

Georgiou and colleagues discuss rapidly evolving methods for high-throughput sequencing of the antibody repertoire, and how the resulting data may be applied to answer basic and translational research questions.
Subject terms: Immunogenetics, B cells, Applied immunology, Antibody isolation and purification
Abstract
Efforts to determine the antibody repertoire encoded by B cells in the blood or lymphoid organs using high-throughput DNA sequencing technologies have been advancing at an extremely rapid pace and are transforming our understanding of humoral immune responses. Information gained from high-throughput DNA sequencing of immunoglobulin genes (Ig-seq) can be applied to detect B-cell malignancies with high sensitivity, to discover antibodies specific for antigens of interest, to guide vaccine development and to understand autoimmunity. Rapid progress in the development of experimental protocols and informatics analysis tools is helping to reduce sequencing artifacts, to achieve more precise quantification of clonal diversity and to extract the most pertinent biological information. That said, broader application of Ig-seq, especially in clinical settings, will require the development of a standardized experimental design framework that will enable the sharing and meta-analysis of sequencing data generated by different laboratories.
Main
A potent adaptive immune system is fundamentally reliant upon the generation of a diverse repertoire of B-lymphocyte antigen receptors (BCRs, the membrane-bound form of antibodies expressed on the surface of B cells). BCRs are assembled by somatic recombination of a large number of immunoglobulin gene segments (Fig. 1), and the repertoire of BCRs expressed in any given individual is continuously shaped by exposure to exogenous antigens and endogenous host factors. Existing mechanisms for BCR diversification can yield an astronomical number of possible BCRs (in theory, >1013 in humans)1,2; this number exceeds the total number of B lymphocytes in the human body (∼1–2 × 1011) (ref. 3). Because of labor and cost considerations, it is completely impractical to analyze such a diverse BCR repertoire using traditional Sanger sequencing. However, Ig-seq (a term coined by Andrew Fire, Stanford University) has allowed us to determine antibody gene repertoires at an unprecedented depth. The information gained by Ig-seq is proving invaluable for understanding antibody responses in health and disease and for diagnostic purposes. In addition, Ig-seq can be combined with other techniques, including expression and isolation of antigen-specific antibodies, sequencing of multiple RNAs from single cells4, and proteomic analyses of antibodies in blood or secretions, to help elucidate the properties of antibodies that mediate protection against infectious diseases or, alternatively, that mediate autoimmune responses. In this Review we describe the experimental approaches and technical challenges related to high-throughput antibody gene sequencing, as well as the ways in which Ig-seq might be applied to advance our understanding of immunology and to address unmet clinical needs related to infectious diseases, immune dysregulation and cancer.
Figure 1. Antibody structure and sequence diversification mechanisms.
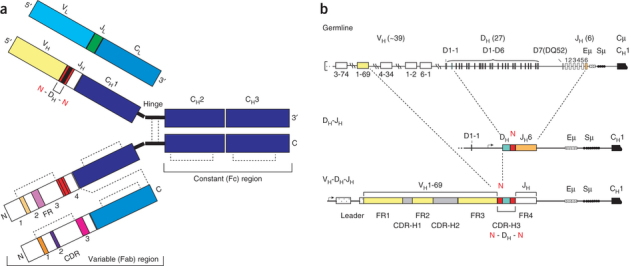
(a) Schematic of IgG structure. In the top chains, domains encoded from germline V, D, J and C segments are indicated. Nontemplated N-nucleotides are shown in red. These top chains delineate the 5′ to 3′ genetic composition of the antibody. In the bottom chains, framework (FR) and complementarity-determining regions (CDRs) are indicated. These bottom chains delineate the N-terminal to C-terminal protein sequence. Dashed lines denote disulfide bonds. (b) Key steps in antibody diversification. The primary antibody heavy chain repertoire is created predominantly by the somatic recombination of variable (V), diversity (D) and joining (J) gene segments, and by the random nontemplated addition of N-nucleotides. The antigen-binding site of a heavy chain is formed by the juxtaposition of the hypervariable complementarity-determining regions (CDR-H1, H2 and H3) and the framework 3 region (FR3). After productive IgH rearrangement, recombination of the light chain (IgL) ensues, and the heterodimeric pairing of H and L chains forms the complete antibody of the IgM isotype that is expressed on the surface of a newly formed immature B cell. Eμ: IgM intronic enhancer; Sμ: tandem repeats critical for class-switch recombination. Numbers in parentheses refer to estimates of human germline VH DH and JH segments.
Generation of the antibody repertoire
Antibodies are produced by a developmentally ordered series of somatic gene rearrangement events that occur exclusively in developing B cells and continue throughout the life of an organism. Antibodies consist of heavy (μ, α, γ, δ, ɛ) and light chains (κ, λ), which are linked by disulfide bonds. The intact antibody contains variable and constant domains (Fig. 1a). Antigen binding occurs in the variable domain, which is generated by recombination of a finite set of tandemly arranged variable (V), diversity (D) and joining (J) germline gene segments (Fig. 1b). This process, called VDJ recombination, often results in the addition and deletion of nucleotides at the junctions between ligated gene segments (Fig. 1b). More specifically, DNA exonucleases can trim the ends of the gene segments, and DNA polymerases and transferases can randomly insert templated palindromic or nontemplated nucleotides, respectively.
During B-cell development, immunoglobulin heavy (IgH) chain gene recombination typically occurs before immunoglobulin light (IgL) chain gene recombination. If both IgH and IgL genes are productively rearranged, the fully assembled antibody heterodimer is expressed on the surface of the B cell. In B cells bearing productively rearranged antibodies, the process of allelic exclusion (and locus exclusion in the case of IgL) ensures that each B cell expresses a single antibody5. After passage through several developmental checkpoints, newly generated mature IgM+IgD+ B cells form the naive B cell (and, therefore, naive antibody) repertoire. Most of the diversity in the naive antibody repertoire is concentrated at the site of IgH VDJ gene segment ligation, also known as the IgH complementarity-determining region 3 (CDR-H3) (Fig. 1b). Because of the combinatorial and nontemplated nature of the mechanisms that generate the CDR-H3, it is the most diverse component in terms of length and sequence of the antibody H-chain repertoire and is a principal determinant of antibody specificity6,7. Nonetheless, there are instances where antigen specificity is dictated solely or predominantly by the L chain.
When a B cell encounters antigen in an environment that provides requisite co-stimulatory signals and T-cell help, BCR stimulation induces B-cell proliferation. This process, known as B-cell clonal expansion, occurs primarily in highly organized areas of secondary lymphoid organs (e.g., spleen, lymph nodes and Peyer's patches8,9) referred to as germinal centers (Fig. 2). Clonal expansion is followed by somatic hypermutation of the variable domains of antibodies; this is mediated by activation-induced cytidine deaminase. B cells expressing BCRs bearing somatic mutations that increase affinity for antigen outcompete other B cells for access to antigen. As a result, the B cells bearing the highest-affinity antibodies undergo preferential expansion and survival, a process referred to as affinity maturation. Somatic hypermutation also results in sequence diversification of the CDR-H1 and CDR-H2 hypervariable regions and of the framework 3 (FR3) region, which was proposed to constitute a fourth hypervariable region of the antibody H chain10. Activation-induced cytidine deaminase also mediates class-switch recombination, which generates antibodies bearing different constant regions. B cells expressing somatically mutated, high-antigen-affinity BCRs can differentiate into long-lived memory B cells, capable of mediating rapid recall responses to the same antigen, or into terminally differentiated plasma cells; the latter downregulate BCR expression, establish residency in the bone marrow, gut lamina propia (and, to a smaller degree, in secondary lymphoid tissues), and secrete protective antibodies at extremely high rates estimated at 10,000–20,000 antibody molecules per second11. Antibody production by long-lived plasma cells in the bone marrow is postulated to proceed for very long times, possibly throughout the entire lifetime of the organism.
Figure 2. Key steps in the development of antigen-specific B cells.
