

Abstract
An active site water molecule coordinated by conserved histidine and asparagine residues seems to serve as the catalytic base in all ent-copalyl diphosphate synthases (CPSs). When these residues are substituted by alanine, the mutant CPSs produce stereochemically novel ent-8-hydroxy-CPP. Given the requisite presence of CPSs in all land plants for gibberellin phytohormone biosynthesis, such plasticity presumably underlies the observed extensive diversification of the resulting labdane-related diterpenoids.
Keywords: enzyme catalysis, terpene synthase, natural products, biosynthesis, hydratases
Plant natural products biosynthesis has been suggested to be largely derived from hormone metabolism.[1] For example, production of labdane-related diterpenoids by plants is derived from gibberellin phytohormone biosynthesis.[2] Giberellin production is initiated by a cycloisomerization reaction that transforms the general diterpenoid precursor (E,E,E)-geranylgeranyl diphosphate (GGPP; 1) into ent-copalyl diphosphate (ent-CPP; 2). This reaction is catalyzed by ent-CPP synthases (CPSs), which are then ubiquitously found in all vascular plants, providing the source for repeated evolution of more specialized labdane-related diterpenoid biosynthesis, as evidenced by the fact that the vast majority of the ~7,000 known such natural products are uniquely produced by plants.[2] Intriguingly, although it is known that evolutionarily derived class II diterpene synthases can incorporate water into their reactions, producing a chemically altered (hydroxylated) analog of CPP, these also all differ in stereochemistry (e.g., 8α-hydroxy-CPP, 3).[3] Accordingly, formation of hydroxylated ent-CPP would represent a novel enzymatic product. Here we report identification of the CPS catalytic base group as a water activated by a histidine-asparagine dyad, with substitution of either residue by alanine leading to production of an epimeric pair of 8-hydroxylated-ent-CPPs (β, 4; α, 5).
The characteristic feature of the labdane-related diterpenoid super-family of natural products is their biosynthetic origins in a bicyclization reaction catalysed by class II diterpene cyclases. This directly results in formation of a labdadienyl hydrocarbon backbone, leading to the unifying nomenclature.[2] The underlying enzymatic mechanism relies on acid-base catalysis, with initiating protonation of the terminal carbon-carbon double bond, sequential cyclization via addition from the internal double bonds in a carbocationic cascade, followed by deprotonation (Scheme 1). While this can simply take the form of a cyclo-isomerization reaction, as indicated by the designation of CPSs as isomerases (EC 5.5.1.13), the reaction also is amendable to the addition of water prior to deprotonation, leading to formal designation of the relevant class II diterpene cyclases as hydratases (EC 4.2.1.133). However, these cyclo-hydratases also differ in sterochemical outcome, which stems from pre-catalysis folding of the substrate 1 in a ‘normal’ pro-chair-chair conformation rather than the antipodal conformation leading to the ent-CPP produced by CPSs en route to gibberellins.[2] Thus, while the CPSs produce 2 (9R,10R), all of the characterized class II diterpene cyclo-hydratases (i.e. those that produce hydroxylated analogs), despite many exhibiting clear homology to CPSs, produce 3 (9S10,S) instead.
Scheme 1.

Reactions mediated by known class II diterpene cyclo-isomerases (e.g., 2) and cyclo-hydratases (i.e., 3), along with the novel products found here (4 and 5).
Class II diterpene cyclases are characterized by a (D,E)xDD motif, wherein the middle aspartic acid residue acts as the catalytic acid.[4] By contrast, there does not appear to be a similarly well-conserved residue that acts as the catalytic base, perhaps in part due to the variation in substrate conformation underlying the production of different stereoisomers of CPP, which presumably requires different relative positioning of the catalytic acid and base.[2] Thus, the catalytic base might be conserved by stereochemical product outcome.
Building on our recently reported crystal structure for the bifunctional abietadiene synthase from Abies grandis (AgAS),[5] we have recently reported that histidine 348 may function as the catalytic base.[3h] However, this is limited to the closely related bifunctional diterpene synthases from gymnosperms (all of which produce normal CPP),[6] as a histidine is not found at this position in other class II diterpene cyclases, including the only known ent-CPP producing CPS from a gymnosperm – i.e., the CPS from Picea glauca (PgCPS).[7] Intriguingly, substitutions for this histidine lead to incorporation of water into the usually catalysed cyclo-isomerization reaction, with mutation to either alanine or, more specifically, aspartate, leading to cyclo-hydratase activity.[3h] However, the AgAS:H348A and H348D mutants presumably catalyse the production of 3, with normal (9S,10S) stereochemistry in common with all of the previously characterized class II diterpene cyclo-hydratases.
