humidifiers and occasionally on the hands of medical personnel [1], [2]. The ubiquitous nature of this bacterium has allowed it to adapt to a broad range of hosts in which it can cause diseases. The role of P. aeruginosa as an opportunistic human pathogen is of particular concern, especially because it is a frequent cause of nosocomial infections such as pneumonia, urinary tract infections, and bacteremia [1], [3], [4]. P. aeruginosa infection in the respiratory tract of cystic fibrosis (CF) patients causes a rapid deterioration in lung function and thus patient survival [5], [6]. The pathogenicity of P. aeruginosa infection in CF patients has been extensively studied in terms of biofilm production [7]–[9] and quorum sensing (QS) controlled virulence [10]–[12]. However, little effort has been placed towards the contribution of P. aeruginosa nutrient acquisition aids high-cell-density (HCD) replication during lung infection.
Previous studies have shown that P. aeruginosa can undergo HCD replication in the lung of CF patients reaching >109 CFU/ml [13]–[15]. HCD replication is highly energy demanding, requiring efficient nutrient acquisition and metabolism. However, evidence showed that the nutrients in the lung environment are lipids and amino acids derived from proteins or polypeptides [16]–[18], to allow P. aeruginosa HCD growth and maintenance in vivo. An in vitro study revealed that P. aeruginosa has directional twitching motility toward phosphatidylethanolamine (PE) and phosphatidylcholine (PC) [19]. Mammalian lungs are naturally coated by indispensible lung surfactant, which is composed of approximately 10% protein and 90% lipids, with about 80% of the lung surfactant lipids being phosphatidylcholine (PC) [20]–[22]. Thus, PC, the most abundant lipid in lung surfactant may provide significant nutrient for HCD bacterial growth in vivo. In accordance with this hypothesis, our initial studies suggest that PC is a major nutrient source of P. aeruginosa during lung infection and supports HCD replication [15], [17], [18].
Our previous in vivo CF sputa study showed that P. aeruginosa produced phospholipase C (heat-labile hemolysin) and lipases that could cleave exogenous PC into three components: a phosphorylcholine headgroup, glycerol and two long-chain fatty acids (LCFAs) [15] (Fig. 1A). These three components can be further metabolized by the choline (bet), glycerol (glp), and fatty acid degradation (Fad) pathways (Fig. 1B), respectively. Choline and glycerol metabolism by P. aeruginosa are well characterized [23]–[27]. However, LCFA degradation by P. aeruginosa and the genes involved in this process remain to be fully elucidated. Our previous in vivo CF sputa study also detected the expression of P. aeruginosa genes involved in each pathway for metabolizing all three PC components [15]. The betAB-operon was induced and glpD and glpK genes were constitutively expressed in vivo [15]. Among genes for Fad (Fig. 1B), the expression of fadBA1 was detected when P. aeruginosa was grown on PC in vitro, and fadBA5 and fadA4 were induced and constitutively expressed in CF sputa [15], suggesting the involvement of fadBA145 in LCFA degradation. We have also shown the reduced ability of the fadD mutants to utilize FAs as nutrients led to their decreased fitness during mouse lung infection [17], [18]. However, further evidence is needed to support the hypothesis that all three components of lung surfactant PC (phosphorylcholine, glycerol and FA) serve as nutrient sources for P. aeruginosa during in vivo lung infection.
Figure 1. Phosphatidylcholine (PC) degradation pathways in Pseudomonas aeruginosa.
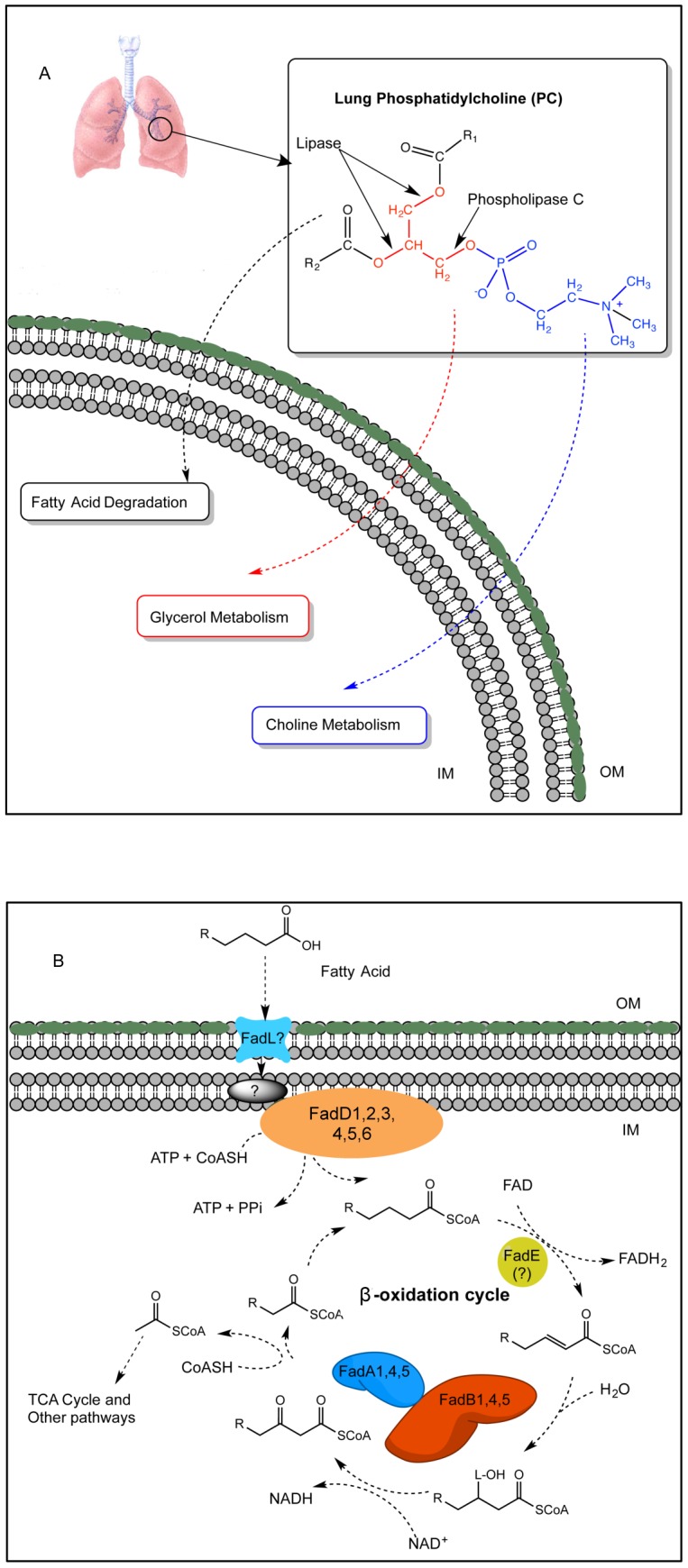
(A) PC is the main component of lung surfactant and can be cleaved by phospholipase C and lipases, producing free fatty acids, glycerol, and phosphorylcholine. Three different pathways then further metabolize each component: the bet pathway for choline head group metabolism, the glp pathway for glycerol metabolism, and the β-oxidation pathway for the degradation of the FAs. (B) The proposed P. aeruginosa FA β-oxidation pathway. Abbreviations: FadA, 3-ketoacyl-CoA thiolase; FadB, cis-Δ3-trans-Δ2-enoyl-CoA isomerase, enoyl-CoA hydratase, 3-hydroxyacyl-CoA epimerase, and 3-hydroxyacyl-CoA dehydrogenase; FadD, fatty acyl-CoA synthetase; FadE, acyl-CoA dehydrogenase; FadL, outer membrane long-chain fatty acid translocase; OM, out membrane; IN, inner membrane.