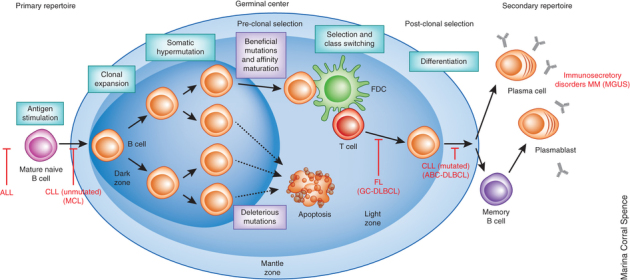
The steps of normal B-cell differentiation and diversification of the antibody repertoire are indicated in black text. Normal B cells are generated in the bone marrow, migrate to the periphery and, following developmental checkpoint selection, comprise the population of IgM+IgD+ mature naive B cells. When these cells are activated by cognate antigen in the presence of T-cell help, they enter a germinal center (GC) reaction where they rapidly proliferate; this results in clonal expansion and subsequent somatic hypermutation catalyzed by activation-induced cytidine deaminase. B cells bearing antibodies with high affinity for cognate antigen and that survive the GC reaction can undergo class-switch recombination to IgG, IgA or IgE isotypes and ultimately differentiate into memory B cells, antibody-secreting plasmablasts or plasma cells. After subsequent encounter with the same cognate antigen, memory B cells can proliferate or differentiate directly into antibody-secreting cells. Steps that proceed abnormally, leading to the development of human B-cell leukemias and lymphomas, are indicated in red text. ALL, acute lymphoblastic leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; GC-DLBCL, germinal center diffuse large B cell lymphoma; FL, follicular lymphoma; ABC-DLBCL, activated B cell–like DLBCL; MGUS, monoclonal gammopathy of undetermined significance; MM, multiple myeloma. B-cell malignancies that have not been analyzed extensively by high-throughput sequencing are shown in parenthesis.
Diversity in the primary antibody repertoire (before exogenous antigen exposure) stems from the allelic diversity in immunoglobulin gene segments, combinatorial diversity introduced during somatic recombination, junctional diversity caused by the imprecision of the recombination process, pairing of IgH and IgL polypeptide chains, and receptor editing, wherein the existing V-gene segment is replaced with another (Fig. 1). In addition, VH replacement, a process resulting from the presence of a cryptic recombination signal sequence in FR3, might influence as much as 5–12% of the human primary B-cell antibody repertoire12. Diversification of the post-antigen-stimulation secondary antibody repertoire stems from somatic hypermutation and class-switch recombination.
Organism age also influences the antibody repertoire13,14. During early ontogeny, the mammalian adult B-cell repertoire is generated in a predictable developmentally programmed fashion, whereas in advanced age humoral immune responsiveness deteriorates; this phenomenon is referred to as immunosenescence and is thought to be attributable in part to a progressive restriction of the antibody repertoire. For example, among the elderly there is an increased prevalence of autoantibodies and, at the serological level, an increased amount of either a single or a small number of serum immunoglobulins that are produced at a high level by benign outgrowths of one or more plasma cell clones15,16.
Low-throughput analysis of the antibody repertoire
In the 1990s, Sanger sequencing enabled the determination of IgH and IgL VDJ recombinants (hereafter simply referred to as V genes) in typically up to a few hundred B cells per experiment17,18,19. Subsequent studies began to clone immunoglobulin genes from single B cells following limiting dilution, and to express and functionally characterize the cloned antibody proteins, thus enabling the interrogation of antibody specificity. This advance proved invaluable for the isolation of antibodies relevant to disease, especially the isolation of pathogen-neutralizing antibodies20. B-cell immortalization (and subsequent sequencing of the V genes in immortalized B clones) provided an alternative route for the expression of small numbers of antibodies21,22,23.
The ability to interrogate the antibodies encoded by small numbers of B cells has yielded numerous important immunological insights. For example, Wardemann et al.24 first used single-cell cloning and antibody expression to demonstrate that a fraction of newly generated B cells in the human bone marrow express self- and polyreactive antibodies and that their development is regulated at two independent self-tolerance checkpoints. Subsequent studies from our laboratory and from others elucidated mechanisms and features of antibodies associated with autoimmune diseases or immunodeficiency syndromes25,26,27,28. B-cell cloning techniques also enabled the isolation of antibodies able to neutralize numerous clinically important pathogens including severe, acute, respiratory syndrome coronavirus (SARS-CoV), influenza and HIV-1, among many others29,30,31,32,33,34,35. Understanding the mechanisms that lead to the elicitation of neutralizing antibodies is, in turn, helping researchers design more effective vaccines20,36,37,38. Nonetheless, one key limitation of low-throughput, B-cell cloning studies has been that they provide only a glimpse of a miniscule slice of the full antibody repertoire.
High-throughput sequencing of the antibody repertoire
Compared with Sanger sequencing, Ig-seq can provide a much broader picture of the antibody repertoire (Fig. 3a). Although conceptually simple, for its proper application, Ig-seq demands thoughtful consideration of experimental design, a detailed understanding of the sources of DNA sequence and quantification errors, the ability to delineate which VH genes are paired with which VL genes in each single B cell, and the use of appropriate data mining and visualization tools to make biological sense of the large amounts of information generated in such experiments.
Figure 3. Methods for high-throughput sequencing of the Ig sequence repertoire.
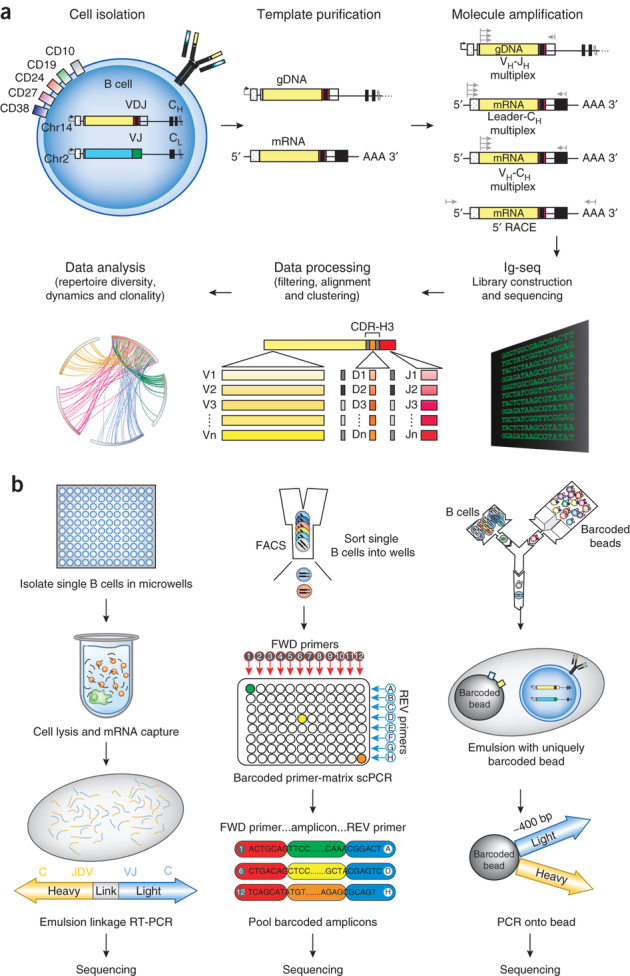
(a) Schematic of steps involved in high-throughput sequencing of Ig genes from bulk B-cell populations of B-cell subsets sorted according to expression of indicated cell-surface markers. Either genomic DNA (gDNA) or mRNA can be used as template, and the choice of template influences the number and location of primers used for subsequent PCR amplification. gDNA amplification is performed using primers complementary to the rearranged V-region gene (VDJ recombinant); amplification of cDNA is performed either using a 5′ primer pool complementary to the leader peptides or FR1s of V-gene segments, and a single 3′ CH1 (or Cκ,Cλ if amplifying light chain genes) primer, or alternatively by 5′ RACE. Although throughput is high, in bulk analysis information regarding which VH and VL chains were paired in the same cell is lost, as cells are lysed in bulk and VH and VL genes are amplified in separate reactions. (b) Schematic of single-cell immunoglobulin repertoire sequencing methods, which preserve information about endogenous VH:VL pairs. Left panel: B-cell lysis and mRNA capture in picoliter well arrays. Middle panel: single-cell PCR following limited B-cell dilution and amplification using barcoded primers. Right panel: microfluidic barcoding of VH and VL cDNAs. Ig, immunoglobulin.
Experimental design.
The first consideration is the source of B cells. Most human antibody sequencing studies have used B cells from peripheral blood because the blood is one of the few readily accessible sources of B cells in humans (tonsils is the other one). However, it is estimated that only 2% of the 1–2 × 1011 B cells in the human body are present in peripheral blood, compared with almost 28% in lymph nodes, 23% in the spleen and on mucosal surfaces, and 17% in the red bone marrow (medulla ossium rubra)3. Thus, the antibody repertoire in peripheral B cells provides a narrow view of the humoral response to antigen challenge.
