

Abstract
The TOR (target of rapamycin) pathway has been convincingly shown to promote aging in various model organisms. In mice, inhibiting mTOR (mammalian TOR) by rapamycin treatment later in life can significantly extend lifespan and mitigate multiple age-related diseases. However, the underlying mechanisms are poorly understood. Cellular senescence is strongly correlated to organismal aging therefore providing an attractive model to examine the mechanisms by which mTOR inhibition contributes to longevity and delaying the onset of related diseases. In this review, we examine the connections between mTOR and cellular senescence and discuss how understanding cellular senescence on the aspect of mTOR signaling may help to fully appreciate its role in the organismal aging. We also highlight the opposing roles of senescence in various human diseases and discuss the caveats in interpreting the emerging experimental data.
Keywords: senescence, aging, mTOR, rapamycin, age-related disease
In vitro cultured primary cells from human tissue do not proliferate indefinitely but instead will reach a cell cycle arrest state after 40–60 cell divisions, known as Hayflick limit [1]. This cell cycle arrest state is called replicative senescence, which is believed to be relevant to human aging. It is an irreversible cell cycle arrest likely due to telomeric attrition accompanied with cell cycle progression [2]. Supporting the role of telomere in cellular senescence, ectopic expression of catalytic subunit of human telomerase hTERT, which is supposed to mitigate the shortening of the telomere during cell division, has been shown to delay replicative senescence [3, 4].
Senescence can also be induced in the absence of telomeric attrition (Figure 1A). Several mitogenic stressors can lead to acute cellular senescence, which is termed premature senescence because it occurs without telomere shortening. For example, overexpression of oncogenic RAS (H-ras V12) or its downstream effector RAF can lead to senescent phenotypes called oncogene-induced senescence (OIS) [5, 6]. The OIS has also been reported in several other cases as well. BRAFV600E mutation in human naevi results in various phenotypes of cellular senescence [7]. In addition, loss of PTEN has been reported to cause cellular senescence, which is termed PTEN-loss-induced cellular senescence (PICS) [8]. Some DNA damage-inducing agents are also known to cause senescence of tumor cells in vitro and in vivo, and this is dependent on the tumor suppressors p16 and p53 [9, 10]. It should be noted that since the definition of senescence is based on limited markers (which will be discussed as follows), whether various senescence inducing conditions converge on a similar pathway at the molecular level is not well understood.
Figure 1.
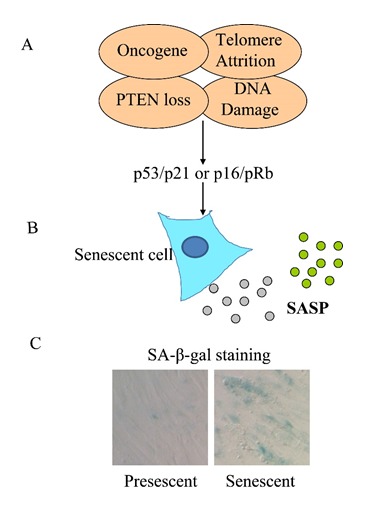
An overview of cellular senescence. (A) A variety of stimuli, such as oncogene expression, PTEN loss of function, DNA damage and telomere attrition can lead to cellular senescence. The establishment of cellular senescence requires the activation of at least one of the two largely independent pathways involving the well-known tumor suppressors p53/p21 and p16/pRb. (B) Senescent cells secrete a plethora of cytokines, chemokines, growth factors and proteases, termed SASP, which is thought to contribute to the organismal aging. (C) SA-β-gal activity is a marker for cellular senescence. Images show the SA-β-gal staining pattern of pre-senescent and replicative senescent primary human fibroblast (IMR90).
Despite the cell cycle arrest, the senescent cells remain metabolically active. It is known that senescent cells can secrete various pro-inflammatory cytokines, chemokines, growth factors and proteases. This process is termed senescence-associated secretory phenotype (SASP) [11–13]. SASP is one of the most striking characteristics of senescent cells (Figure 1B). Other than this, senescent cells also display flattened morphology and senescence-associated heterochromatin foci (SAHF) [14]. In addition, SA-β-gal (senescence-associated β-galactosidase) activity is also increased in senescent cells [15]. This characteristic has been broadly used as a histochemical marker for cellular senescence due to the ease of examination (Figure 1C). How senescent cells contribute to organismal aging remains poorly understood. However, the senescent markers have been detected in various animal tissues and are correlated very well with chronological aging [16–20]. In addition, it has been shown that senescent metabolites and cell factors such as SASP contribute to various physiological malfunctions. It is therefore likely that these “senescent factors” may play a causative role in aging and age-related diseases. However, how organismal aging is caused has been explained by many other theories but none of them seems to be fully satisfactory [21]. Nevertheless, and strikingly enough, removing senescent cells by genetic manipulation in a progerial mice model delays the aging phenotype and related diseases [22], demonstrating a significant relevance of cellular senescence to organismal aging.