In this study, up to five potential fadBA-operons (encoding 3-hydroxyacyl-CoA dehydrogenase and acyl-CoA thiolase) were identified, and three of them fadBA1 (PA1737, PA1736), fadBA4 (PA4786, PA4785) and fadBA5 (PA3014, PA3013) were shown to be significantly involved in medium- and long-chain Fad. Coupling of the fadBA145 mutations with mutations in choline and/or glycerol degradation were investigated to determine the importance of these pathways to degrade PC in vitro and in vivo. Competition studies were initiated to analyze the competitive fitness of these mutants relative to strains with intact pathways.
Results and Discussion
P. aeruginosa has five fadBA-operons potentially involved in fatty acid degradation
The well-established aerobic fatty acid degradation (Fad) pathway in E. coli was used as a model to characterize P. aeruginosa Fad. E. coli possesses only a single copy of each fad gene for aerobic Fad [28], [29], and the cyclic degradation of fatty acids by two carbons per cycle is primarily catalyzed by an acyl-CoA dehydrogenase coded by fadE, and the products of the fadBA-operon, 3-hydroxyacyl-CoA dehydrogenase and acyl-CoA thiolase, respectively. Up to five potential fadBA-operons were identified in P. aeruginosa by BLAST analysis of the E.coli fadBA sequence against the P. aeruginosa genome, including fadBA1 (PA1737, PA1736), fadBA2 (PA3590, PA3589), fadBA3 (PA2554, PA2553), fadBA4 (PA4786, PA4785), and fadBA5 (PA3014, PA3013) (Fig. S1). Of these five FadBAs, FadBA5 showed the greatest homology to the E.coli FadBA with FadB5 having 72% similar (54% identical) and FadA5 having 76% similar (61% identical) to the E.coli FadBA, respectively [30], [31].
Considering the larger size of the P. aeruginosa genome (6.29 Mb) [32] and its wide range of environmental niches and metabolic capabilities, it is not surprising that P. aeruginosa has up to five fadBA-operon homologues. Therefore, it is important next to narrow down which of these five operons are important in Fad, using a mutagenesis approach.
P. aeruginosa fadBA145-operons are important for degrading PC and medium- and long-chain fatty acids
Our previous work showed that the ΔfadBA5 mutant has a reduced ability to utilize LCFA as a sole carbon source, but this ΔfadBA5 mutant can still grow on LCFAs as a carbon source, indicating the existence of other potential fadBA-operons in P. aeruginosa for LCFA degradation [31]. Further supporting this idea, the fadBA1-operon was shown to be induced by medium-chain fatty acids (MCFAs) and to a lesser extent by LCFAs [33]. The fadBA5-operon plays the most significant role in LCFA degradation, because neither the single ΔfadBA1 mutant, nor the single ΔfadBA4 mutant, showed much defects in their ability to utilize MCFA and LCFA as sole carbon sources (Fig. S2). It is possible that the FadBA1 and/or other FadBA(s) might have overlapping functions with FadBA5 in the metabolism of different chain length FAs, but these activities are masked by the more dominant FadBA5. Evidence for the involvement of other FadBAs is lacking, and needs to be addressed.
Because it is too overwhelming to test all possible double, triple, and quadruple fadBA-mutant combinations and the complicated dominance of fadBA5, we demonstrated the involvement of each fadBA-operon by testing different triple mutant combinations. In this study, we generated several triple mutants (ΔfadBA125, ΔfadBA135, ΔfadBA145, ΔfadBA235, ΔfadBA245, ΔfadBA345) and a quintuple mutant ΔfadBA1-5 (Table 1) for growth analysis on MCFA and LCFAs as sole carbon sources, to further characterize the function of these fadBA-operons. The growth defects were previously defined by the slower growth rate and lower overall final cell densities compared to wildtype strain PAO1, which suggest a reduced ability to metabolize these FAs, presumably due to the accumulation of growth inhibiting intermediates [17], [31]. Significant growth defects were observed for any combinations of triple mutants in which both the fadBA5- and fadBA1-operons were deleted (i.e., ΔfadBA125, ΔfadBA135 and ΔfadBA145 mutants), revealing the importance of fadBA1 and fadBA5 contributing to growth on MCFA and LCFAs (Fig. 2A–D). Although the level of defects is different for each type of FA used, the trend is consistent for these mutants in all FAs (Fig. 2A–D). Only the ΔfadBA145 triple mutant showed the same growth defect as the ΔfadBA1-5 quintuple mutant on all fatty acids tested, suggesting the importance of all three fadBA1, fadBA4, and fadBA5 operons and the minor role of fadBA2 and fadBA3-operons in metabolizing MCFA and LCFAs. The importance of fadBA4 was further confirmed by the fact that the ΔfadBA245 and ΔfadBA345 triple mutants showed an additional growth defect on all FAs compared to the ΔfadBA235 mutant (Fig. 2A–D). However, the fadBA4-operon displays less of an involvement in metabolizing all FAs tested compared to both fadBA1 and fadBA5-operons (i.e., comparing ΔfadBA245 to ΔfadBA125 or ΔfadBA345 to ΔfadBA135). Knowing that only fadBA1, fadBA4, and fadBA5 are important for degrading MCFA and LCFA of PC, we continued with further experiments from this point forward in our study by using the ΔfadBA145 mutant background, rather than the ΔfadBA1-5 quintuple mutant.
Table 1. Bacterial strains used in this studya.