Second, it is important to consider whether to use genomic DNA (gDNA) or mRNA for immunoglobulin sequencing analyses (Fig. 3a). Whether or not one should use gDNA or mRNA depends on what question is being asked. Sequencing gDNA facilitates estimation of the clonality of a given Ig sequence (in other words, the number of B cells expressing that antibody) because the number of sequence reads will, in general, be proportional to the number of gDNA template molecules (assuming no primer biases, as discussed below). On the other hand, using mRNA as a template can provide an estimate of the relative expression level of various immunoglobulin sequences in the repertoire. However, because immunoglobulin transcription varies dramatically (up to 100-fold) between naive B cells and plasma cells (Fig. 2)39, using unsorted bulk B cells from peripheral blood as the source of mRNA makes it challenging to deduce cellular clonal frequencies. When unsorted peripheral blood mononuclear cells are used as a source of mRNA, the degree of somatic hypermutation in sequencing data sets may be employed ipso facto to distinguish V-gene transcripts derived from antigen-experienced versus naive B cells40. Alternatively, cell-surface markers can be used to sort B-cell subsets of interest before mRNA isolation41,42. Further complicating comparisons between repertoires from different studies, recent work43,44,45,46 has unveiled an unexpectedly high degree of polymorphism in the human IGH locus. Understanding the degree of genetic variation is critical for assigning VDJ segment usage, estimating somatic hypermutation and comparing antibody responses among individuals.
Another experimental design consideration relates to the large number of B cells in humans (∼2–4 × 109 B cells in the blood alone). The minimal sampling depth needed to cover the antibody repertoire to an extent sufficient to answer a particular biological question needs to be considered. Clearly the sequencing depth must be greater than the number of B cells in the sample (which means that enumeration of B cells by fluorescence-activated cell sorting is essential). Further, the required sequencing depth is also dependent on the approach used to minimize PCR and base-calling errors (see below).
Distinguishing error from true biological variation.
There are two sources of sequence errors: those that arise from sample preparation (reverse transcription and PCR) and those inherent to the DNA sequencing platform.
Relative clonal frequencies can be estimated from gDNA by preparing and sequencing libraries from multiple aliquots from the same sample (technical replicates)41,47. In this approach, each aliquot is used to generate a barcoded library, and the resulting libraries from different aliquots are sequenced. The barcode prevents artifacts due to the contamination of one library with DNA from a different aliquot. Because antibody genes found in multiple libraries can arise only if the respective clonal B cells are present in each of the starting aliquots, this method can reveal clonal expansions41,47. However, using gDNA as the templates is not without complications. First, amplification of VDJ segments from gDNA necessitates the use of primer sets that anneal to all the individual germline V-gene segments (Fig. 3a). Another drawback of gDNA-derived antibody gene libraries is that they contain productive and nonproductive VDJ rearrangements (the latter are also present in cDNA libraries but at a much lower frequency due to the decay of nonsense RNAs)48,49. Lastly, the lower concentration of template in gDNA necessitates a greater number of PCR cycles; this increases error frequencies and further confounds quantification.
Using mRNA as the starting material enables amplification with reverse transcription and 5′RACE (5′ rapid amplification of cDNA ends) with 3′ primers that anneal to the constant region of IgH or IgL, thus circumventing the need for complex V-gene-specific primer sets (Fig. 3a). If starting with mRNA, errors introduced by reverse transcriptase can be minimized using commercial high-fidelity retroviral reverse transcriptases or thermostable group II intron reverse transcriptases50.
Regardless of whether gDNA or cDNA is used as the template, PCR introduces amplification artifacts owing to the differential amplification of some DNA templates over others (even in 5′RACE), base misincorporation and template switching; the latter results in chimeras from the joining of fragments encoded by two or more template DNAs. Nucleotide misincorporation by PCR cannot generally be distinguished from most types of base-calling errors introduced during sequencing, but the latter generally occur at higher frequency and hence they are a greater concern. Chimeras resulting from template switching generate sequences that either cannot be assigned to a germline V-gene segment by standard VDJ identification algorithms, or are interpreted as having an artifactually very high rate of somatic hypermutation. The presence of chimeras makes discernment of true VH gene replacement events particularly challenging. However, the fact that gene replacement by definition has to involve recombination to upstream VH gene segments in the chromosome (because downstream VH gene segments will have been deleted during the primary recombination event) can be used to assist the identification of true VH replacement events. Quantification biases introduced by PCR may be minimized by amplifying and then sequencing the same sample with two or more different primer sets, followed by informatics comparison of the similarities in the respective datasets51; another possible solution is using emulsion PCR to sequester individual DNA molecules for amplification52.
Several of the complications underlying DNA sequencing error have been recently reviewed53,54,55,56,57 so here we focus only on the issues most pertinent to Ig-seq, and on recent technological advances aimed at increasing sequencing accuracy of antibody repertoires. Sequencing errors relate to the particular DNA sequencing technology used in each experiment and encompass incorrect base assignment, insertion/deletions (collectively known as indels) and ambiguous base calls. Pyrosequencing-based technologies (Roche 454 and IonTorrent) are dominated by indels, whereas dye-labeled reversible terminator technology (Illumina HiSeq and MiSeq) is dominated by substitution errors55,58,59,60,61. Indels generated by pyrosequencing methods arise at frequencies around 5 × 10−3 and can be computationally accounted for with various degrees of success56,62,63. According to a recent analysis, the frequency of base substitutions varies from 0.3% to 0.9%, depending on the platform61. Overall, the Illumina platform is most suitable for Ig-seq applications because of its combination of relatively low base-calling error rates and relatively low cost.
It should be noted that the importance of sequencing errors depends on the objective of the antibody repertoire sequencing experiment. For example, if the objective is to generate CDR-H3 length distribution statistics or V-J segment use statistics, then sequencing errors are less of a problem, as meaningful information can be obtained using clustering algorithms that group together highly homologous sequences and minimize the effects of sequencing error51,64,65,66,67. For other applications where sequence accuracy is critical, a number of recently developed approaches can help reduce sequencing errors. For example, up to tenfold higher sequencing accuracy can be attained using circularized V-gene DNA that is sequenced multiple times using the PacBio platform68; however, this accuracy comes at a higher cost and much lower throughput. Various DNA barcoding techniques, whereby a nucleotide barcode is appended to each DNA template molecule before PCR amplification, can also improve sequencing accuracy69,70,71. Barcoding enables more accurate quantification of the DNA template molecules in the original library (by counting barcodes instead of sequence reads), and error correction (by generating consensus reads having the same barcode). In a recent application of barcoding to Ig-seq, the Quake laboratory72 achieved 95% identity in the V-gene repertoire (for sequences present at >5 reads) between two technical replicates. Another method73 that relies on barcoding and sequencing both strands can achieve very high accuracy (<10−8 error rate), although at the expense of throughput.
Identifying endogenous VH:VL pairs.
As discussed above, until recently, native VH:VL pairs could be identified only after single-cell cloning by limiting dilution and Sanger sequencing of the individual VH and VL genes. This process is inherently low throughput, expensive (due to high reagent usage) and yields a very limited set of antibody sequences. A modest increase in single-cell cloning throughput (from ∼500 to 2,000 B cells) was achieved using overlap extension PCR to produce single VH:VL amplicons (as opposed to separate VH and VL cDNAs) for Sanger sequencing74. In a study from the Quake laboratory, Weinstein et al.4 sorted ∼200 mouse B cells into microwells, followed by quantitative RT-PCR on a microfluidic chip using primers specific for genes of interest that also contained sequencing adapters, thus enabling the correlation between expression of multiple genes and the degree of somatic hypermutation in the antibody heavy and light chain genes.
Additional approaches for sequencing endogenous VH:VL pairs were more recently developed in the Wardemann and Georgiou laboratories. Busse et al.75 used a two-dimensional, bar-coded primer matrix to combine single-cell VH and VL gene amplification with high-throughput sequencing; this increases throughput up to a total of 50,000 individual B cells (Fig. 3b). Importantly, this approach also enabled the facile cloning of IgH and Igκ,λ sequences into expression vectors for further antibody characterization. DeKosky et al.76 developed a VH:VL pairing technology that relies on sequestering single B cells into subnanoliter volume wells, lysing the cells, capturing RNA on poly-dT beads and generating amplicons encoding linked VH:VL segments by emulsion overlap extension PCR (Fig. 3b). Yields of up to 6–7 × 103 unique VH:VL pairs from 7 × 104 activated memory B cells in a one-day experiment, with >96% validated pairing accuracy, have been reported. This method was adapted to detect co-expression of V genes with transcription factors of interest (e.g., BLIMP1) in antibody-secreting cells (B.J. DeKosky, personal communication).