Signaling pathways involved in cellular senescence
The signaling pathways underlying the cellular senescence remain poorly characterized. To date, experimental evidence from many labs collectively suggests that although many stimuli can induce senescence response, they converge on two main pathways, p53 and pRb [23]. However, this could be just the tip of the iceberg. For example, gene expression profile by microarray shows that the characteristics of the replicative senescence response are highly cell-type specific [24], suggesting multiple mechanisms to induce cellular senescence.
Early studies by inactivating p53 using the SV40 virus large T antigen shows that DNA synthesis is reinitiated and the progress of replicative aging is delayed in cultured human fibroblasts, establishing a role of p53 in cellular senescence [25, 26]. The full characterization of the function of p53 in cellular senescence comes from several recent studies. It is found that senescent human fibroblasts display phosphorylated H2AX nuclear foci at chromosome termini, which are co-localized with DNA damage checkpoint factors such as 53BP1 (p53 binding protein 1), MDC1 and NBS1. This indicates that DNA double-strand break response (DDR) occurs when cells progress to senescent state [2]. The senescent fibroblasts also have elevated amount of Ser15-phosphorylated p53 and p21 and the integrity of p53 pathway is necessary for the maintenance of senescence phenotypes [27, 28]. p53 also contributes to chemotherapy-induced senescence. For example, primary Eμ-myc lymphomas undergo cyclophosphamide (CTX) cytostatic state, which turns out to be p53 dependent [10]. In addition, ectopic expression of the p53 target gene p21 is sufficient to induce senescence program in HT1080 human fibrosarcoma cells [29, 30]. All these studies argue for a significant role of p53 in establishing and maintaining senescent program in the cell.
Senescence programs also engage another important signaling pathway, the p16-pRb pathway (Figure 1). p16 is a cell cycle factor that functions as an inhibitor of cyclin-dependent kinases Cdk4 and Cdk6 [5, 6, 31]. It has been reported that p16 expression is elevated in premature senescence induced by mitogenic activation of Ras or Raf [5, 6, 32]. Furthermore, ectopic expression of p16 in human diploid fibroblast is sufficient to induce senescence phenotypes including altered cell size and shape, appearance of SA-β-gal staining and reduced proliferation capacity [33]. This line of evidence establishes p16 as another yet to be fully characterized pathway in the regulation of cellular senescence. Overexpression of p16 activates the tumor suppressor pRB, which is required for maintaining senescent state. Replicative aging can be delayed by overexpressing p53 and RB in an additive manner, suggesting that these two pathways are largely independent [34]. However, there is no doubt that many crosstalk exist between these two pathways [35].
Emerging role of mTOR pathway in cellular senescence
The mTOR pathway
mTOR, the mammalian TOR protein, is the intracellular target of rapamycin, a pharmacological compound whose derivatives have been approved by FDA (Food and Drug Administration, USA) for various types of cancers. TOR is a large protein kinase belonging to phosphatidylinosital 3 kinase-related kinase (PI3KK) family [36]. TOR protein is present in all eukaryotic species examined to date including algae, slime mold, plants, worms, flies [36], indicating that the function of TOR is highly conserved throughout evolution. There are two conserved TOR complexes in a cell, which is usually referred to as TORC1 (TOR complex 1) and TORC2 (TOR complex 2) [37, 38]. They can be differentiated by the distinct associated proteins, for example Raptor for mTORC1, and Rictor for mTORC2. The two TOR complexes have distinct roles in cell biology, which remain not fully understood. However, experimental data in the past two decades suggest that TORC1 is the main mediator of nutrient signaling and is central to growth regulation. TORC2, although poorly characterized, may regulate the spatial organization of cytoskeleton, which coordinates the TORC1 machinery to expand the cytoplasmic volume. Rapamycin is highly specific and inhibits mTORC1 activity in a nanomolar concentration in cultured cells. However, it may also inhibit mTORC2 activity in long term treatment [39].