| Strain | Lab IDb | Genotype/Description | Reference |
| E.coli | |||
| EP-Max10B | E1231 | F− λ− mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZ ΔM15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara, leu)7697 galU galKrpsL nupG | BioRad |
| SM10 | E006 | thi thr leu tonA lacY supE recA::RP4-2Tc::Mu Kmr | [48] |
| P. aeruginosa | |||
| PAO1 | P007 | Prototroph | [49] |
| PAO1- mucA− | P447 | Cbr, PAO1 with pUC18 inserted in mucA gene | This study |
| ΔfadBA125 | P122 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA2::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA135 | P124 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA3::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA145 | P319 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA4::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA235 | P317 | Gmr, PAO1-ΔfadBA2::FRT, ΔfadBA3::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA245 | P130 | Gmr, PAO1-ΔfadBA2::FRT, ΔfadBA3::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA345 | P126 | Gmr, PAO1-ΔfadBA3::FRT, ΔfadBA4::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA1-5 | P102 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA2::FRT, ΔfadBA3::FRT,ΔfadBA4::FRT, ΔfadBA5::Gm | This study |
| ΔfadBA145 ΔglpD | P539 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA4::FRT, ΔfadBA5::FRT,ΔglpD::Gm-FRT1 | This study |
| ΔfadBA145 ΔbetAB | P555 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA4::FRT, ΔfadBA5::FRT,ΔbetAB::Gm-FRT3 | This study |
| ΔfadBA145 ΔbetAB ΔglpD | P561 | Gmr, PAO1-ΔfadBA1::FRT, ΔfadBA4::FRT, ΔfadBA5::FRT,ΔbetAB::FRT3, ΔglpD::Gm-FRT1 | This study |
| ΔfadBA145/complement | P965 | Gmr, Tetr ; ΔfadBA145 complemented with miniCTX2-fadBA5 | This study |
| ΔfadBA145 ΔglpD/complement | P1015 | Gmr, Tetr ; ΔfadBA145 ΔglpD complemented with miniCTX2-fadBA5/glpD | This study |
| ΔfadBA145 ΔbetAB/complement | P1017 | Gmr, Tetr ; ΔfadBA145 ΔbetAB complemented with miniCTX2-fadBA5/betAB | This study |
| ΔfadBA145 ΔbetABΔglpD/complement | P1019 | Gmr, Tetr ; ΔfadBA145 ΔbetAB ΔglpD complemented withminiCTX2-fadBA5/betAB/glpD | This study |
| ΔfadBA145-mucA− | P576 | Gmr, Cbr; ΔfadBA145 with pUC18 inserted in mucA gene | This study |
| ΔfadBA145 ΔglpD-mucA− | P570 | Gmr, Cbr; ΔfadBA145 ΔglpD with pUC18 inserted in mucA gene | This study |
| ΔfadBA145 ΔbetAB-mucA− | P572 | Gmr, Cbr; ΔfadBA145 ΔbetAB with pUC18 inserted in mucA gene | This study |
| ΔfadBA145 ΔbetABΔglpD-mucA− | P574 | Gmr, Cbr; ΔfadBA145 ΔbetAB ΔglpD with pUC18 inserted in mucA gene | This study |
| ΔfadBA145- mucA−/complement | P584 | Gmr, Cbr, Tetr; ΔfadBA145-mucA− complemented with miniCTX2-fadBA5 | This study |
| ΔfadBA145 ΔglpD- mucA−/complement | P578 | Gmr, Cbr, Tetr; ΔfadBA145 ΔglpD-mucA− complemented withminiCTX2-fadBA5/glpD | This study |
| ΔfadBA145 ΔbetAB- mucA−/complement | P580 | Gmr, Cbr, Tetr; ΔfadBA145 ΔbetAB-mucA− complemented withminiCTX2-fadBA5/betAB | This study |
| ΔfadBA145 ΔbetAB ΔglpD- mucA−/complement | P582 | Gmr, Cbr, Tetr; ΔfadBA145 ΔbetAB ΔglpD-mucA− complemented withminiCTX2-fadBA5/betAB/glpD | This study |
For strains constructed in this study, please see text for further details.
Please use Lab ID for requesting strains.
Figure 2. Growth analysis of different fadBA mutant combinations on medium (C12∶0) and long chain-length fatty acid (C14∶0, C16∶0 and C18∶1 Δ9).

Along with the wildtype PAO1 strain, mutants were grown in 1×M9 minimal medium supplemented with 0.4% different test FAs (A to D) or 1% control casamino acids (CAA, E) as sole carbon sources. Although fadBA mutants showed various defects when grown with FAs of different chain-lengths, no growth defects were observed for any of the mutants when grown with CAA as a control.
We next tested the ΔfadBA145 mutant for its ability to grow on PC. The ΔfadBA145 triple mutant displayed a reduced growth rate and lower final cell density as compared to wildtype PAO1 when grown with PC as a sole carbon source (Fig. 3A). There was no growth defect observed for this mutant when grown in control LB medium (Fig. 3B). The ΔfadBA145 mutant strain is competitively less fit than the complement in the in vitro competition study when grown in PC or FA (C18∶1 Δ9) (Fig. 4A panel i). No competitive defect was observed when the ΔfadBA145 mutant was grown on LB, glucose, casamino acids (CAA), glycerol or choline controls (Fig. 4A panel i). We concluded that the reduced growth of the ΔfadBA145 mutant strain on PC observed in figure 3A is due to a decreased ability to degrade LCFAs of PC and not glycerol or choline. All the evidence we have here strongly suggests the involvement of the three fadBA1,4,5-operons in Fad and PC degradation. We complemented ΔfadBA145 mutant strain by integrating miniCTX2-fadBA5 (i.e., the dominant fadBA operon as explained above) as a single copy into the ΔfadBA145 mutant background (Table 1). The complemented strain was fully restored to wildtype growth on PC (Fig. 3A) and FAs (not shown). Hence, all complementation experiments in this study for any ΔfadBA145 mutant background were performed with only the dominant fadBA5-operon.
Figure 3. Growth analysis on phosphatidylcholine.

(A) Some mutants exhibited growth defects on PC as a sole carbon source. The growth defects were fully recovered in complemented strains, as they had identical growth rates compared to the wildtype PAO1 strain. (B) No growth defects in control LB medium were observed.
Figure 4. Competition studies of pathway mutants.

(A) In vitro competition studies of the various mutants and their complemented strains in different growth media (n equals the number of independent in vitro competition experiments performed with each carbon source). (B) In vivo lung competition of the various mutants and their complemented strains after 24 h, where n equals the number of mice in each group that were inoculated with a total of 6×106 CFU/mouse. The solid red line indicates the geometric mean of the competitive indices (CI) in each competition group. CI<1 indicates the mutant was less competitive than its complemented strain in various growth media (A) or within the lungs (B). Numbers above the red line represent the average total recovered CFU/mouse for each competition group.
Mutants blocked in FA, glycerol, and choline degradation displayed dramatically reduced ability to utilize PC in vitro
The enzymatic activity of phospholipase C on PC releases the phosphorylcholine headgroup and the diacylglycerol (DAG) molecule (Fig. 1A). The phosphorylcholine headgroup is first transported across the cell membrane and dephosphorylated by a phosphatase [23], [34], [35] to yield choline, which has previously been shown to be sufficient for P. aeruginosa to grow on as a sole carbon, nitrogen, and energy source [36]. P. aeruginosa BetAB (encoding choline dehydrogenase and a glycine betaine aldehyde dehydrogenase) catalyzes the conversion of choline to glycine betaine [23]. Glycine betaine is successively demethylated to form dimethylglycine (DMG), sarcosine (monomethylglcine), and finally glycine [24], [37]. The DAG molecule is cleaved by the P. aeruginosa lipase, liberating a glycerol molecule and two LCFAs. Glycerol metabolism has been well characterized in P. aeruginosa. The operon primarily consists of glpD (a sn-glycerol-3-phosphate dehydrogenase [38]), glpF (a membrane-associated glycerol diffusion facilitator [27], [39]), glpK (a glycerol kinase [27], [39]), glpM (a membrane protein affecting alginate synthesis [26]), and glpR (a regulator of the glp operon [25]).