A much higher-throughput method (>2 × 106 B cells per experiment) under development in our laboratory relies on cell encapsulation within controlled microdroplet diameter emulsions (an important consideration because reverse transcriptase is inhibited in droplets of <5 nl volume) (unpublished data). Alternatively, single cells have been encapsulated in water-in-oil emulsion together with uniquely barcoded beads using a microfluidic device. After reverse transcriptase and PCR, every RNA molecule originating from a single cell is effectively loaded onto the uniquely barcoded beads. PCR products are sequenced on the Illumina platform and correctly paired VH and VL sequences are identified by virtue of their shared barcode77 (Fig. 3c). Given the rapid pace of technology advancement, it may be expected that in the very near future all antibody repertoire analyses will report natively paired VH and VL genes.
Bioinformatic analysis of antibody sequences.
Several established methods for VDJ assignment and CDR-H3 identification are available (e.g., IMGT/V-Quest, IgBLAST or iHMMune-align) and faster, more precise algorithms continue to be developed54,57,78,79. Estimation of the size of the antibody repertoire can be accomplished by rarefaction analysis, maximum entropy and Poisson log-normal distribution models51,80,81. There are also many approaches for clustering V genes likely to have originated from the expansion and somatic hypermutation of a single B cell encoding an unmutated antibody precursor57. However, clustering becomes particularly problematic as the sequencing depth and repertoire diversity increases. Likewise, inference of VH evolution (i.e., how somatically mutated V genes arise from precursor sequences initially present in the naive primary repertoire) is a major challenge67,82. Finally, it should be noted that researchers working in this area generally use custom-made bioinformatics pipelines. The lack of standardization and shared computational resources makes it extremely difficult to carry out meta-analyses of published data generated in different laboratories; this is a key issue that will need to be addressed as the field matures. Interchangeable data formats deposited in a central database, validated open-source algorithms for data analysis and standards for Ig-seq experimental description analogous to the minimum information about a microarray experiment83 are critically needed84.
Applications of antibody repertoire sequencing
Ig-seq is finding a wide range of basic and applied immunology applications.
Antibody discovery.
Screening of large combinatorial libraries by ribosomal, phage, bacterial or yeast display is widely employed for the isolation of antibodies capable of binding virtually any ligand85,86,87. Combinatorial libraries are typically generated by the random pairing of very large (typically >107 each) ensembles of VH and VL genes isolated from mammals (naive or immune libraries, depending on whether the animal was immunized); alternatively one can use synthetic libraries in which a single, or a small set, of VH and VL genes are diversified by mutagenesis of the CDRs. Screening involves sequential rounds of binding to antigen, a process that progressively restricts the diversity of the library to very few antibody clones with the requisite affinity and specificity. We and others have used high-throughput sequencing as a means of evaluating the initial diversity of antibodies encoded in a library64,88,89 and to determine how this diversity declines as binders to antigen are progressively enriched89,90,91,92. During library screening, antibody diversity is reduced as antigen-binding clones are enriched over the background of unrelated antibodies in the starting library. However, the expression of some antibodies having high affinity often adversely affects the growth of the cells that encode them (nota bene: antibodies with poor affinity are equally likely to have an adverse effect on cell growth, but obviously such clones are of no interest in library screening). As a result, the respective antibody genes are enriched during early rounds of screening for antigen binding but are then progressively depleted because they are outcompeted by faster growing cells expressing lower quality antibodies87,90,92,93. High-throughput sequencing of libraries following one or two rounds of selection for antigen binding has therefore been used to rescue clones encoding high-affinity antibodies that could not have been discovered otherwise.
Three different approaches that exploit antibody repertoire sequencing analysis for antibody isolation directly from animals or humans, without library screening, have been developed. First, in work from the Georgiou laboratory, Reddy et al.94 observed that in mice the antibody repertoire encoded by CD138+ antibody-secreting B cells in the bone marrow (bone marrow plasma cells) becomes highly polarized seven days after secondary immunization with antigen, with the most abundant VH and VL genes representing 2–30% of all V-gene sequences. To discover antigen-specific antibodies, Reddy et al.94 paired VH and VL genes based on their relative rank frequency in the respective repertoires and thus obtained antibodies with nanomolar affinity for antigen. Subsequently, Saggy et al.95 showed that VH genes present at high frequencies in the splenic V-gene repertoire of immunized mice encode antibodies capable of binding the immunizing antigen. However, the types of bone marrow or spleen samples used in these mouse analyses are rarely available from humans.
A different approach, pioneered by Shapiro, Kwong and coworkers, builds on the observation that the co-evolution of heavy and light chains in broadly neutralizing HIV-1–specific antibodies is reflected in the matching topology of VH and VL phylogenetic trees as gleaned from Ig-seq63,96. VH and VL genes within matching branches of the respective phylogenetic trees, that is, displaying similar patterns of mutation accumulation, were paired to yield novel broadly neutralizing antibodies. This method is particularly useful for the identification of antibodies with high somatic hypermutation levels (such as those elicited by persistent infection) where a deep phylogenetic tree can be constructed.
Ig-seq has also enabled the proteomic identification of antibodies from biological fluids. Even though the main effector function of B cells is the secretion of antibodies into blood or mucosal and respiratory epithelia, the composition of the antibody repertoire in these bodily fluids remained elusive. We and others realized that shotgun liquid chromatography–tandem mass spectrometry (LC-MS/MS) proteomic identification of the antibodies that comprise the humoral response requires a matched personal antibody sequence database, obtained by Ig-seq, to interpret the MS/MS spectra (Fig. 4)97,98,99. In this manner, Polakiewicz and coworkers98,99 identified high-affinity, antigen-specific antibodies from immunized animals, from a human vaccinated with hepatitis B and from a cytomegalovirus-infected volunteer98,99. In more recent studies, we determined the monoclonal antibody repertoire that comprises the serum polyclonal response in rabbits and in humans following vaccination (unpublished data and ref. 66). The advent of antibody discovery by proteomic mining of the serum antibody response has in turn opened the way for the isolation of biologically relevant antibodies from convalescent patients. Finding the serum antibodies that were responsible for the resolution of disease states in patients is likely to be of great relevance for drug discovery because such antibodies will have already been established to be of therapeutic value.
Figure 4. Deconvoluting the serum antibody repertoire.
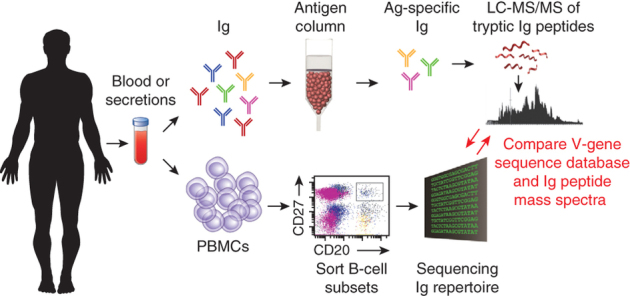
B cells from peripheral blood or other tissues are sorted and subjected to high-throughput immunoglobulin V-gene sequencing, resulting in generation of a personal antibody sequence database. Antigen-specific antibodies from serum are then isolated by affinity chromatography, digested into peptides, and subjected to LC-MS/MS analysis. The MS/MS data are interpreted using the antibody sequence database, thereby allowing identification of CDRH3-derived peptides and the genes encoding the repertoire of antigen-specific antibodies in the serum. Ig, immunoglobulin.
Understanding immune repertoire development.
The use of Ig-seq to map out the global antibody repertoire was first accomplished by Quake and coworkers in the zebrafish, which exhibit the basic features of adaptive immunity but have fewer possible VDJ combinations than mice or humans51,100. Ig-seq has subsequently been used to analyze the repertoires of a variety of other species, including mice, chickens and cattle51,66,68,94,101,102,103,104,105,106,107.
More importantly, however, Ig-seq is providing unprecedented insights into the mechanisms responsible for shaping the human naive BCR repertoire. As noted above, Ig-seq analysis of the VH repertoire revealed the high degree of allelic diversity in the human immunoglobulin locus43,45,108. Analysis by our laboratory and by others of germline VH, Vκ and Vλ segment usage and frequencies of recombination between particular V-D and D-J segments in the naive BCR repertoire revealed a marked skewing that in turn then shapes the repertoire in mature, antigen-experienced B cells7,43,47,49,64,102,109,110,111,112. A single D-segment reading frame is overwhelmingly preferred in VDJ recombination, inversions of D segments are extremely rare113 and the CDR-H3 region exhibits universal categorical constraints with respect to amino acid composition, average hydrophobicity, charge distribution and length. Antibodies with long or charged CDR-H3s are of great clinical interest because, although they are more likely to be autoreactive, they often enable binding to occluded sites on pathogens and mediate pathogen neutralization. In the human repertoire, long CDR-H3s arise by D-D joining (which occurs at frequencies from 0.125% to >0.5% in the naive repertoire but is reduced in antigen-experienced B cells) and by extensive nucleotide addition combined with preferential usage of longer D segments and of the longer J6 segment in the germline47,49,114,115. The conservation of hydrophobicity, which results in the selection of a neutral CDR-H3 and against hydropathically extreme (charged or hydrophobic) antibody-binding sites, is a common theme across vertebrate evolution7,116. Collectively, these findings indicate that the CDR-H3 loop in humans explores vast sequence space, but within boundaries40,53,64,102,105.