TORC1 activity is regulated by nutrient availability, especially amino acids [40]. However, there is no evidence that TORC1 is a direct nutrient sensor. In higher organisms, insulin and insulin-like growth factor (IGF) are critically important to signal nutrient cues and activate TORC1 [41]. The upstream and downstream of TOR has been nicely delineated using cultured mammalian cells. In the upstream, there are multiple inputs including insulin signaling through PI3K and AKT, energy signaling (ATP:AMP ratio) through AMPK and stress signaling through poorly characterized pathways. These signals converge on TSC1-TSC2 complex. TSC complex serves as a GEF (GTPase Exchange Factor) for Rheb while Rheb in GTP-bound form activates mTOR through direct binding [42]. Amino acid signaling does not require Rheb but goes through different sets of small GTPase RagA/B and RagC/D. In the downstream of mTOR, S6K1 and 4EBP1 are two most well-studied effectors, which are phosphorylated by mTORC1 but not mTORC2 [43]. The phosphorylation of these two effectors, especially S6K1 phosphorylation at Threonine 389 has been used broadly as readout for mTORC1 activity. These two effectors are involved in regulation of translational initiation. Other than this, ULK1-ATG13, the autophagic initiation complex is also regulated by mTOR through direct phosphorylation. Recent studies also identify another conserved effector Maf1 [44]. Maf1 is a transcriptional repressor of RNA polymerase III (Pol III) that synthesize 5S rRNA and tRNA. Maf1 is directly phosphorylated by TORC1 in both yeast [45] and human cells [46] and is critical in growth control as demonstrated by genetic data in Drosophila [47].
TOR and cellular senescence
Cell growth and senescence are seemingly two antagonistic biological processes, because cell growth is a process of mass accumulation while senescence is state of arrest. However, emerging data show that these two cellular processes are linked together [48, 49]. Specifically, these newly emerging data argue for a role of mTOR in promoting cellular senescence. This, in light of the extensive literatures on lifespan extension through attenuating TOR activity (which will be discussed in point 4), is consistent with the idea that cellular senescence may reflect some if not all aspects of organismal aging. It is therefore worth examining the literatures concerning the cellular senescence that are linked to mTOR pathway.
Recent data have begun to uncover the important role of mTOR in the senescence of various cell types derived from human or mice in vitro. For example, in HT-p21 cells, a human fibrosarcoma cell line, stimulating growth while inhibiting cell cycle simultaneously increases SA-β-gal activity, a well-known marker for cellular senescence (Table 1). Interestingly, treating these cells with rapamycin diminishes the senescence markers [30]. What makes these studies especially intriguing is that these arrested cells can re-enter cell cycle when rapamycin and IPTG (used to confer cell cycle arrest by inducing p21 expression) are removed. Note that in the control, treatment of IPTG alone for 3 days has already caused irreversible cell cycle arrest [50]. These data suggest that mTOR inhibition by rapamycin can delay the progression of cellular senescence. Supporting this idea, in another normal human fibroblast WI-38, rapamycin treatment prevents or attenuates senescence induced by chemotherapy drug doxorubicin (DOX) [30]. In addition, in ARPE-19 cells (a human retinal pigment epithelial cell line), rapamycin not only decreases hydrogen peroxide (H2O2)-induced senescence, but also prevents the permanent loss of proliferation capacity [50]. Several other cell lines have also been tested and the results are strikingly consistent (Table 1), arguing for a critical role of mTOR in establishing the senescent phenotypes.
Table 1.