Since our previous data showed that betAB and glpD were expressed in vivo [15], they may potentially be involved in PC degradation during lung infection. However, before we could address the in vivo aspect of PC degradation, further experiments are needed to characterize P. aeruginosa PC degradation in vitro. We engineered double pathway mutants ΔfadBA145ΔbetAB (blocked in FA and choline degradation) and ΔfadBA145ΔglpD (blocked in FA and glycerol degradation) and a triple pathway mutant ΔfadBA145ΔbetABΔglpD (blocked in FA, choline, and glycerol degradation) (Table 1), to further determine whether these mutants are deficient in growth on PC. As expected, all three mutants experienced various growth defects with decreased cell density and delayed log-phase when grown on PC (Fig. 3A). The triple pathway mutant ΔfadBA145ΔbetABΔglpD exhibited the most significant reduced ability to utilize PC, reaching to only about one-third of the wildtype final cell density. We complemented these mutants (i.e., ΔfadBA145ΔbetAB, ΔfadBA145ΔglpD, and ΔfadBA145ΔbetABΔglpD) by integrating the respective miniCTX2-fadBA5/betAB, miniCTX2-fadBA5/glpD or miniCTX2-fadBA5/betAB/glpD as a single copy on the P. aeruginosa chromosome and the complemented strains fully recovered these mutants’ ability to grow on PC as compared to wildtype PAO1 (Fig. 3A). No mutants or complement showed any growth defects on control LB medium (Fig. 3B).
We performed an in vitro competition study between pathway mutants (ΔfadBA145ΔbetAB, ΔfadBA145ΔglpD, ΔfadBA145ΔbetABΔglpD) and their complements to exam whether the mutation reduced their ability to metabolize various carbon sources. As expected, all the pathway mutants were less competitive than their respective complements in media containing PC and other sole carbon sources involved in the respective pathways (Fig. 4A panels ii-iv). For example, the ΔfadBA145ΔbetAB mutant was less competitive than its complement when grown using PC, FA, or choline, as sole carbon source (Fig. 4A panel ii), which is not the case in other control media (e.g., LB, glucose, CAA or glycerol). Likewise, the ΔfadBA145ΔglpD mutant was less competitive than its complement only if PC, FA, or glycerol was used as sole carbon source (Fig. 4A panel iii). The triple pathway mutant was almost completely outcompeted by its complemented strain with the competitive indices (CI) dramatically reduced to ∼0.1 when growing in the media containing PC, choline, glycerol, or oleate FA (Fig. 4A panel iv). Overall, these in vitro data showed that we have three mutants (ΔfadBA145ΔbetAB, ΔfadBA145ΔglpD, ΔfadBA145ΔbetABΔglpD) and their complement that could be used to assess the utilization of PC in vivo, through competitive index experiments within the mouse lung.
Blocking FA, glycerol, and choline degradation significantly reduces replication fitness of P. aeruginosa in vivo
Since PC is the major component of lung surfactant in mammals, including mice [21], a mouse lung infection model [40] was utilized for our in vivo competition study to evaluate the fitness of the PC degradation pathway mutants within the lung environment. The mucoid, exopolysaccharide alginate-overproducing phenotype is a distinguishing feature of P. aeruginosa isolated from CF patients [40], [41]. An alginate-overproducing strain carrying a mucA insertional mutation, which allows the mucoid strain to survive and replicate in the lung, has been successfully used in BALB/c mouse model to establish the connection between nutrient acquisition and in vivo lung fitness for P. aeruginosa [17]. Therefore, we constructed various mucA− alginate-overproducing strains, such as ΔfadBA145-mucA−, ΔfadBA145ΔglpD-mucA−, ΔfadBA145ΔbetAB-mucA−, ΔfadBA145ΔbetABΔglpD-mucA− and their complemented strains for this study. Prior to the animal study, the phenotypes of all mucA strains were confirmed by patching on minimal media plates with FA, choline, or glycerol as sole carbon sources along with all appropriate controls (all mucA wild-type strains and complemented strains). As expected, the mucA mutation did not affect the metabolism of any of these carbon sources (Fig. S3). As previously described [17], [40], BALB/c mice were inoculated via intratracheal intubation with equal ratios of each mutant and its complement pair (6×106 CFU/animal). At 24 h post-infection, bacterial CFU recovered from the lungs were determined, followed by CI calculations. For all the strains, the average total CFU per mouse lung recovered at 24 hours post-infection was greater than the initial inoculum (Fig. 4B), indicating that all these P. aeruginosa strains maintained the ability to replicate within the mouse lung. The ΔfadBA145 mutant still replicated significantly in vivo compared to its complement. Surprisingly, the ΔfadBA145 CI is quite high compared to other FAD mutants (i.e., fadD mutants) we have previously published where CI is approximately 0.5 [17], [18]. The ΔfadBA145ΔbetAB, ΔfadBA145ΔglpD in vivo CI is lower than the ΔfadBA145 when compared to their respective complements (Fig. 4B), showing the importance of glycerol and choline degradation as potential nutrient sources in vivo. Most significantly, the mean CI value for the triple pathway mutant (i.e., ΔfadBA145ΔbetABΔglpD) showed that the triple pathway mutant had a significantly reduced ability to survive and multiply in the lungs of mice compared to its complement.
We monitored all strains tested in vivo for different virulence expression, including proteases, rhamnolipid, hemolysins and lipases (Fig. S4). With similar level of these common secreted virulence factors observed between strains (Fig. S2), the low CI is most likely due to its inability to metabolize PC and the three components of PC (LCFAs, glycerol, and phosphorylcholine) as a nutrient source, rather than resulting from altered virulence expression. Overall, the altered ability for the pathway mutants to metabolize PC as nutrient in vitro was clearly mirrored by their competitive fitness within the lung.
In summary, P. aeruginosa possesses an impressive repertoire of virulence factors, and the expression of most of these only occurs during the HCD replication and their timely expression is regulated by QS [42], which occurs at HCD. P. aeruginosa requires large amount of readily available energy to reach and maintain HCD and produce the high-energy dependent virulence structure like biofilm. Thus, exploration of the nutrient sources supporting such an energy intensive processes is of importance, especially for chronic P. aeruginosa lung infections in CF patients. In addition, the identification of the genes and pathways for P. aeruginosa HCD replication in CF lungs provides fundamental knowledge for possibly developing new therapeutic strategies targeting bacterial nutrient metabolism in the lung, thereby preventing bacterial HCD. The expression of genes involved in P. aeruginosa PC degradation within the lungs of CF patients has been previously demonstrated [15]. Our study focused on providing further evidence to determine whether PC serves as a significant nutrient source during P. aeruginosa lung infection. In order to decipher the role of PC in vivo, we first characterized PC degradation pathways in vitro. Of the three components released by the enzymatic cleavage of PC by bacterial phospholipase C and lipases (phosphorylcholine, LCFAs, and glycerol), LCFAs are highly reduced and yield the most carbon and energy. In our study, five potential fadBA-operons were investigated and three of them (i.e., fadBA1,4,5-operons) proved to be significantly involved in Fad. The in vitro growth analysis of different pathway mutants (ΔfadBA145, ΔfadBA145ΔbetAB, ΔfadBA145ΔglpD, ΔfadBA145ΔbetABΔglpD) on PC provided direct evidence to support that P. aeruginosa utilizes the FA, glycerol and choline degradation pathways to degrade individual components of PC in vitro. Our in vivo competition study was performed utilizing a mouse lung infection model [40] to evaluate the fitness of the pathway mutants within the lung environment. The triple pathway mutant ΔfadBA145ΔbetABΔglpD exhibited the greatest growth defect on relevant carbon sources in vitro and was outcompeted by its complement in vivo. Since no altered expression of virulence factors was observed for all the pathway mutants and their complement pairs compared to wildtype PAO1, it is highly likely that the decreased ability to utilize PC resulted in lower replication fitness in the lung environment. This study strongly supports the hypothesis that P. aeruginosa utilizes lung surfactant PC as one of the nutrient sources for chronic lung infection.