Recent studies suggest determinism in VDJ recombination frequencies in the primary antibody repertoire. VDJ segment usage in the antibody repertoire of homozygotic twins is indistinguishable, suggesting that it is determined by genetic factors40. Nonetheless, it is important to note that diversification in the CDR-H3 results in a highly private repertoire that shows very little overlap among individuals. Further, the human naive antibody repertoire that developed in immunocompromised mice engrafted with human CD34+ hematopoietic stem cells105 showed V-J usage patterns and, even more strikingly, checkpoint depletion of antibodies displaying autoreactivity signatures similar to those in humans24.
Important insights are also being gleaned from sequencing of antibody repertoires encoded by B cells at different anatomical sites. One particularly noteworthy study analyzed the murine intestinal IgA repertoire117. Another study of mouse mucosal B-cell repertoires in the intestinal lamina propria raised the intriguing possibility that, contrary to long-standing dogma, the bone marrow might not be the exclusive site of adult B-cell development. This was because B cells in the gut expressed Rag-1 recombinase, displayed a pre-B or immature B phenotype and importantly, encoded a distinctive VL repertoire; these features are consistent with B-cell development and selection in situ in the lamina propria118. The IgG VH repertoire in the cerebrospinal fluid has also been examined119 and found to have differences in somatic hypermutation compared to that in the peripheral blood, further suggesting that B-cell maturation might have occurred independently in the central nervous system or the periphery.
Several studies have attempted to estimate the total diversity of the human antibody repertoire by Ig-seq of peripheral B cells40,47,102. This task is very challenging owing to the large size of the antibody repertoire, variable sampling depth, transcriptional differences among B-cell subsets, sequencing errors and, last but not least, the fact that peripheral blood contains <2% of all B cells3. The overwhelming majority of published studies105,120 as well as our unpublished data are consistent with the notion that, on the one hand, the VH gene repertoire is highly private (unique to an individual), although a small number of CDR-H3 appear to be shared (in other words, they are stereotypical or public) among different individuals. However, a sizable fraction of shared sequences are found in light chains, due to the lower diversity of the VL gene repertoires121,122. It is intriguing that shared IgL chain genes also tend to be the most abundantly expressed123.
Infectious diseases.
Ig-seq is also providing insights into the adaptive immune responses elicited by pathogen challenge or vaccination. Pathogen challenge can affect the BCR repertoire of responding B as well as the naive repertoire. Many pathogens produce superantigens, which are proteins that bind to certain antibody V domains, resulting in BCR cross-linking and subsequent B-cell deletion. Predictably, superantigen exposure results in a skewed naive antibody repertoire103. Surprisingly, however, depletion of V genes bound by superantigen was not observed in the naive repertoire of transgenic mice constitutively expressing superantigen48. Interestingly, skewed naive B-cell antibody repertoire was also reported for patients with chronically evolving hepatitis C infection124. Changes in the overall antibody repertoire are also evident following vaccination or infection120,125,126,127. Notably, Boyd, Fire and coworkers125 observed convergent antibody signatures (stereotyped CDR-H3 sequences) in patients experiencing acute dengue infection. This observation raises the possibility that Ig-seq aimed at detecting stereotypical responses may be used as a diagnostic tool for predicting infectious disease severity.
Another exciting potential application of Ig-seq is the identification of V genes clonally related to those encoded by protective antibodies isolated from an individual; one can then infer the antibody lineages that led to the evolution of these protective antibodies, starting from an unmutated common ancestor IgH germline sequence. This approach is particularly relevant to understanding the evolution of broadly neutralizing antibodies (bNAbs) during infection with rapidly evolving viral pathogens such as influenza and HIV-1 (refs. 67, 96, 127, 128, 129, 130). Time-ordered sequencing of evolving virus populations and antibody responses in the same host, together with isolation of bNAbs, is helping delineate the dynamic between adaptive immune responses that exert selective pressure on the virus and the emergence of viral escape mutations131. Tracing the evolutionary paths that lead to the generation of bNAbs is also critical for the design of immunogens and vaccination schedules that will elicit an immune response by first activating naive, germline, antibody-expressing B cells and then steering B clonal selection toward an affinity maturation pathway that leads to the production of bNAbs36,38. Multidonor analysis of VRC01-class anti-HIV-1 bNAbs confirmed that the elicitation of such antibodies from a single ancestor B cell indeed occurs in multiple individuals132. Ig-seq is also useful for evaluating how innate immune responses elicited by adjuvants, such as toll-like receptor 4 (TLR4) or TLR7/8 agonists, affect the diversity of the antibody response and possibly antibody functionality133.
Ig-seq might also be applied to answer the long-standing question of why people in certain age groups, usually the elderly, exhibit higher susceptibility to infectious disease and/or are less well-protected by vaccination. For example, by analyzing antibody lineage structure, isotype and mutational load in the VH repertoire of volunteers in various age groups before and after influenza vaccination, our laboratory detected a higher IgM mutational load before vaccination and a lower degree of repertoire diversity after vaccination in elderly individuals120. Another study also detected a smaller degree of IgM and IgA CDR-H3 diversification in elderly individuals before and after receiving the influenza or the 23-valent pneumococcal vaccine126.
Immune dysregulation.
We expect that deep sequencing of the antibody repertoire in patients with autoimmunity or primary immunodeficiency will provide important mechanistic insights that in turn may guide the development of appropriate therapies. However, very few analyses of antibody repertoires in patients with immune dysregulation have been published thus far; we believe this dearth reflects the very recent development of Ig-seq. That said, a few studies have been reported. In multiple sclerosis, the cerebrospinal fluid VH gene repertoire is biased and shows strong evidence of B-cell activation119. It will be interesting to determine whether the activated B cells in the cerebrospinal fluid are reactive towards KIR4.1, a recently discovered dominant antigen in multiple sclerosis134. In idiopathic IgG4-associated cholangitis (an autoimmune disease associated with abnormal levels of IgG4 in serum), Ig-seq revealed the presence of large clonal IgG4 expansions in affected tissues and peripheral blood; these clonal expansions disappeared after corticosteroid treatment, indicating that determination of IgG4 clonality is a distinguishing feature of the disease and therefore constitutes a useful tool for differential diagnosis135.
Cancer.
B-cell leukemias, lymphomas and multiple myeloma are malignancies that arise at different stages of B-cell development (Fig. 2). As such, BCRs on malignant B cells constitute a biomarker for the abundance of the malignant cell population. Ig-seq of the V-gene repertoire in peripheral B cells, bone marrow samples, tumors and even blood-borne free DNA has been used both for disease detection and for delineating the degree to which antibody evolution and diversification in malignant cells correlates with disease progression or relapse47,58,136,137,138,139,140. For example, Ig-seq of V genes in peripheral blood facilitated detection of cancerous cells and minimal residual disease following treatment of B-cell chronic lymphocytic leukemia (CLL), the most common leukemia in adults47,58,137. The use of Ig-seq to determine the V-gene repertoire en masse and detect the presence of antibodies encoded by leukemic clones (whose V-gene sequences are established from analyses of cancer cell samples before initiation of treatment) circumvents the need to develop a personalized PCR assay for the CLL clonotype in each patient to determine whether relapse has occurred. Ig-seq was also used to detect minimal residual disease in pediatric patients with B-cell acute lymphoblastic leukemia (B-ALL)138 and as a marker of non-Hodgkins lymphoma136.
Ig-seq also revealed that B-ALL patients display various degrees of clonotypic diversity, which arises predominantly from VH gene replacement and appears to be related to relapse frequency138,139. A small degree of B-cell clonal heterogeneity was also observed in CLL (with the degree varying based on whether the disease originated from unmutated or somatically hypermutated B cells)58,137. In contrast, the malignant clonotype in multiple myeloma, a disease which arises from terminally differentiated plasma cells in the bone marrow (Fig. 2) that lack active mechanisms of antibody diversification, displayed little evidence of heterogeneity140. Of note, in CLL and other hematologic malignancies, the same (stereotypic) CDR-H3 is detected in many patients141. The wider application of high-throughput sequencing for the detection of malignancy-stereotypic clonotypes, which may be present at a low frequency in subjects that do yet not display clinical disease, may prove to be a useful, early diagnostic tool. Lastly, Ig-seq may also be applied to understand gammopathies, which are conditions that result in abnormally high levels of antibodies in serum; these are more prevalent in the elderly and can develop into multiple myeloma15,16.