Crystal structures also have been reported for the CPS from Arabidopsis thaliana (AtCPS).[8] Careful inspection of these revealed the presence of an active site histidine (H263 in AtCPS), which differs in positioning from the critical histidine characterized in AgAS, but is conserved in all the known ent-CPP producing CPSs, including those from the monocot rice (Oryza sativa; e.g., OsCPS1),[9] as well as the gymnosperm PgCPS,[7] and even a bifunctional diterpene cyclase from the moss Physcomitrella patens (PpCPSKS),[10] representing over 450 million years of evolutionary separation.[11]
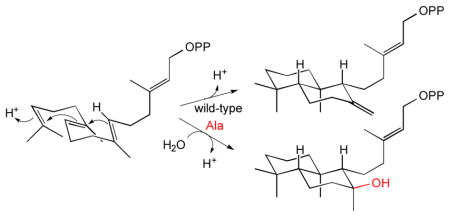
Much as with AgAS,[3h] we initially hypothesized that this histidine might be the catalytic base. Accordingly, we substituted this histidine in AtCPS with alanine, creating an AtCPS:H263A mutant. Rather than the loss of catalytic activity that might have been expected, this substitution led to significant production of two novel hydroxylated products, along with some production of 2 as well, either from in vitro reactions with 1, or upon expression in E. coli metabolically engineered to produce 1[12] (see Figure 2). The two new observed compounds, following dephosphorylation, by GC-MS exhibited identical retention times and mass spectra to the previously reported pair of 8-hydroxy epimers of labda-13E-en-8,15-diol (Figure S1),[3i] demonstrating cyclo-hydratase activity. This was further verified by NMR analysis of the epimer corresponding to the major product 4 (Table S1 and Figures S2 & 3).
Figure 2.

Effect of H263A, N322A and H263A/N322A mutations on AtCPS product outcome. Chromatograms from GC-MS analysis of the dephosphorylated products (numbering as in text).
Our previous results with AgAS suggested that it might be possible to divert AtCPS more completely to cyclo-hydratase activity by substitution with aspartate instead of alanine.[3h] Accordingly, we substituted aspartate for H263 in AtCPS. However, the resulting AtCPS:H263D mutant failed to exhibit any cyclo-hydratase activity, although it retained the ability to produce 2 (Figure S4). This result clearly differentiates the role of the CPS specific critical histidine identified here from that previously found in AgAS.
Careful inspection of AtCPS crystal structure revealed that H263 is hydrogen bonded to a water molecule that also is held in place by a hydrogen bond to an active site asparagine (N322 in AtCPS). This asparagine also is well conserved in ent-CPP producing CPSs (Figure 1), consistent with a role in catalysis. Substitution of this conserved asparagine with alanine led to cyclo-hydratase activity with AtCPS:N322A, as well as AtCPS:H263A/N322A, both of which similarly largely produce the 8-hydroxy epimers observed with AtCPS:H263A (Figure 2). These results indicate that the actual base in the cyclo-isomerization reaction catalysed by CPSs is the tightly bound water (i.e., this water is activated by a catalytic His-Asn dyad, forming a catalytic base group). We hypothesize that, in the absence of either or both ligating residues, this water is then directly added to the labda-13E-en-8-yl carbocation intermediate, with the resulting oxacarbenium ion quenched by deprotonation catalysed by H263 in the case of the AtCPS:N322A mutant, or by an additional water molecule in the case of alanine substitution for AtCPS:H263 (i.e., with H263A or H263A/N322A). To verify that the observed cyclo-hydratase activity was enabled by this latter hypothesized steric effect (i.e., opening up of the active site), we substituted valine and isoleucine for AtCPS:H263. While the AtCPS:H263V mutant retained some ability to make 4 as well as 2, the AtCPSH263I mutant is largely inactive, although it does make trace amounts of 4 (Figure S5), consistent with the hypothesis that cyclo-hydratase activity requires suitable space for an additional water molecule to bind (i.e., while sufficient steric volume might be available with these latter mutants, the hydrophobic effect of these larger aliphatic side chains makes occupation by a water increasingly unlikely).
Figure 1.