Materials and Methods
Ethic Statement
All animal experiments were performed in compliance with the NIH (National Institutes of Health) Guide for the Care and Use of Laboratory Animals and were approved by the University of Hawaii Institutional Animal Care and Use Committee (protocol no. 06-023-04).
Bacterial strains and growth conditions
Bacterial strains and plasmids utilized in this study are listed in Table 1 and 2. E. coli EP-Max10B was used as cloning strains and cultured in Luria-Bertani (LB) medium (Difco). Pseudomonas Isolation Agar or Broth (PIA or PIB; Difco) or LB medium were used to culture P. aeruginosa strain PAO1 and derivatives. All fatty acids (FAs) stocks were made as previously described [17]. Strains for growth analyses were cultured in 1× M9 minimal medium +0.2% (w/v) Brij-58 (Sigma) +1% (w/v) casamino acids (CAA) or 0.4% (w/v) of the individual FA, C12∶0 to C16∶0, or C18∶1 Δ9 (Sigma; Fig. 2) and 1× M9 minimal medium +0.2% (w/v) Brij-58 (Sigma) +0.4% (w/v) phosphatidylcholine (PC, Sigma; Fig. 3A), at 37°C with a shaking speed of 200 r.p.m. Since most of FAs hydrolyzed from in vivo PC are C16∶0 (50–60%), with ∼10–20% of each of C14∶0, C16∶1, C18∶1 Δ9, and C18∶2 constituting the rest [43], the growth analysis was performed in 1×M9 minimal medium supplied with each of C12∶0 (medium-chain fatty acid), C14∶0, C16∶0, and C18∶1 Δ9 (LCFAs) as a sole carbon source. Accordingly, the PC we used in these in vitro experiments contains mostly LCFAs, approximately 33% C16∶0, 13% C18∶0, 31% C18∶1 Δ9, and 15% C18∶2. The in vitro competition studies (Fig. 4A) were performed under the growth condition mentioned above as previously described [17].
Table 2. Plasmids used in this studya.
| Plasmids | Lab IDb | Relevant properties | Reference |
| pFlp2 | E0067 | Apr, sacB +; Flp-containing plasmid | [45] |
| pPS856 | E0050 | Apr; Gmr; plasmid with Gmr-FRT-cassette | [45] |
| pUC18 | E0135 | Apr; cloning vector | [50] |
| pUC18-‘mucA’ | E1907 | Apr; mucA internal region cloned into pUC18 | This study |
| pUC19 | E0014 | Apr; cloning vector with Plac | [50] |
| pUC19-glpD | E1843 | Apr; pUC19 with glpD gene cloned in downstream of Plac | This study |
| pEX18T | E0055 | Apr, oriT+, sacB +; gene replacement vector | [45] |
| pEX18TΔfadBA1::Gm | E0202 | Apr, Gmr; pEX18T with ΔfadBA1 operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔfadBA2::Gm | E0224 | Apr, Gmr; pEX18T with ΔfadBA2 operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔfadBA3::Gm | E0225 | Apr, Gmr; pEX18T with ΔfadBA3 operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔfadBA4::Gm | E0226 | Apr, Gmr; pEX18T with ΔfadBA4 operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔfadBA5::Gm | E0461 | Apr, Gmr; pEX18T with ΔfadBA5 operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔglpD::Gm | E1066 | Apr, Gmr; pEX18T with ΔglpD operon with Gmr-FRT-cassette insertion | This study |
| pEX18TΔbetAB::Gm | E1070 | Apr, Gmr; pEX18T with ΔbetAB operon with Gmr-FRT-cassette insertion | This study |
| miniCTX2 | E0076 | Tetr; site-specific integration vector | [46] |
| miniCTX2-fadBA5 | E1765 | Tetr; miniCTX2 with cloned fadBA5 | This study |
| miniCTX2-fadBA5/glpD | E2035 | Tetr; miniCTX2 with cloned fadBA5/glpD | This study |
| miniCTX2-fadBA5/betAB | E1953 | Tetr; miniCTX2 with cloned fadBA5/betAB | This study |
| miniCTX2-fadBA5/betAB/glpD | E1992 | Tetr; miniCTX2 with cloned fadBA5/betAB/glpD | This study |
For plasmids constructed in this study, please see text for further details.
Please use Lab ID for requesting plasmids.
General molecular methods
Oligonuceotides were synthesized through Integrated DNA Technology and are listed in Table 3. All molecular methods and their components utilized were employed as previously described [44].
Table 3. Primers used in this study.
| Primer number and name | Sequencea |
| 186; fadBA1-upstream | 5′-CGAAAGCTTGCATGGTGCTATCTTCC-3′ |
| 187; fadBA1-downstream | 5′-GCGGAATTCGCCCTACCCGTGGCG-3′ |
| 218; fadBA2-upstream | 5′-CGGTGAAGCTTTCGCGCACC-3′ |
| 219; fadBA2-downstream | 5′-GGGGAATTCGGTGTCTCATCGGCAGCGC-3′ |
| 220; fadBA3-upstream | 5′-GCGAAGCTTATTCAGCAGGAGAAAACGACG-3′ |
| 221; fadBA3-downstream | 5′-TGCGGAATTCGACGGATAGTCGCCGCTAC-3′ |
| 211; fadBA4-upstream | 5′-CGTAAGCTTGCCGGGGAGTCAGGGGC-3′ |
| 212; fadBA4-downstream | 5′-CCCGAATTCGCACGGCACCGCCCAAG-3′ |
| 272; fadBA5-HindIII | 5′-AGTTCAAGCTTCCATAATAGC-3′ |
| 273; fadBA5-EcoRI | 5′-CCCGGAATTCCCCCTTCGAGAACGCTTAG-3′ |
| 518; glpK-BamHI | 5′-AGCTGAAGTGGATCCTCGACAA-3′ |
| 519; glpKD-SacI | 5′-CTGGCGAGCTCAGGCCGCATGCACCCG-3′ |
| 522; betA-SacI | 5′-CAACGAGCTCGGCGATATCTACGGCGG-3′ |
| 523; betB-HindIII | 5′-GCCAAAGCTTCCAGGACAAGAACGGCT3′ |
| 888; Xho-fadB5 | 5′-CCTGCGCAGAGGGCCTCGAGGAGGGC-3′ |
| 889; fadB5-Bam | 5′-GGGCACGAGGATCCCCGGCTTTCCCC-3′ |
| 895; Spe-betB | 5′-CGGATTCAGACTAGTACCTGCTCG-3′ |
| 896; Hind-glpD | 5′-GCCTGGTGAAGCTTCGGGCTGGTC-3′ |
| 927; SacI-Plac-glpD | 5′-CGCTCGCCGGAGCTCGAACGACCGAGC-3′ |
Restriction enzyme sites utilized in this study are underlined.