Conclusions
The humoral immune system has evolved to encode an astonishing diversity of antibodies that collectively comprise the antibody repertoire and provide a potent arsenal of recognition reagents (or anticipatory receptors) that can recognize virtually any organic macromolecule of biological significance. The B lymphocyte and its 'anticipatory' receptor, a term used to emphasize the ability of the naive repertoire to bind biologically relevant antigens, is as ancient as the last common vertebrate ancestor (500 million years ago)142. The extraordinary complexity of the vertebrate adaptive immune system has been likened to the Titan brothers Epimetheus (hindsight) and Prometheus (foresight) of Greek mythology143. The germline conservation of antibody genes carries the imprint of ancient adaptations to pathogens humans were exposed to during evolution and, hence, is Epimethean in scope; on the other hand, the immunologic foresight represented by the repertoire encoded by antigen-experienced, mature B cells within an individual enables adaptations to future pathogen challenges and is a Promethean and anticipatory feature. The advent of high-throughput DNA sequencing has enabled the determination of the antibody gene repertoire at unprecedented depth that was inconceivable, even half-a-dozen years ago. We can begin to decipher both the Epimethean and the Promethean compartments of humoral immunity and how each is shaped by the other. Technologies to improve sequence precision and data analysis are being developed at a breakneck pace, reshaping our understanding of many important aspects of B-cell immunology and increasingly affecting clinical diagnosis, antibody drug discovery and vaccine development. However, realizing the full impact of Ig-seq will require the implementation of standards for experimental annotation and data analysis, as well as the creation of databases facilitating deposition and sharing of these important data.
Acknowledgements
We thank B. DeKosky, J. Lavinder & K. Hon Hoi for reading this manuscript; J. Lavinder and C. Chrysostomou for help with the figures; and A. Fire for coining the term Ig-seq. We are also very grateful to the Nature Biotechnology Editorial staff for their meticulous editing and for numerous useful suggestions. This work was supported by grants from the Clayton Foundation, Defense Threat Reduction Agency and US National Institutes of Health (G.G.).
Competing interests
The authors declare no competing financial interests.
References
- 1.Schroeder HW., Jr. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev. Comp. Immunol. 2006;30:119–135. doi: 10.1016/j.dci.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Janeway, C.A. Immunobiology (Garland Science, 2004).
- 3.Apostoaei, A.J. & Trabalka, J.R. Review, Synthesis, and Application of Information on the Human Lymphatic System to Radiation Dosimetry for Chronic Lymphocytic Leukemia (SENES Oak Ridge, Inc., Oak Ridge, TN, 2010).
- 4.Weinstein JA, Zeng X, Chien YH, Quake SR. Correlation of gene expression and genome mutation in single B-cells. PLoS ONE. 2013;8:e67624. doi: 10.1371/journal.pone.0067624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brady BL, Steinel NC, Bassing CH. Antigen receptor allelic exclusion: an update and reappraisal. J. Immunol. 2010;185:3801–3808. doi: 10.4049/jimmunol.1001158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi: 10.1016/S1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 7.Ippolito GC, et al. Forced usage of positively charged amino acids in immunoglobulin CDR-H3 impairs B cell development and antibody production. J. Exp. Med. 2006;203:1567–1578. doi: 10.1084/jem.20052217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2012;12:24–34. doi: 10.1038/nri3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Victora GD, Nussenzweig MC. Germinal centers. Annu. Rev. Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 10.Capra JD, Kehoe JM. Variable region sequences of five human immunoglobulin heavy chains of the VH3 subgroup: definitive identification of four heavy chain hypervariable regions. Proc. Natl. Acad. Sci. USA. 1974;71:845–848. doi: 10.1073/pnas.71.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibi T, Dosch HM. Limiting dilution analysis of the B cell compartment in human bone marrow. Eur. J. Immunol. 1986;16:139–145. doi: 10.1002/eji.1830160206. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z. VH replacement in mice and humans. Trends Immunol. 2007;28:132–137. doi: 10.1016/j.it.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Schroeder HW, Jr., Hillson JL, Perlmutter RM. Early restriction of the human antibody repertoire. Science. 1987;238:791–793. doi: 10.1126/science.3118465. [DOI] [PubMed] [Google Scholar]
- 14.Boyd SD, Liu Y, Wang C, Martin V, Dunn-Walters DK. Human lymphocyte repertoires in ageing. Curr. Opin. Immunol. 2013;25:511–515. doi: 10.1016/j.coi.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varettoni M, et al. Clues to pathogenesis of Waldenstrom macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance provided by analysis of immunoglobulin heavy chain gene rearrangement and clustering of B-cell receptors. Leuk. Lymphoma. 2013;54:2485–2489. doi: 10.3109/10428194.2013.779689. [DOI] [PubMed] [Google Scholar]
- 16.Kyle RA, et al. Prevalence of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2006;354:1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 17.Ehlich A, Martin V, Muller W, Rajewsky K. Analysis of the B-cell progenitor compartment at the level of single cells. Curr. Biol. 1994;4:573–583. doi: 10.1016/S0960-9822(00)00129-9. [DOI] [PubMed] [Google Scholar]
- 18.Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J. Exp. Med. 1998;188:1679–1689. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuppers R, Zhao M, Hansmann ML, Rajewsky K. Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO J. 1993;12:4955–4967. doi: 10.1002/j.1460-2075.1993.tb06189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson PC, Andrews SF. Tools to therapeutically harness the human antibody response. Nat. Rev. Immunol. 2012;12:709–719. doi: 10.1038/nri3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tiller T, et al. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26:205–213. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 23.Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu. Rev. Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- 24.Wardemann H, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 25.Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr. Opin. Immunol. 2008;20:632–638. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Scheid JF, et al. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc. Natl. Acad. Sci. USA. 2011;108:18044–18048. doi: 10.1073/pnas.1113395108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Niro R, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat. Med. 2012;18:441–445. doi: 10.1038/nm.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amara K, et al. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J. Exp. Med. 2013;210:445–455. doi: 10.1084/jem.20121486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Traggiai E, et al. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat. Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wrammert J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu X, et al. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455:532–536. doi: 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheid JF, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458:636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- 34.Beltramello M, et al. The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe. 2010;8:271–283. doi: 10.1016/j.chom.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muellenbeck MF, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J. Exp. Med. 2013;210:389–399. doi: 10.1084/jem.20121970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwong PD, Mascola JR. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity. 2012;37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burton DR, et al. A Blueprint for HIV Vaccine Discovery. Cell Host Microbe. 2012;12:396–407. doi: 10.1016/j.chom.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein U, Kuppers R, Rajewsky K. Evidence for a large compartment of IgM-expressing memory B cells in humans. Blood. 1997;89:1288–1298. [PubMed] [Google Scholar]
- 40.Glanville J, et al. Naive antibody gene-segment frequencies are heritable and unaltered by chronic lymphocyte ablation. Proc. Natl. Acad. Sci. USA. 2011;108:20066–20071. doi: 10.1073/pnas.1107498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maecker HT, et al. New tools for classification and monitoring of autoimmune diseases. Nat. Rev. Rheumatol. 2012;8:317–328. doi: 10.1038/nrrheum.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in human B cell phenotypic profiling. Front. Immunol. 2012;3:302. doi: 10.3389/fimmu.2012.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyd SD, et al. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J. Immunol. 2010;184:6986–6992. doi: 10.4049/jimmunol.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, et al. Genomic screening by 454 pyrosequencing identifies a new human IGHV gene and sixteen other new IGHV allelic variants. Immunogenetics. 2011;63:259–265. doi: 10.1007/s00251-010-0510-8. [DOI] [PubMed] [Google Scholar]