Identification of catalytic base group, consisting of water activated by conserved active site histidine and asparagine in CPSs. A) Active site of AtCPS (cartoon format) with side chains of the identified histidine and asparagine with bound water molecule (red sphere) as well as the aspartates of the DxDD catalytic acid motif, in stick representation. B) Conservation of the histidine and asparagine (boxed) in CPSs spanning land plant evolution, as indicated by alignment of the representative examples described in text.
Given that AtCPS produces ent-CPP, we hypothesized that the compounds observed here were actually enantiomeric – i.e., ent-labda-13E-en-8,15-diols (e.g., eperu-13-en-8β,15-diol[13]), resulting from dephosphorylation of 8-hydroxy-ent-CPP, rather than the previously observed normal (9S,10S) isomers (e.g., 3). This stereochemistry was investigated by enzymatic feeding studies, using the previously reported specificity of the bifunctional cis-abienol synthase from Abies balsamea (AbCAS), as this has been shown to specifically react with only 3.[3g] Based on previous work,[4, 14] the endogenous class II activity of AbCAS was blocked by mutation of the aspartate that acts as the catalytic acid (i.e., the ‘middle’ aspartate from the highly conserved DxDD motif[4]) to an alanine. The resulting AbCAS:D405A mutant does not react with 1, but will react with 3 produced by either a previously characterized class II diterpene cyclo-hydratase from Nicotiana glutinosa (NgCLS), or the AgAS:H348D mutant (with additional D621A mutation to prevent any further reaction from its endogenous subsequently acting class I activity),[3h] but does not react with any product of the AtCPS cyclo-hydratase mutants (Figure S6). These results demonstrate the expected retention of stereochemistry for the AgAS and, more critically, AtCPS cyclo-hydratase mutants as well. Notably, the hydroxylated products from the AtCPS mutants then exhibits enantiomeric configuration, which appears to be the first example of a class II diterpene cyclo-hydratase producing this 9R,10R stereoisomer, specifically both 8β-hydroxy-ent-CPP (4) and 8α-hydroxy-ent-CPP (5).
Although there is a substrate analog bound in the active site of the AtCPS crystal structures, this is clearly not in a reactive conformation.[8] Nevertheless, analysis of the products of the AtCPS cyclo-hydratase mutants presented here provides some indication of the positioning of the water molecule relative to the labda-13E-en-8-yl carbocation intermediate (Scheme 2). The production of both 8-hydroxy epimers demonstrates access to both faces of this final carbocation, with the relatively greater epimeric specificity exhibited by AtCPS:N322A relative to either AtCPS:H263A or AtCPS:H263A/N322A, indicating that H263 hinders access to the ‘bottom’ face of the labda-13E-en-8-yl carbocation intermediate. This contrasts to the product outcome mediated by the AgAS:H348A mutant, which more selectively produces the 8α-epimer 3, along with appreciable amounts of the 7,8-double bond isomer of CPP,[3h] which then indicates a subtle difference in relative positioning of the base in the CPSs versus gymnosperm enzymes. Intriguingly, these results suggest it may be possible to make other product variants through other active site substitutions (e.g., the 8,9-double bond isomer, which has not been seen before).
Scheme 2.

The role of water in the reactions catalysed by wild type AtCPS or H263A/N322A mutant.
In any case, the ability of simple alanine substitutions for the histidine and/or asparagine of the catalytic base group identified here to fundamentally alter product chemical outcome demonstrates striking plasticity for the CPSs ubiquitously found in all land plants. Coupled to the previously demonstrated plasticity of the subsequently acting class I diterpene synthases,[15] this highlights the ability of simple single residue changes to generate new chemical diversity from the enzymes required for gibberellin phytohormone biosynthesis. For example, the ability of the CPS mutants characterized here to produce novel hydroxylated versions of CPP hold the promise of leading to the further production of more elaborated compounds, such as enantiomeric forms of cis-abienol, sclareol or manoyl oxide (Scheme 3). Indeed, this plasticity presumably led to the observed extensive diversification of labdane-related diterpenoids in plants.
Scheme 3.

Potential diterpenoids that might be produced from 4.