Targeting mTOR to delay cellular senescence
| Model | Description | Senescence inducing methods | Effect of rapamycin | Ref. |
|---|---|---|---|---|
| HT1080-p21 | Human fibrasarcoma | IPTG-induced p21 expression | Reduce SA-β-gal activity | [30] |
| HT1080-p21 | Human fibrasarcoma | IPTG-induced p21 expression | Re-enter cell cycle when inducing agents are removed. Remain large size | [50] |
| HT1080-p16 | Human fibrasarcoma | IPTG-induced p16 expression | Preserve the proliferative capacity | [50] |
| ERas | Rodent fibroblast | Sodium butyrate-induced p21 | Marginal decrease in SA-β-gal, no change in morphology, yet prevent the loss of proliferative potential | [50] |
| WI-38 | Human primary lung fibroblast | DNA damage by Doxorubicin | Partially prevented senescent phenotype | [30] |
| ARPE-19 | human retinal pigment epithelial cell | H2O2 | Reduce SA-β-gal activity, did not change flat morphology, prevent the permanent loss of proliferation | [50] |
| BJ | Human skin fibroblasts | Continuous passage | Suppress IL-8 and p21 but not SA-β-gal activity and flattened morphology in senescent cells | [52] |
| BJ | Human skin fibroblasts | Continuous passage | Treatment of pre-senescent cells delay SA-β-gal, no change in cell morphology | [52] |
| BJ | Human skin fibroblasts | RAS overexpression | Higher proliferation rate and less SA-β-gal | [52] |
| MEFs | Mouse embryonic cells | Continuous passage | Partially suppress senescent marker. Cells adapt to rapmycin, not useful for long term treatment. | [53] |
| REFs | Rat embryonic cells | Continuous passage | Reverse SA-β-gal and DNA damage marker H2AX and 53BP1 | [53] |
| Wnt1 transgenic mice | Doxycycline-inducible K5rtTA/tet-Wnt1 mice | Persistent activation of Wnt1 | Partially suppressed disappearance of the epidermal stem cell compartment and subsequent hair loss | [54] |
Apart from chemical-induced senescence, the effect of rapamycin on attenuating senescence is also demonstrated in several other conditions, such as replicative senescence and oncogene-induced senescence (OIS) [51–53]. In a study on replicative senescence of human skin fibroblasts (BJ cells), 3-day rapamycin treatment suppresses the induction of IL-8 and p21, but not other senescent markers such as SA-β-gal activity and flattened morphology [52]. Nevertheless, rapamycin can still significantly slow down the process of replicative senescence if treated from early passage (cells are in presenescent stage). Untreated BJ cells with additional 30 passages (total 60 passages) will assume flattened morphology and SA-β-gal staining, while cells with rapamycin treatment not only show higher proliferative rate, but also have higher numbers of population doubling [52]. In addition, these rapamycin-treated BJ cells display less SA-β-gal activity and less expression of p21 and IL8 [52]. In the case of OIS that is induced by oncogene RAS, rapamycin-treated cells show a higher proliferation rate and less SA-β-gal staining compared to non-treated cells [52]. Similar observations have been reported in rat embryonic cells, although proliferation-inhibiting effect of rapamycin on rodent cells masks its effect on replicative life span [53]. Persistent activation of Wnt1causes disappearance of the epidermal stem cell compartment and subsequent hair loss in a mouse model, apparently due to stem cell senescence [54]. Later in vitro experiments show that Wnt1 activates mTOR signaling in cultured keratinocytes from such transgenic mice and leads to subsequent cellular senescence. Significantly, this phenotype can be partially suppressed by rapamycin treatment [54]. All the above studies suggest that rapamycin treatment, which decreases the activity of mTOR, can decelerate cellular senescence caused by different stimuli.
Potential mechanisms
Although cell cycle is arrested, senescent cells are active in metabolism and protein synthesis [11–13, 55]. A common feature of senescent cells is the enlarged cell morphology (hypertrophy) [56]. Consistently, senescent cells contain more cell mass [57]. It has also been shown that during therapy-induced senescence (TIS), protein synthesis rate is elevated as judged by the increased incorporation of fluorescence-labeled methionine [55]. The increase in cell mass hence enlarged morphology is consistent with the role of mTOR in promoting cellular senescence, as mTOR promotes protein synthesis and cell growth[48]. Presumably, mTOR is activated in senescent cells, which may be necessary for progression of cellular senescence. Indeed, evidence has been shown that S6 phosphorylation, a well-established marker for mTORC1 activity, is enhanced in these cells [53]. Furthermore, mTOR inhibition by rapamycin treatment attenuates the activation of some if not all senescent markers. These data collectively suggest an intriguing role of mTOR in establishing cellular senescence (Figure 2A). A variety of oncogenic proteins, such as RAF and RAS can activate mTOR pathway. Consistently, these oncogenic proteins are well known to cause cellular senescence [5, 6].
Figure 2.
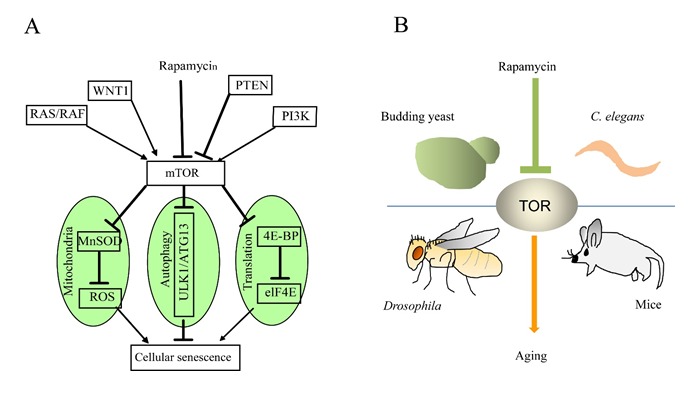
Emerging role of mTOR in cellular senescence and organismal aging. (A) mTOR integrates different signaling pathways to cellular senescence. mTOR regulates cellular senescence through modulation of mitochondrial metabolism, autophagy and protein translation. (B) mTOR homologs in many model organisms promote organismal aging through poorly characterized mechanisms.