- 45.Kidd MJ, et al. The inference of phased haplotypes for the immunoglobulin H chain V region gene loci by analysis of VDJ gene rearrangements. J. Immunol. 2012;188:1333–1340. doi: 10.4049/jimmunol.1102097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson CT, et al. Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation. Am. J. Hum. Genet. 2013;92:530–546. doi: 10.1016/j.ajhg.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyd SD, et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel VDJ pyrosequencing. Sci. Transl. Med. 2009;1:12ra23. doi: 10.1126/scitranslmed.3000540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aoki-Ota M, Torkamani A, Ota T, Schork N, Nemazee D. Skewed primary Igkappa repertoire and V-J joining in C57BL/6 mice: implications for recombination accessibility and receptor editing. J. Immunol. 2012;188:2305–2315. doi: 10.4049/jimmunol.1103484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larimore K, McCormick MW, Robins HS, Greenberg PD. Shaping of human germline IgH repertoires revealed by deep sequencing. J. Immunol. 2012;189:3221–3230. doi: 10.4049/jimmunol.1201303. [DOI] [PubMed] [Google Scholar]
- 50.Mohr S, et al. Thermostable group II intron reverse transcriptase fusion proteins and their use in cDNA synthesis and next-generation RNA sequencing. RNA. 2013;19:958–970. doi: 10.1261/rna.039743.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinstein JA, Jiang N, White RA, III, Fisher DS, Quake SR. High-throughput sequencing of the zebrafish antibody repertoire. Science. 2009;324:807–810. doi: 10.1126/science.1170020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rubelt F, et al. Onset of immune senescence defined by unbiased pyrosequencing of human immunoglobulin mRNA repertoires. PLoS ONE. 2012;7:e49774. doi: 10.1371/journal.pone.0049774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prabakaran P, Streaker E, Chen W, Dimitrov DS. 454 antibody sequencing - error characterization and correction. BMC Res. Notes. 2011;4:404. doi: 10.1186/1756-0500-4-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benichou J, Ben-Hamo R, Louzoun Y, Efroni S. Rep-Seq: uncovering the immunological repertoire through next-generation sequencing. Immunology. 2012;135:183–191. doi: 10.1111/j.1365-2567.2011.03527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baum PD, Venturi V, Price DA. Wrestling with the repertoire: the promise and perils of next generation sequencing for antigen receptors. Eur. J. Immunol. 2012;42:2834–2839. doi: 10.1002/eji.201242999. [DOI] [PubMed] [Google Scholar]
- 56.Bolotin DA, et al. Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms. Eur. J. Immunol. 2012;42:3073–3083. doi: 10.1002/eji.201242517. [DOI] [PubMed] [Google Scholar]
- 57.Mehr R, Sternberg-Simon M, Michaeli M, Pickman Y. Models and methods for analysis of lymphocyte repertoire generation, development, selection and evolution. Immunol. Lett. 2012;148:11–22. doi: 10.1016/j.imlet.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 58.Campbell PJ, et al. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. USA. 2008;105:13081–13086. doi: 10.1073/pnas.0801523105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen P, et al. Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire. BMC Genomics. 2011;12:106. doi: 10.1186/1471-2164-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kircher M, Heyn P, Kelso J. Addressing challenges in the production and analysis of illumina sequencing data. BMC Genomics. 2011;12:382. doi: 10.1186/1471-2164-12-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loman NJ, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 62.Michaeli M, Noga H, Tabibian-Keissar H, Barshack I, Mehr R. Automated cleaning and pre-processing of immunoglobulin gene sequences from high-throughput sequencing. Front. Immunol. 2012;3:386. doi: 10.3389/fimmu.2012.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu J, et al. Mining the antibodyome for HIV-1-neutralizing antibodies with next-generation sequencing and phylogenetic pairing of heavy/light chains. Proc. Natl. Acad. Sci. USA. 2013;110:6470–6475. doi: 10.1073/pnas.1219320110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glanville J, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA. 2009;106:20216–20221. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z, Collins AM, Wang Y, Gaeta BA. Clustering-based identification of clonally-related immunoglobulin gene sequence sets. Immunome Res. 2010;6(suppl. 1):S4. doi: 10.1186/1745-7580-6-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wine Y, et al. Molecular deconvolution of the monoclonal antibodies that comprise the polyclonal serum response. Proc. Natl. Acad. Sci. USA. 2013;110:2993–2998. doi: 10.1073/pnas.1213737110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu J, et al. Somatic populations of PGT135–137 HIV-1-neutralizing antibodies identified by 454 pyrosequencing and bioinformatics. Frontiers Microbiol. 2012;3:315. doi: 10.3389/fmicb.2012.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larsen PA, Smith TP. Application of circular consensus sequencing and network analysis to characterize the bovine IgG repertoire. BMC Immunol. 2012;13:52. doi: 10.1186/1471-2172-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fu GK, Hu J, Wang PH, Fodor SP. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc. Natl. Acad. Sci. USA. 2011;108:9026–9031. doi: 10.1073/pnas.1017621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shiroguchi K, Jia TZ, Sims PA, Xie XS. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc. Natl. Acad. Sci. USA. 2012;109:1347–1352. doi: 10.1073/pnas.1118018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vollmers C, Sit RV, Weinstein JA, Dekker CL, Quake SR. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:13463–13468. doi: 10.1073/pnas.1312146110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meijer PJ, et al. Isolation of human antibody repertoires with preservation of the natural heavy and light chain pairing. J. Mol. Biol. 2006;358:764–772. doi: 10.1016/j.jmb.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 75.Busse, C.E., Czogiel, I., Braun, P., Arndt, P.F. & Wardemann, H. Single-cell based high-throughput sequencing of full-length immunoglobulin heavy and light chain genes. Eur. J. Immunol. 10.1002/eji.201343917 (22 October 2013). [DOI] [PubMed]
- 76.DeKosky BJ, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 2013;31:166–169. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Church, G.M., Vigneault, F., Laserson, U. & Bachelet, I. High-throughput immune sequencing. US patent US20130296535 (2013).
- 78.Lakhani KR, et al. Prize-based contests can provide solutions to computational biology problems. Nat. Biotechnol. 2013;31:108–111. doi: 10.1038/nbt.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kidd BA, Peters LA, Schadt EE, Dudley JT. Unifying immunology with informatics and multiscale biology. Nat. Immunol. 2014;15,:118–127. doi: 10.1038/ni.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rempala GA, Seweryn M, Ignatowicz L. Model for comparative analysis of antigen receptor repertoires. J. Theor. Biol. 2011;269:1–15. doi: 10.1016/j.jtbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mora T, Walczak AM, Bialek W, Callan CG., Jr. Maximum entropy models for antibody diversity. Proc. Natl. Acad. Sci. USA. 2010;107:5405–5410. doi: 10.1073/pnas.1001705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yaari G, Uduman M, Kleinstein SH. Quantifying selection in high-throughput Immunoglobulin sequencing data sets. Nucleic Acids Res. 2012;40:e134. doi: 10.1093/nar/gks457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edgar R, Barrett T. NCBI GEO standards and services for microarray data. Nat. Biotechnol. 2006;24:1471–1472. doi: 10.1038/nbt1206-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brusic, V., Gottardo, R., Kleinstein, S.H., Davis, M. M. & the HIPC Steering Committee. Computational resources for high-dimension immune analysis from the Human Immunology Project Consortium. Nat. Biotechnol. 10.1038/nbt.2777 (19 January 2014). [DOI] [PMC free article] [PubMed]
- 85.Bradbury AR, Sidhu S, Dubel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol. 2011;29:245–254. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010;10:301–316. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 87.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat. Rev. Cancer. 2012;12:278–287. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 88.Ge X, Mazor Y, Hunicke-Smith SP, Ellington AD, Georgiou G. Rapid construction and characterization of synthetic antibody libraries without DNA amplification. Biotechnol. Bioeng. 2010;106:347–357. doi: 10.1002/bit.22712. [DOI] [PubMed] [Google Scholar]
- 89.Mahon CM, et al. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J. Mol. Biol. 2013;425:1712–1730. doi: 10.1016/j.jmb.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 90.Fischer N. Sequencing antibody repertoires: the next generation. MAbs. 2011;3:17–20. doi: 10.4161/mabs.3.1.14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ravn U, et al. Deep sequencing of phage display libraries to support antibody discovery. Methods. 2013;60:99–110. doi: 10.1016/j.ymeth.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 92.Ravn U, et al. By-passing in vitro screening–next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010;38:e193. doi: 10.1093/nar/gkq789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Larman HB, Xu GJ, Pavlova NN, Elledge SJ. Construction of a rationally designed antibody platform for sequencing-assisted selection. Proc. Natl. Acad. Sci. USA. 2012;109:18523–18528. doi: 10.1073/pnas.1215549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reddy ST, et al. Monoclonal antibodies isolated without screening by analyzing the variable-gene repertoire of plasma cells. Nat. Biotechnol. 2010;28:965–969. doi: 10.1038/nbt.1673. [DOI] [PubMed] [Google Scholar]
- 95.Saggy I, et al. Antibody isolation from immunized animals: comparison of phage display and antibody discovery via V gene repertoire mining. PEDS. 2012;25:539–549. doi: 10.1093/protein/gzs060. [DOI] [PubMed] [Google Scholar]
- 96.Zhu J, et al. De novo identification of VRC01 class HIV-1-neutralizing antibodies by next-generation sequencing of B-cell transcripts. Proc. Natl. Acad. Sci. USA. 2013;110:E4088–E4097. doi: 10.1073/pnas.1306262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lavinder, J.J. et al. Proteomic identification of antibodies. US patent 20130178370. (2013).