Construction of mutants and complementation strains
All mutants were constructed as described previously [45]. Briefly, the fadBA (fadBA1, fadBA2, fadBA3, fadBA4, fadBA5) operons, betAB operon, and glpD gene were amplified by PCR using respective upstream and downstream primer pair listed in Table 3. The PCR products were purified from the gel, digested with appropriate restriction enzymes, and cloned into the gene replacement vector pEX18T, digested with the same restriction enzymes, to yield each of the pEX18T-target gene constructs. After deletions were made on plasmid in each of the fadBA-operons, the glpD gene, and the betAB-operon through restriction digestion (fadBA1: PstI, BamHI; fadBA2: StuI, BamHI; fadBA3: NotI, SmaI; fadBA4: EcoRV; fadBA5: SphI, PstI) and blunt-ended (except for glpD, which was blunt-ended using SmaI), the 1.1 kb FRT-GmR-FRT cassette obtained from pPS856 digested with SmaI was inserted into each gene. These newly constructed pEX18T vectors were transformed into E.coli SM10 or ER2566-mob, and conjugated into PAO1 to engineer the unmarked mutations as previously described [45]. To obtain all triple mutants, we invested an enormous amount of work to first create all single and double mutants with the proper confirmed Flp/FRT-excision of the gentamycin antibiotic resistance cassette to recycle this resistance marker for subsequent mutagenesis (data not shown).
The single copy integration vector, miniCTX2, was used to engineer the complemented strains for each triple-pathway mutant as previously described [46]. Briefly, fadBA5 and betAB were PCR amplified with primers 888/889 and 522/895, respectively. The miniCTX2-fadBA5 was derived by inserting fadBA5 fragment into miniCTX2, both digested with XhoI and BamHI. The betAB gene was sub-cloned in using SacI and SpeI, yielding miniCTX2-fadBA5-betAB. glpD was first cloned into pUC19 by digesting the PCR product with HindIII and SacI, which was amplified using primers 896/519. Plac-glpD fragment was amplified using primers 519/927 from pUC19-glpD and cloned into miniCTX2-fadBA5 and miniCTX2-fadBA5-betAB to yield miniCTX2-fadBA5-glpD and miniCTX2-fadBA5-betAB-glpD, respectively.
The newly engineered mutant strains ΔfadBA145, ΔfadBA145ΔglpD, ΔfadBA145ΔbetAB, and ΔfadBA145ΔbetABΔglpD, were complemented using the relevant gene(s) on the miniCTX2 single copy integration vector as preciously described [46]. The resulting strains ΔfadBA145/complement, ΔfadBA145ΔglpD/complement, ΔfadBA145ΔbetAB/complement, ΔfadBA145ΔbetABΔglpD/complement were used in the growth curve experiment (Table 1 and Fig. 3A).
Growth characterization of mutants and complementation strains
Growth curve analyses have been described previously [17]. Briefly, all strains utilized were initially grown overnight in Pseudomonas Isolation Broth (PIB). The overnight cultures were centrifuged and the cell pellets were washed twice with 1×M9 minimal medium, and then a 1∶50 dilution was made into 25 ml of the respective media (described above) for different growth curves. To clarify any insoluble FA, individual cultures were diluted 4-fold in 4% Brij-58, pre-incubated at 42°C for 2 minutes, prior to taking OD540 measurement at each time point (Fig. 2 and 3). To obtain the growth curves in Figure S2, all of the strains were grown overnight at 37°C in LB broth. The overnight cultures were centrifuged and the cell pellets were washed twice with 1×M9 minimal medium, and then a 1∶400 dilution was made into respective media (described above) for different growth curves. 125 µl aliquots of the diluted cultures were transferred to a sterile, polystyrene 96-well assay plate (Falcon Microtest flat bottom plate, catalog no. 35–1172; Becton-Dickinson Labware). Growth was recorded using an ELx808 Absorbance Microplate Reader (BioTek Instruments, Winooski, VT) under the following conditions: temperature37°C, and shaking at a low speed. The plate was read at 630 nm every 30 min for 40 h. All of the data was transferred and plotted using Prism.
Virulence factors detection
Strains used for virulence factors detection were grown in LB medium. At each time point, aliquots of individual culture were used for OD540 measurement (Fig. 3B). The detection of proteases, hemolysins, lipases, and rhamnolipid was performed as described elsewhere [17]. All assays were conducted in triplicate, and the data were analyzed as previously described [17].
Growth Phenotype Confirmation of Mucoid and Non-mucoid Strains
To confirm that mutations in mucA do not have additional effects on nutrient metabolism of the pathway mutant strains, all of the pathway mutants and complement strains were purified on LB plate or LB plate supplemented with 250 µl/ml carbenicillin (Cb250) for mucA− strains. After 24 h incubation at 37°C, single colony of each strain was patched on 1× M9 solid medium +1% (w/v) Brij-58 supplemented with 0.2% (w/v) C18∶1 Δ9, 40 mM glycerol or 30 mM choline as sole carbon source. They were also patched on LB plate, which served as a control. The growth pattern was observed after 24–36 h incubation at 37°C (Fig. S3).
In vitro and in vivo competition studies
In vitro and in vivo competition studies were performed as previously described [17]. Briefly, seven growth media with different carbon sources, including Luria-Bertani (LB) medium, casamino acids (CAA), glucose, PC, C18∶1 Δ9, choline, and glycerol, were used in this study. The bacterial CFU were determined after inoculation into each of the medium for 24–48 h. The CI was calculated as the CFU ratio of mutant/wildtype recovered at each time point divided by the CFU ratio of mutant/wildtype in the input inoculum [47]. The smaller the CI value, the more the significant reduction in fitness of the mutant.
Various alginate-overproducing strains, ΔfadBA145-mucA−, ΔfadBA145ΔglpD-mucA−, ΔfadBA145ΔbetAB-mucA−, ΔfadBA145ΔbetABΔglpD-mucA− and the complement strains for each mutant utilized in this study are listed in Table 1. The use of the mucA− mutation is essential in this animal model as previously described [40].
Supporting Information
Five potential fadBA -operon homologues of P. aeruginosa. (A) Genes of operons (GenBank accession numbers in parentheses) are shown in light purple with percent of identity and similarity to the E. coli FadBA. fadBA1 is 3.363 kb; fadBA2 is 2.760 kb; fadBA3 is 2.346 kb; fadBA4 is 2.887 kb; and fadBA5 is 3.353 kb, (B) Alignment of P. aeruginosa FadAs and FadBs with E. coli FadA and FadB motifs. Amino acids with similar properties are assigned the same colors using CLC Sequence Viewer 6.
(TIF)
Growth analysis of different single fadBA mutants on medium (C12∶0) and long chain-length fatty acid (C14∶0, C16∶0 and C18∶1Δ9). Along with the wildtype PAO1 strain, mutants ΔfadBA1, ΔfadBA2, ΔfadBA3, ΔfadBA4 and ΔfadBA5 were grown in 1×M9 minimal medium supplemented with 0.05% different test FAs (A to D) and 1% Brij-58 or LB broth as a control (E). Only the ΔfadBA5 mutant showed various defects when grown with FAs of different chain-lengths, no significant growth defects were observed for the rest of single fadBA mutants. All of the mutants grew to the same level as wildtype when grown in LB.