mTOR positively regulates protein synthesis and negatively regulates autophagy pathway [48]. Excessive activity of mTOR during aging may increase abnormal proteins in senescent cells, as a result of both increased protein synthesis and decreased autophagic activity. This in turn, might lead to protein aggregation in degeneration disease. Supporting this argument, ATF4, an unfolded protein response (UPR) marker, is activated by therapy-induced senescence (TIS) [55]. Senescent cells rely on autophagy and lysosome pathway to eliminate mis-folded proteins and relieve proteotoxic stress. Inhibiting autophagy and lysosome activity is known to cause senescent cell death [55]. This work suggests that the autophagy-lysosome pathway likely serves to delay the progression of senescence [55]. Interestingly, as also mentioned early in this review, autophagy is negatively regulated by mTORC1 [58–60]. In senescent cells, high levels of mTOR activity may mitigate the effect of autophagy on clearing excessive, damaged proteins and organelles, therefore accelerating the progression of senescence (Figure 2A). To the contrary however, inhibiting mTOR by rapamycin activates autophagy, protects cells from proteotoxicity, therefore delaying cellular senescence. This argument is also supported by recent data showing that TOR inhibition decreases the mitochondrial UPR marker HSP-60 in C. elegans [61].
Mitochondria dysfunction has been suggested to be an important mechanism leading to cellular senescence, as decreased mitochondrial function, impaired ATP generation and increased reactive oxygen species (ROS) levels are implicated in cellular senescence [62, 63]. Mitochondrial biogenesis is essential for maintaining healthy and normal function of mitochondria in the cell. PPARγ and its co-activator 1(PGC1-α) are master regulators of mitochondrial metabolism and biogenesis. Intriguingly, mTORC1 is linked to mitochondrial biogenesis through regulating the transcriptional activity of PGC1-α [64]. Supporting this, studies elsewhere show that reducing mTORC1 function by rapamycin treatment enhances mitochondrial membrane potential, reduces ROS levels, and increases replicative life span [65]. In a radiation-induced senescence model, rapamycin treatment prevents the ROS induction by irradiation in primary normal oral keratinocytes (NOK) [66]. The prevention of ROS is mainly mediated by the induction and stability of MnSOD protein, as knockdown of MnSOD abrogates the increased clonogenic capacity of rapamycin-treated NOKs [66]. These data suggest that mTOR activity contributes to the radiation-induced senescence by accelerating intracellular ROS accumulation, which is mediated through the inhibition of mitochondrial superoxide dismutase MnSOD (Figure 2A).
TOR pathway in regulation of lifespan in model organisms
The role of mTOR in cellular senescence is consistent with a large body of work that has been done in model organisms (Figure 2B), where inhibition of mTOR homologues consistently extends lifespan [41]. The direct evidence showing the role of TOR in modulating lifespan is initially found in C. elegans, where lowering TOR activity increases lifespan more than 2 folds, one of the most striking records that have been made. Note that this specific experiment was done at a very specific temperature (25.5 °C) [67], which may account for the only mild lifespan extension observed in later studies. Nevertheless, shortly after this initial observation, experiments in Drosophila and budding yeast confirm the important role of TOR in lifespan regulation. Down-regulation of TOR activity in Drosophila through expressing a dominant-negative TOR allele extends mean lifespan around 15% [68]. In yeast, replicative lifespan can be extended when TORC1 activitry is decreased [69]. In subsequent studies, both genetic and pharmacological manipulations in various model organisms consistently show that TOR inhibition can extend lifespan [70–72]. Impressively, rapamycin administration starting at 600 days in mice, an age analogous to ∼50 years in human, extends lifespan up to 14% in female and 9% in male mice [72]. Since TOR is sensitive to nutrients, it is believed that TOR inhibition underlies the effect of calorie restriction to extend lifespan. It has been shown that rapamycin inhibits only TORC1 but not TORC2 [73]. However, later experiments demonstrate that chronic rapamycin treatment can also inhibit TORC2 activity [39]. It is therefore unclear in situations involving long term rapamycin treatment, especially those related to research on senescence and organismal aging, whether TORC2 is also playing a role. Nevertheless, the results obtained from the mice study suggest that rapamycin derivatives as FDA-approved drugs hold promise in delaying aging and alleviating age-related disease in human.