- 98.Cheung WC, et al. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat. Biotechnol. 2012;30:447–452. doi: 10.1038/nbt.2167. [DOI] [PubMed] [Google Scholar]
- 99.Sato S, et al. Proteomics-directed cloning of circulating antiviral human monoclonal antibodies. Nat. Biotechnol. 2012;30:1039–1043. doi: 10.1038/nbt.2406. [DOI] [PubMed] [Google Scholar]
- 100.Jiang N, et al. Determinism and stochasticity during maturation of the zebrafish antibody repertoire. Proc. Natl. Acad. Sci. USA. 2011;108:5348–5353. doi: 10.1073/pnas.1014277108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ota M, et al. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J. Immunol. 2010;185:4128–4136. doi: 10.4049/jimmunol.1002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnaout R, et al. High-resolution description of antibody heavy-chain repertoires in humans. PLoS ONE. 2011;6:e22365. doi: 10.1371/journal.pone.0022365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gronwall C, Kosakovsky Pond SL, Young JA, Silverman GJ. In vivo VL-targeted microbial superantigen induced global shifts in the B cell repertoire. J. Immunol. 2012;189:850–859. doi: 10.4049/jimmunol.1200245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang F, et al. Reshaping antibody diversity. Cell. 2013;153:1379–1393. doi: 10.1016/j.cell.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ippolito GC, et al. Antibody repertoires in humanized NOD-scid-IL2Rgamma(null) mice and human B cells reveals human-like diversification and tolerance checkpoints in the mouse. PLoS ONE. 2012;7:e35497. doi: 10.1371/journal.pone.0035497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Castro R, et al. Teleost fish mount complex clonal IgM and IgT responses in spleen upon systemic viral infection. PLoS Pathog. 2013;9:e1003098. doi: 10.1371/journal.ppat.1003098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu L, et al. Fundamental characteristics of the immunoglobulin VH repertoire of chickens in comparison with those of humans, mice, and camelids. J. Immunol. 2012;188:322–333. doi: 10.4049/jimmunol.1102466. [DOI] [PubMed] [Google Scholar]
- 108.Wang Y, et al. IgE sequences in individuals living in an area of endemic parasitism show little mutational evidence of antigen selection. Scand. J. Immunol. 2011;73:496–504. doi: 10.1111/j.1365-3083.2011.02525.x. [DOI] [PubMed] [Google Scholar]
- 109.Wu YC, et al. High-throughput immunoglobulin repertoire analysis distinguishes between human IgM memory and switched memory B-cell populations. Blood. 2010;116:1070–1078. doi: 10.1182/blood-2010-03-275859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Prabakaran P, et al. Origin, diversity, and maturation of human antiviral antibodies analyzed by high-throughput sequencing. Front. Microbiol. 2012;3:277. doi: 10.3389/fmicb.2012.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vollmers C, Sit RV, Weinstein JA, Dekker CL, Quake SR. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc. Natl. Acad. Sci. USA. 2013;110:13463–13468. doi: 10.1073/pnas.1312146110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Briney BS, Willis JR, Hicar MD, Thomas JW, II, Crowe JE., Jr. Frequency and genetic characterization of V(DD)J recombinants in the human peripheral blood antibody repertoire. Immunology. 2012;137:56–64. doi: 10.1111/j.1365-2567.2012.03605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Benichou J, et al. The restricted DH gene reading frame usage in the expressed human antibody repertoire is selected based upon its amino acid content. J. Immunol. 2013;190:5567–5577. doi: 10.4049/jimmunol.1201929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Briney BS, Willis JR, McKinney BA, Crowe JE., Jr. High-throughput antibody sequencing reveals genetic evidence of global regulation of the naive and memory repertoires that extends across individuals. Genes Immun. 2012;13:469–473. doi: 10.1038/gene.2012.20. [DOI] [PubMed] [Google Scholar]
- 115.Briney BS, Willis JR, Crowe JE., Jr. Location and length distribution of somatic hypermutation-associated DNA insertions and deletions reveals regions of antibody structural plasticity. Genes Immun. 2012;13:523–529. doi: 10.1038/gene.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zemlin M, et al. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J. Mol. Biol. 2003;334:733–749. doi: 10.1016/j.jmb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 117.Lindner C, et al. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 2012;209:365–377. doi: 10.1084/jem.20111980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wesemann DR, et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature. 2013;501:112–115. doi: 10.1038/nature12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.von Budingen HC, et al. B cell exchange across the blood-brain barrier in multiple sclerosis. J. Clin. Invest. 2012;122:4533–4543. doi: 10.1172/JCI63842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang N, et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci. Transl. Med. 2013;5:171ra119. doi: 10.1126/scitranslmed.3004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jackson KJ, et al. Divergent human populations show extensive shared IGK rearrangements in peripheral blood B cells. Immunogenetics. 2012;64:3–14. doi: 10.1007/s00251-011-0559-z. [DOI] [PubMed] [Google Scholar]
- 122.Hoi KH, Ippolito GC. Intrinsic bias and public rearrangements in the human immunoglobulin Vlambda light chain repertoire. Genes Immun. 2013;14:271–276. doi: 10.1038/gene.2013.10. [DOI] [PubMed] [Google Scholar]
- 123.Schoettler N, Ni D, Weigert M. B cell receptor light chain repertoires show signs of selection with differences between groups of healthy individuals and SLE patients. Mol. Immunol. 2012;51:273–282. doi: 10.1016/j.molimm.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Racanelli V, et al. Antibody V(h) repertoire differences between resolving and chronically evolving hepatitis C virus infections. PLoS ONE. 2011;6:e25606. doi: 10.1371/journal.pone.0025606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parameswaran P, et al. Convergent antibody signatures in human dengue. Cell Host Microbe. 2013;13:691–700. doi: 10.1016/j.chom.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ademokun A, et al. Vaccination-induced changes in human B-cell repertoire and pneumococcal IgM and IgA antibody at different ages. Aging Cell. 2011;10:922–930. doi: 10.1111/j.1474-9726.2011.00732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Krause JC, et al. Epitope-specific human influenza antibody repertoires diversify by B cell intraclonal sequence divergence and interclonal convergence. J. Immunol. 2011;187:3704–3711. doi: 10.4049/jimmunol.1101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat. Rev. Immunol. 2013;13:693–701. doi: 10.1038/nri3516. [DOI] [PubMed] [Google Scholar]
- 129.Wu X, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu J, et al. Mining the antibodyome for HIV-1-neutralizing antibodies with next-generation sequencing and phylogenetic pairing of heavy/light chains. Proc. Natl. Acad. Sci. USA. 2013;110:6470–6475. doi: 10.1073/pnas.1219320110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liao HX, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496:469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhou T, et al. Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity. 2013;39:245–258. doi: 10.1016/j.immuni.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wiley SR, et al. Targeting TLRs expands the antibody repertoire in response to a malaria vaccine. Sci. Transl. Med. 2011;3:93ra69. doi: 10.1126/scitranslmed.3002135. [DOI] [PubMed] [Google Scholar]
- 134.Srivastava R, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N. Engl. J. Med. 2012;367:115–123. doi: 10.1056/NEJMoa1110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.de Buy Wenniger LJ, et al. IgG4+ clones identified by next-generation sequencing dominate the b-cell receptor repertoire in IgG4-associated cholangitis. Hepatology. 2013;57:2390–2398. doi: 10.1002/hep.26232. [DOI] [PubMed] [Google Scholar]
- 136.He J, et al. IgH gene rearrangements as plasma biomarkers in Non- Hodgkin's lymphoma patients. Oncotarget. 2011;2:178–185. doi: 10.18632/oncotarget.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Logan AC, et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc. Natl. Acad. Sci. USA. 2011;108:21194–21199. doi: 10.1073/pnas.1118357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Faham M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–5180. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gawad C, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–4417. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tschumper RC, et al. Comprehensive assessment of potential multiple myeloma immunoglobulin heavy chain V-D-J intraclonal variation using massively parallel pyrosequencing. Oncotarget. 2012;3:502–513. doi: 10.18632/oncotarget.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Darzentas N, Stamatopoulos K. Stereotyped B cell receptors in B cell leukemias and lymphomas. Methods Mol. Biol. 2013;971:135–148. doi: 10.1007/978-1-62703-269-8_8. [DOI] [PubMed] [Google Scholar]
- 142.Hirano M, et al. Evolutionary implications of a third lymphocyte lineage in lampreys. Nature. 2013;501:435–438. doi: 10.1038/nature12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Silverstein AM. History of immunology. Cell. Immunol. 1979;42:1–2. doi: 10.1016/0008-8749(79)90215-6. [DOI] [PubMed] [Google Scholar]