(TIF)
Growth Phenotype Confirmation of Mucoid and Non-mucoid Strains. Along with the wildtype PAO1 and PAO1-mucA− strains, all of the pathway mutants and their corresponding complement strains were patched on 1× M9 solid medium +1% (w/v) Brij-58 supplemented with 0.2% (w/v) C18∶1 Δ9 (B), 40 mM glycerol (C), or 30 mM choline (D). (A) Growth on LB was performed as a control. Alginate over-producing strains show a light sheen surface indicated by white arrow in panel A. Similar growth defects were shown between mucoid and non-mucoid strains on different plates. A detailed plate layout is shown in panel E with strains identification of Table 1 in parentheses.
(TIF)
Analyses of proteases, hemolysins, lipases, and rhamnolipid productions by P. aeruginosa various pathway mutant. No mutants displayed significant (P≤0.05, based on student t-test) decrease in productions of proteases (A), rhamnolipid (B), hemolysins (C), and lipases (D).
(TIF)
Acknowledgments
We would like to thank previous (Asha S. Nayar, and Joon Kim) and current (Jan Zarzycki-Siek) lab members for their assistance in some mutagenesis experiments, as well as current lab members (Andrew Bluhm and Ian McMillan) for their critical reading of this manuscript.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funding for this research was provided by grant number P20GM103516 from the National Institute of General Medical Sciences of the National Institutes of Health. The study design and content are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wilson R, Dowling RB (1998) Pseudomonas aeruginosa and other related species. Thorax 53: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Driscoll J, Brody SL, Kollef MH (2007) The epidemiology, pathogenesis and treatemnt of Pseudomonas aeruginosa infection. Drugs 67: 351–368. [DOI] [PubMed] [Google Scholar]
- 3. Richards MJ, Edwards JR, Culver DH, Gaynes RP (1999) Nosocomial infections in medical intensive care units in the United States. Crit Care Med 27: 887–892. [DOI] [PubMed] [Google Scholar]
- 4. Lode H, Raffenberg M, Erbes R, Geerdes-Fenge H, Mauch H (2000) Nosocomial pneumonia: epidemiology, pathogenesis, diagnosis, treatment and prevention. Curr Opin Infect Dis 13: 377–384. [DOI] [PubMed] [Google Scholar]
- 5. Pompilio A, Crocetta V, Scocchi M, Pomponio S, Di Vincenzo V, et al. (2012) Potential novel therapeutic strategies in cystic fibrosis: antimicrobial and anti-biofilm activity of natural and designed alpha-helical peptides against Staphylococcus aureus, Pseudomonas aeruginosa, and Stenotrophomonas maltophilia . BMC Microbiol 12: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL (2002) Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 34: 91–100. [DOI] [PubMed] [Google Scholar]
- 7. Wagner VE, Iglewski BH (2008) P. aeruginosa biofilms in CF infection. Clin Rev Allergy Immunol 35: 124–134. [DOI] [PubMed] [Google Scholar]
- 8. Fricks-Lima J, Hendrickson CM, Allgaier M, Zhuo H, Wiener-Kronish JP, et al. (2011) Differences in biofilm formation and antimicrobial resistance of Pseudomonas aeruginosa isolated from airways of mechanically ventilated patients and cystic fibrosis patients. Int J Antimicrob Agents 37: 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coban AY, Ciftci A, Onuk EE, Erturan Z, Tanriverdi Cayci Y, et al. (2009) Investigation of biofilm formation and relationship with genotype and antibiotic susceptibility of Pseudomonas aeruginosa strains isolated from patients with cystic fibrosis. Mikrobiyol Bul 43: 563–573. [PubMed] [Google Scholar]
- 10. Pesci EC, Iglewski BH (1997) The chain of command in Pseudomonas quorum sensing. Trends Microbiol 5: 132–134. [DOI] [PubMed] [Google Scholar]
- 11. Bjarnsholt T, Jensen PO, Jakobsen TH, Phipps R, Nielsen AK, et al. (2010) Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS One 5: e10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith RS, Iglewski BH (2003) P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol 6: 56–60. [DOI] [PubMed] [Google Scholar]
- 13. Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, et al. (2000) Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407: 762–764. [DOI] [PubMed] [Google Scholar]
- 14. Storey DG, Ujack EE, Rabin HR (1992) Population transcript accumulation of Pseudomonas aeruginosa exotoxin A and elastase in sputa from patients with cystic fibrosis. Infect Immun 60: 4687–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Son MS, Matthews Jr WJ, Kang Y, Nguyen DT, Hoang TT (2007) In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infection and immunity 75: 5313–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmer KL, Mashburn LM, Singh PK, Whiteley M (2005) Cystic fibrosis sputum supports growth and cues key aspects of Pseudomonas aeruginosa physiology. J Bacteriol 187: 5267–5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kang Y, Zarzycki-Siek J, Walton CB, Norris MH, Hoang TT (2010) Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa . PLoS One 5: e13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zarzycki-Siek J, Norris MH, Kang Y, Sun Z, Bluhm AP, et al. (2013) Elucidating the Pseudomonas aeruginosa Fatty Acid Degradation Pathway: Identification of Additional Fatty Acyl-CoA Synthetase Homologues. PLoS One 8: e64554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miller RM, Tomaras AP, Barker AP, Voelker DR, Chan ED, et al. (2008) Pseudomonas aeruginosa twitching motility-mediated chemotaxis towards phospholipids and fatty acids: specificity and metabolic requirements. Journal of Bacteriology 190: 4038–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernhard W, Wang J-Y, Tschernig T, Tummler B, Hedrich HJ, et al. (1997) Lung surfactant in a cystic fibrosis animal model: increased alveolar phospholipid pool size without altered composition and surface tension function in cftr m1HGU/m1HGU mice. Thorax 52: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bernhard W, Hoffmann S, Dombrowsky H, Rau GA, Kamlage A, et al. (2001) Phosphatidylcholine molecular species in lung surfactant: composition in relation to respiratory rate and lung development. Am J Respir Cell Mol Biol 25: 725–731. [DOI] [PubMed] [Google Scholar]
- 22. Hite RD (2002) Surfactant deficiency in adults. Clin Pulm Med 9: 39–45. [Google Scholar]
- 23. Velasco-Garcia R, Mujica-Jimenez C, Mendoza-Hernandez G, Munoz-Clares RA (1999) Rapid purification and properties of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa . J Bacteriol 181: 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wargo MJ, Szwergold BS, Hogan DA (2008) Identification of two gene clusters and a transcriptional regulator required for Pseudomonas aeruginosa glycine betaine catabolism. J Bacteriol 190: 2690–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schweizer HP, Po C (1996) Regulation of glycerol metabolism in Pseudomonas aeruginosa: characterization of the glpR repressor gene. J Bacteriol 178: 5215–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schweizer HP, Po C, Bacic MK (1995) Identification of Pseudomonas aeruginosa GlpM, whose gene product is required for efficient alginate biosynthesis from various carbon sources. JBacteriol 177: 4801–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schweizer HP, Po C, Jump R (1997) Structure and gene-polypeptide relationships of the region encoding glycerol diffusion facilitator (glpF) and glycerol kinase (glpK) of Pseudomonas aeruginosa . Microbiology 143: 1287–1297. [DOI] [PubMed] [Google Scholar]