Other than TOR kinase, many TOR regulators and effectors are also lifespan regulators. For example, C. elegans lifespan can be lengthened by RNAi knockdown of Rheb homolog [74], or by expressing a dominant negative form Rag [75]. In addition, worms heterozygous for daf-15, the homolog of mammalian Raptor, extends lifespan about 30% [76]. The TOR downstream effectors including S6K1, 4EBP1 and ULK1 are also implicated in lifespan regulation. For example, deletion of the gene encoding Sch9, the yeast homolog of human S6K1, gives as much as 90% in lifespan extension [77]. RNAi knockdown of rsks-1/S6K1 in C. elegans causes up to 50% increase in mean lifespan. In flies, expression of a dominant-negative dS6K1 increases mean lifespan around 15% [68]. Consistently, S6K1 knockout in mice extends mean lifespan up to 19% [78]. Another TOR effector 4EBP1 is negatively regulated by TORC1. Overexpression of 4EBP1 in flies increases lifespan up to 11% in male and 22% in female [79]. Atg1 is the yeast homolog of ULK1 and deletion of ATG1 not only shortens WT lifespan but also blocks the life-prolonging effect of rapamycin [80]. It would be interesting to see if these TOR pathway regulators and effectors are also implicated in cellular senescence and related diseases.
Opposing roles of senescence in diseases
Although evidence has been shown that cellular senescence can contribute to organismal aging and age-related diseases [16–20], paradoxically, senescence is well known to protect cells from cancerous transformation. For example, senescence has been proposed to be an anticancer mechanism that prevents the neoplastic transformation. Such ideas find plenty of experimental supports in the literature [81]. For example, human naevi are in a senescent state and refractory to oncogenic transformation caused by mutations in the RAS pathway [7]. In addition, acute loss of PTEN, a tumor suppressor in mouse induces cell cycle arrest both in vitro and in vivo but does not initiate tumorigenesis as expected [8]. In another setting, RAS-induced lymphomagenesis is accelerated by the loss of cellular senescence [82]. The positive role of senescence in disease has also been reported in wound healing. In a mouse model of liver injury, senescent cells from activated hepatic stellate cells are found to accumulate in the injured liver [83]. Loss of senescence in these cells results in significant increase in fibrosis after injury. Mechanistically, senescent stellate cells secrete extracellular matrix-degrading enzyme and enhance immune surveillance, thus may contribute to fibrosis resolution [83].
Despite the beneficial role of cellular senescence mentioned above, senescent phenotype is well correlated to normal aging and several other related diseases (Figure 3). For example, degeneration at the organismal level is at least in part due to altered secretion of cell factors from senescent cells [11–13]. These secreted factors can disrupt the normal tissue structure and function in cell culture [49], suggesting a similar role in vivo. Matrix metalloproteinases (MMPs), which are prominent SASP components, can disrupt alveolar morphogenesis, differentiation and branching of mammalian breast epithelial cells [84]. In addition, evidence has been shown that in the aging brain, astrocytes undergo cellular senescence and express p16 and matrix metalloproteinase-1(MMP-1). Furthermore, the frontal cortex of AD patients shows more senescent astrocytes than that of normal people of similar age [85]. The senescent astrocytes can poorly support in vitro cultured neurons, suggesting that astrocytes senescence may contribute to neuronal degeneration in vivo [86].
Figure 3.
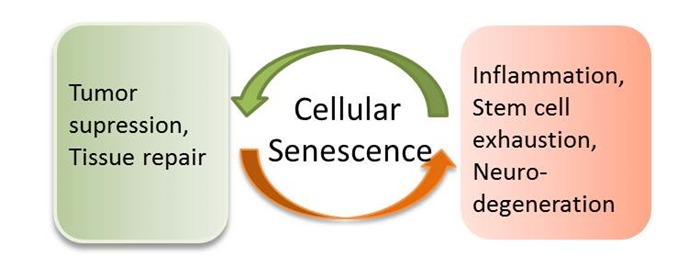
Opposing roles of cellular senescence in disease. Cellular senescence on the one hand can cause chronic inflammation, decrease stem cell renewal ability and promote progerial syndrome, but on the other hand serves to inhibit cancerous transformation and enhance tissue repair. The role of cellular senescence in disease, especially age-related disease awaits further investigation.