Experimental Section
The recombinant AtCPS used here was expressed as a previously described pseudomature construct.[16] Mutants were generated by whole plasmid PCR amplification with overlapping mutagenic primers of pENTR/SD/D-TOPO (Invitrogen) clones, and verified by complete gene sequencing prior to transfer by directional recombination to expression vectors (pDEST17 and pGG-DEST). The resulting constructs were heterologously expressed in the C41 OverExpress strain of Escherichia coli (Lucigen), much as previously described.[17] Briefly, the recombinant E. coli were grown in liquid NZY media to 0.6 A600 at 37 °C, then shifted to 16 °C for an hour prior to induction with 0.5 mM IPTG, followed by fermentation at 16 °C. For in vitro assays, the enzymes were expressed as pDEST17 6xHis tagged constructs for ease of purification, which was accomplished much as previously described.[18] Briefly, cells from overnight fermentation were harvested by centrifugation, lysed by gentle sonification in lysis buffer (50 mM Bis-Tris, pH 6.8, 150 mM KCl, 10 mM MgCl2, 1 mM DTT, 10% glycerol), with the resulting lysate clarified by centrifugation (15,000g x 20 min. at 4 °C). The tagged enzymes were purified over Ni-NTA His-bind resin (Novagen), in batch mode, washing with 20 mM imidazole and elution by 250 mM imidazole in column buffer (50 mM Bis-Tris, pH 6.8, 1 mM DTT). Enzymatic assays for class II activity were carried out much as previously described.[19] Enzymatic products also were investigated by expression from pGG-DEST based constructs in our previously described modular metabolic engineering system,[12] which couples production of GGPP in E. coli with further engineering to flux into isoprenoid metabolism,[20] as depicted in Figures 2 and S1–S4. Briefly, the products resulting from 3 day fermentations of 50 mL cultures were extracted with an equal volume of hexanes, which was dried under N2, resuspended in 1 mL fresh hexanes, and then filtered prior to analysis by gas chromatography with mass spectra detection (GC-MS), using a 3900 GC with Saturn 2100T ion trap MS (Varian) equipped with HP5-ms column (Agilent), as previously described.[21]
Supplementary Material
Footnotes
This work was supported by a grant from the NIH (GM076324) to R.J.P., who also gratefully acknowledges sabbatical fellowship support from the Alexander von Humboldt Foundation during preparation of this manuscript.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Chu HY, Wegel E, Osbourn A. Plant J. 2011;66:66–79. doi: 10.1111/j.1365-313X.2011.04503.x. [DOI] [PubMed] [Google Scholar]
- 2.Peters RJ. Nat Prod Rep. 2010;27:1521–1530. doi: 10.1039/c0np00019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Falara V, Pichersky E, Kanellis AK. Plant Physiol. 2010;154:301–310. doi: 10.1104/pp.110.159566. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caniard A, Zerbe P, Legrand S, Cohade A, Valot N, Magnard JL, Bohlmann J, Legendre L. BMC Plant Biol. 2012;12:119. doi: 10.1186/1471-2229-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schalk M, Pastore L, Mirata MA, Khim S, Schouwey M, Deguerry F, Pineda V, Rocci L, Daviet L. J Am Chem Soc. 2012;134:18900–18903. doi: 10.1021/ja307404u. [DOI] [PubMed] [Google Scholar]; d) Sallaud C, Giacalone C, Topfer R, Goepfert S, Bakaher N, Rosti S, Tissier A. Plant J. 2012;72:1–17. doi: 10.1111/j.1365-313X.2012.05068.x. [DOI] [PubMed] [Google Scholar]; e) Gunnewich N, Higashi Y, Feng X, Choi KB, Schmidt J, Kutchan TM. Phytochemistry. 2012;91:93–99. doi: 10.1016/j.phytochem.2012.07.019. [DOI] [PubMed] [Google Scholar]; f) Zerbe P, Hamberger B, Yuen MM, Chiang A, Sandhu HK, Madilao LL, Nguyen A, Hamberger B, Bach SS, Bohlmann J. Plant Physiol. 2013;162:1073–1091. doi: 10.1104/pp.113.218347. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Zerbe P, Chiang A, Yuen M, Hamberger B, Draper JA, Britton R, Bohlmann J. J Biol Chem. 2012;287:12121–12131. doi: 10.1074/jbc.M111.317669. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Criswell J, Potter K, Shephard F, Beale MB, Peters RJ. Org Lett. 2012;14:5828–5831. doi: 10.1021/ol3026022. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Shephard F. University of Bristol; Bristol: 2002. [Google Scholar]; j) Pateraki I, Andersen-Ranberg J, Hamberger B, Heskes AM, Martens HJ, Zerbe P, Bach SS, Moller BL, Bohlmann J, Hamberger B. Plant Physiol. 2014;164:1222–1236. doi: 10.1104/pp.113.228429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prisic S, Xu J, Coates RM, Peters RJ. ChemBioChem. 2007;8:869–874. doi: 10.1002/cbic.200700045. [DOI] [PubMed] [Google Scholar]