- 28. Clark D (1981) Regulation of fatty acid degradation in Escherichia coli: anaylsis by operon fusion. J Bacteriol 148: 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pramanik A, Pawar S, Antonian E, Schulz H (1979) Five different enzymatic acitivities are associated with the multienzyme complex of fatty acid oxidation in Escherichia coli . J Bacteriol 137: 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campbell JW, Cronan Jr JE (2002) The enigmatic Escherichia coli fadE gene is yafH. . J Bacteriol 184: 3759–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kang Y, Nguyen DT, Son MS, Hoang TT (2008) The Pseudomonas aeruginosa PsrA responds to long-chain fatty acid signals to regulate the fadBA5 β-oxidation operon. Microbiology 154: 1584–1598. [DOI] [PubMed] [Google Scholar]
- 32. Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406: 959–964. [DOI] [PubMed] [Google Scholar]
- 33. Son MS, Nguyen DT, Kang Y, Hoang TT (2008) Engineering of FRT-lacZ fusion constructs: induction of the Pseudomonas aeruginosa fadAB1 operon by medium and long chain-length fatty acids. Plasmid 59: 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beassoni PR, Otero LH, Massimelli MJ, Lisa AT, Domenech CE (2006) Critical active-site residues identified by site-directed mutagenesis in Pseudomonas aeruginosa phosphorylcholine phosphatase, a new member of the haloacid dehalogenases hydrolase superfamily. Curr Microbiol 53: 534–539. [DOI] [PubMed] [Google Scholar]
- 35. Massimelli ML, Beassoni PR, Forrellad MA, Barra JL, Garrido MN, et al. (2005) Identification, cloning, and expression of Pseudomonas aeruginosa phosphorylcholine phosphatase gene. Curr Microbiol 50: 251–256. [DOI] [PubMed] [Google Scholar]
- 36. Nagasawa T KY, Tani Y, Ogata K (1976) Purification and characterization of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa A-16. Agric Biol Chem 40: 1743–1749. [Google Scholar]
- 37. Wargo MJ, Ho TC, Gross MJ, Whittaker LA, Hogan DA (2009) GbdR regulates Pseudomonas aeruginosa plcH and pchP transcription in response to choline catabolites. Infect Immun 77: 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schweizer HP, Po C (1994) Cloning and nucleotide sequence of the glpD gene encoding sn-glycerol-3-phosphate dehydrogenase from Pseudomonas aeruginosa . J Bacteriol 176: 2184–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weissenborn DL, Larson TJ (1992) Structure and regulation of the glpFK operon encoding glycerol diffusion facilitator and glycerol kinase of Escherichia coli K-12. JBiolChem 267: 6122–6131. [PubMed] [Google Scholar]
- 40. Hoffmann N, Rasmussen TB, Jensen PO, Stub C, Hentzer M, et al. (2005) Novel mouse model of chronic Pseudomonas aeruginosa lung infection mimicking cystic fibrosis. Infect Immun 73: 2504–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boucher JC, Yu H, Mudd MH, Deretic V (1997) Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect Immun 65: 3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Darch SE, West SA, Winzer K, Diggle SP (2012) Density-dependent fitness benefits in quorum-sensing bacterial populations. Proc Natl Acad Sci U S A 109: 8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Postle AD, Mander A, Reid KBM, Wang J-Y, Wright SM, et al. (1999) Deficient hydrophilic lung surfactant protein A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol 20: 90–98. [DOI] [PubMed] [Google Scholar]
- 44. Kang Y, Norris MH, Barrett AR, Wilcox BA, Hoang TT (2009) Engineering of tellurite-resistant genetic tools for single-copy chromosomal analysis of Burkholderia spp. and characterization of the Burkholderia thailandensis betBA operon. Applied and Environmental Microbiology 75: 4015–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212: 77–86. [DOI] [PubMed] [Google Scholar]
- 46. Hoang TT, Kutchma AJ, Becher A, Schweizer HP (2000) Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43: 59–72. [DOI] [PubMed] [Google Scholar]
- 47. Brickman TJ, Vanderpool CK, Armstrong SK (2006) Heme transport contributes to in vivo fitness of Bordetella pertussis during primary infection in mice. Infect Immun 74: 1741–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Simon R, Priefer U, Puehler A (1983) A broad-host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1: 784–791. [Google Scholar]
- 49. Holloway BW, Roemling U, Tuemmler B (1994) Genomic mapping of Pseudomonas aeruginosa PAO. Microbiol 140: 2907–2929. [DOI] [PubMed] [Google Scholar]
- 50. Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 cloning vectors and host strains: nucleotide sequences of the M13 mp18 and pUC19 vectors. Gene 33: 103–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Five potential fadBA -operon homologues of P. aeruginosa. (A) Genes of operons (GenBank accession numbers in parentheses) are shown in light purple with percent of identity and similarity to the E. coli FadBA. fadBA1 is 3.363 kb; fadBA2 is 2.760 kb; fadBA3 is 2.346 kb; fadBA4 is 2.887 kb; and fadBA5 is 3.353 kb, (B) Alignment of P. aeruginosa FadAs and FadBs with E. coli FadA and FadB motifs. Amino acids with similar properties are assigned the same colors using CLC Sequence Viewer 6.
(TIF)
Growth analysis of different single fadBA mutants on medium (C12∶0) and long chain-length fatty acid (C14∶0, C16∶0 and C18∶1Δ9). Along with the wildtype PAO1 strain, mutants ΔfadBA1, ΔfadBA2, ΔfadBA3, ΔfadBA4 and ΔfadBA5 were grown in 1×M9 minimal medium supplemented with 0.05% different test FAs (A to D) and 1% Brij-58 or LB broth as a control (E). Only the ΔfadBA5 mutant showed various defects when grown with FAs of different chain-lengths, no significant growth defects were observed for the rest of single fadBA mutants. All of the mutants grew to the same level as wildtype when grown in LB.
(TIF)
Growth Phenotype Confirmation of Mucoid and Non-mucoid Strains. Along with the wildtype PAO1 and PAO1-mucA− strains, all of the pathway mutants and their corresponding complement strains were patched on 1× M9 solid medium +1% (w/v) Brij-58 supplemented with 0.2% (w/v) C18∶1 Δ9 (B), 40 mM glycerol (C), or 30 mM choline (D). (A) Growth on LB was performed as a control. Alginate over-producing strains show a light sheen surface indicated by white arrow in panel A. Similar growth defects were shown between mucoid and non-mucoid strains on different plates. A detailed plate layout is shown in panel E with strains identification of Table 1 in parentheses.
(TIF)
Analyses of proteases, hemolysins, lipases, and rhamnolipid productions by P. aeruginosa various pathway mutant. No mutants displayed significant (P≤0.05, based on student t-test) decrease in productions of proteases (A), rhamnolipid (B), hemolysins (C), and lipases (D).
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.