Another line of evidence supporting the role of cellular senescence in causing aging is from the study on stem cell renewal. The contribution of stem cell renewal in delaying aging has become increasingly appreciated [87]. In higher organisms, adult stem cell is essential to replenish the loss of somatic cells due to damage or disease. It is found that p16-dependent stem cell senescence results in decreased proliferation and regeneration potential [88–90]. In old mice, the prosenescence protein p16 is accumulated in the stem and progenitor cells in the brain, bone marrow and pancreas. Significantly, depleting p16 through genetic engineering of corresponding chromosomal locus suppresses the decline in stem cell proliferation and tissue regeneration. However, as p16 functions as a tumor suppressor, these mice die earlier of cancer instead of living healthier and younger. Despite the broad interest in stem cell research in the aging field, significant amount of work awaits to be done in order to exploit the beneficial side of stem cell therapy for aging and age-related disease.
Recently, a genetically engineered mouse model where p16-expressing senescent cells are eliminated by apoptosis [22], demonstrates delayed onsets of progeroid phenotypes in different tissues, such as adipose tissue, skeletal muscle and eyes. However, lifespan is not extended in these mice. Note that the benefits on age-related disease are obtained in a progerial model and whether removing senescent cells could improve healthy span or even life span in normal mice remains to be determined. However, this work has provided the basis for further study on similar topics, which may promise novel strategy to delay aging and improve healthy span in human.
Conclusion
Although the role of TOR in lifespan regulation has been supported by numerous studies in various model organisms, the underlying signaling circuits remain poorly understood, especially those related to mammals. Cellular senescence has been used as a nice aging model to delineate the mechanisms by which mTOR exerts its pro-aging function. However, the contribution of cellular senescence to organismal aging has not been fully established. When it comes to age-related disease, for example cancer, senescence can function to protect against malignant transformation. However, at the organismal level, accumulating data suggest that senescent cells contribute to multiple age-related diseases. The opposing roles of senescence in age-related disease reflect the highly complex interaction of multiple signaling pathways in the pathology of these diseases. Nevertheless, future study on how mTOR modulates senescence of in vitro cultured cells and aging at the organismal level will definitely facilitate the design of strategy to delay aging and mitigate age-related disease.
Acknowledgments
We apologize for not being able to cite all the relevant publications due to space limit. This work is supported by National Natural Science Foundation of China (NSFC 81200248).
References
- [1].Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- [2].d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- [3].Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- [4].Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol. 1998;8:279–282. doi: 10.1016/s0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- [5].Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- [6].Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- [8].Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- [11].Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- [12].Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- [14].Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- [15].Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KE, Wustefeld T, et al. Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. Embo J. 2003;22:4003–4013. doi: 10.1093/emboj/cdg367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lechel A, Satyanarayana A, Ju Z, Plentz RR, Schaetzlein S, Rudolph C, et al. The cellular level of telomere dysfunction determines induction of senescence or apoptosis in vivo. EMBO Rep. 2005;6:275–281. doi: 10.1038/sj.embor.7400352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- [19].Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jin K. Modern Biological Theories of Aging. Aging Dis. 2010;1:72–74. [PMC free article] [PubMed] [Google Scholar]
- [22].Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- [24].Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- [25].Ide T, Tsuji Y, Ishibashi S, Mitsui Y. Reinitiation of host DNA synthesis in senescent human diploid cells by infection with Simian virus 40. Exp Cell Res. 1983;143:343–349. doi: 10.1016/0014-4827(83)90060-5. [DOI] [PubMed] [Google Scholar]
- [26].Ide T, Tsuji Y, Nakashima T, Ishibashi S. Progress of aging in human diploid cells transformed with a tsA mutant of simian virus 40. Exp Cell Res. 1984;150:321–328. doi: 10.1016/0014-4827(84)90575-5. [DOI] [PubMed] [Google Scholar]
- [27].d’Adda di Fagagna F, Teo SH, Jackson SP. Functional links between telomeres and proteins of the DNA-damage response. Genes Dev. 2004;18:1781–1799. doi: 10.1101/gad.1214504. [DOI] [PubMed] [Google Scholar]
- [28].Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur J Biochem. 2001;268:2784–2791. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- [29].Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–2170. doi: 10.1038/sj.onc.1203573. [DOI] [PubMed] [Google Scholar]