- 5.Zhou K, Gao Y, Hoy JA, Mann FM, Honzatko RB, Peters RJ. J Biol Chem. 2012;287:6840–6850. doi: 10.1074/jbc.M111.337592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen F, Tholl D, Bohlmann J, Pichersky E. Plant J. 2011;66:212–229. doi: 10.1111/j.1365-313X.2011.04520.x. [DOI] [PubMed] [Google Scholar]
- 7.Keeling CI, Dullat HK, Yuen M, Ralph SG, Jancsik S, Bohlmann J. Plant Physiol. 2010;152:1197–1208. doi: 10.1104/pp.109.151456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Koksal M, Hu H, Coates RM, Peters RJ, Christianson DW. Nat Chem Biol. 2011;7:431–433. doi: 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koksal M, Potter K, Peters RJ, Christianson DW. Biochim Biophys Acta. 2014;1840:184–190. doi: 10.1016/j.bbagen.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Prisic S, Xu M, Wilderman PR, Peters RJ. Plant Physiol. 2004;136:4228–4236. doi: 10.1104/pp.104.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Otomo K, Kenmoku H, Oikawa H, Konig WA, Toshima H, Mitsuhashi W, Yamane H, Sassa T, Toyomasu T. Plant J. 2004;39:886–893. doi: 10.1111/j.1365-313X.2004.02175.x. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi K, Kawaide H, Notomi M, Sakigi Y, Matsuo A, Nozaki H. FEBS Lett. 2006;580:6175–6181. doi: 10.1016/j.febslet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Kenrick P, Crane PR. Nature. 1997;389:33–39. [Google Scholar]
- 12.Cyr A, Wilderman PR, Determan M, Peters RJ. J Am Chem Soc. 2007;129:6684–6685. doi: 10.1021/ja071158n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Gonzalez A. Phytochemistry. 1975 [Google Scholar]; b) Jefferies P, Payne T. Australian Journal of Chemistry. 1965 [Google Scholar]; c) Zdero C, Bohlmann F, Niemeyer H. Phytochemistry. 1990 doi: 10.1016/0031-9422(90)85198-o. [DOI] [PubMed] [Google Scholar]
- 14.Peters RJ, Ravn MM, Coates RM, Croteau RB. J Am Chem Soc. 2001;123:8974–8978. doi: 10.1021/ja010670k. [DOI] [PubMed] [Google Scholar]
- 15.a) Keeling CI, Weisshaar S, Lin RPC, Bohlmann J. Proc Natl Acad Sci USA. 2008;105:1085–1090. doi: 10.1073/pnas.0709466105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. J Am Chem Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]; c) Zhou K, Peters RJ. ChemComm. 2011;47:4074–4080. doi: 10.1039/c0cc02960b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xu M, Wilderman PR, Peters RJ. Proc Natl Acad Sci USA. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wilderman PR, Peters RJ. J Am Chem Soc. 2007;129:15736–15737. doi: 10.1021/ja074977g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prisic S, Peters RJ. Plant Physiol. 2007;144:445–454. doi: 10.1104/pp.106.095208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters RJ, Flory JE, Jetter R, Ravn MM, Lee HJ, Coates RM, Croteau RB. Biochemistry. 2000;39:15592–15602. doi: 10.1021/bi001997l. [DOI] [PubMed] [Google Scholar]
- 18.Zhou K, Peters RJ. Phytochemistry. 2009;70:366–369. doi: 10.1016/j.phytochem.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann FM, Prisic S, Davenport EK, Determan MK, Coates RM, Peters RJ. J Biol Chem. 2010;285:20558–20563. doi: 10.1074/jbc.M110.123307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrone D, Lowry L, Determan MK, Hershey DM, Xu M, Peters RJ. Appl Microbiol Biotechnol. 2010;85:1893–1906. doi: 10.1007/s00253-009-2219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrone D, Hillwig ML, Mead ME, Lowry L, Fulton DB, Peters RJ. Biochem J. 2011;435:589–595. doi: 10.1042/BJ20101429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.