- [30].Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–3361. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- [31].Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- [32].Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, et al. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature. 2001;409:1067–1070. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- [33].McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–354. doi: 10.1016/s0960-9822(98)70137-x. [DOI] [PubMed] [Google Scholar]
- [34].Shay JW, Wright WE, Werbin H. Defining the molecular mechanisms of human cell immortalization. Biochim Biophys Acta. 1991;1072:1–7. doi: 10.1016/0304-419x(91)90003-4. [DOI] [PubMed] [Google Scholar]
- [35].Hara E, Tsurui H, Shinozaki A, Nakada S, Oda K. Cooperative effect of antisense-Rb and antisense-p53 oligomers on the extension of life span in human diploid fibroblasts, TIG-1. Biochem Biophys Res Commun. 1991;179:528–534. doi: 10.1016/0006-291x(91)91403-y. [DOI] [PubMed] [Google Scholar]
- [36].Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- [37].Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo J, Bonenfant D, et al. Two TOR Complexes, Only One of which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- [38].Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- [39].Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- [40].Wei Y, Zheng XF. Nutritional control of cell growth via TOR signaling in budding yeast. Methods Mol Biol. 2011;759:307–319. doi: 10.1007/978-1-61779-173-4_18. [DOI] [PubMed] [Google Scholar]
- [41].Wei Y, Zhang YJ, Cai Y. Growth or longevity: the TOR’s decision on lifespan regulation. Biogerontology. 2013;14:353–363. doi: 10.1007/s10522-013-9435-6. [DOI] [PubMed] [Google Scholar]
- [42].Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–576. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- [43].Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- [44].Wei Y, Zheng XS. Maf1 regulation: a model of signal transduction inside the nucleus. Nucleus. 2010;1:162–165. doi: 10.4161/nucl.1.2.11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wei Y, Tsang CK, Zheng XF. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009;28:2220–2230. doi: 10.1038/emboj.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010;30:3749–3757. doi: 10.1128/MCB.00319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012;109:1139–1144. doi: 10.1073/pnas.1113311109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. doi: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- [51].Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012;11:2231–2232. doi: 10.4161/cc.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–2401. doi: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11:2402–2407. doi: 10.4161/cc.20882. [DOI] [PubMed] [Google Scholar]
- [54].Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5:279–289. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dorr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Dabritz JH, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501:421–425. doi: 10.1038/nature12437. [DOI] [PubMed] [Google Scholar]
- [56].Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY) 2009;1:1008–1016. doi: 10.18632/aging.100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Blagosklonny MV. Cell senescence: hypertrophic arrest beyond the restriction point. J Cell Physiol. 2006;209:592–597. doi: 10.1002/jcp.20750. [DOI] [PubMed] [Google Scholar]
- [58].Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- [65].Lerner C, Bitto A, Pulliam D, Nacarelli T, Konigsberg M, Van Remmen H, et al. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell. 2013. [DOI] [PMC free article] [PubMed]
- [66].Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401–414. doi: 10.1016/j.stem.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- [68].Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- [70].Wanke V, Cameroni E, Uotila A, Piccolis M, Urban J, Loewith R, et al. Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol. 2008;69:277–285. doi: 10.1111/j.1365-2958.2008.06292.x. [DOI] [PubMed] [Google Scholar]
- [71].Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46. doi: 10.1016/j.cmet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- [74].Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009;457:726–730. doi: 10.1038/nature07583. [DOI] [PubMed] [Google Scholar]
- [75].Schreiber MA, Pierce-Shimomura JT, Chan S, Parry D, McIntire SL. Manipulation of behavioral decline in Caenorhabditis elegans with the Rag GTPase raga-1. PLoS Genet. 2010;6:e1000972. doi: 10.1371/journal.pgen.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- [77].Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- [78].Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Zid BM, Rogers AN, Katewa SD, Vargas MA, Kolipinski MC, Lu TA, et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Alvers AL, Wood MS, Hu D, Kaywell AC, Dunn WA, Jr, Aris JP. Autophagy is required for extension of yeast chronological life span by rapamycin. Autophagy. 2009;5:847–849. doi: 10.4161/auto.8824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- [83].Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–496. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS One. 2012;7:e45069. doi: 10.1371/journal.pone.0045069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J Neurochem. 2007;101:794–805. doi: 10.1111/j.1471-4159.2006.04369.x. [DOI] [PubMed] [Google Scholar]
- [87].Smith JA, Daniel R. Stem cells and aging: a chicken-or-the-egg issue? Aging Dis. 2012;3:260–268. [PMC free article] [PubMed] [Google Scholar]
- [88].Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- [89].Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- [90].Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]