

Abstract
The nervous system has the amazing capacity to transform sensory experience from the environment into changes in neuronal activity that, in turn, cause long-lasting alterations in neuronal morphology. Recent findings illustrate a somewhat surprising result: sensory experience concurrently activates molecular signaling pathways that both promote and inhibit dendritic complexity. Historically, a number of positive regulators of activity-dependent dendritic complexity have been described, while the list of identified negative regulators of this process is much shorter. In recent years, there has been an emerging appreciation of the importance of the Rad/Rem/Rem2/Gem/Kir (RGK) GTPases as mediators of activity-dependent structural plasticity. In the following review, we discuss the traditional view of RGK proteins, as well as our evolving understanding of the role of these proteins in instructing structural plasticity.
Keywords: activity, dendrite, plasticity, RGK
I. Activity-Dependent Regulation of Neuronal Plasticity
An essential property of the central nervous system (CNS) is the ability to respond to sensory input with corresponding changes to neuronal structure and function. At the behavioral level, this plasticity allows an organism to respond to a changing environment appropriately in order to survive. At the level of neuronal networks, this sensory experience is reflected in changes in neuronal activity that, in turn, mediate structural plasticity: experience-dependent alterations in a variety of neuronal processes including synaptic function and neuronal morphology [1, 2]. It is well-established that sensory input during critical periods of development has a profound effect on neuronal networks, as illustrated by classic experiments exploring the development of ocular dominance and receptive fields in the visual system [3]. However, over the past twenty years or so, it has become abundantly clear that neuronal architecture remains quite plastic throughout development and into adulthood, with acute changes in sensory experience and neuronal activity continuing to elicit profound effects on synaptic plasticity and neuronal morphology [4].
One of the major consequences of increased activity in the nervous system is the increased transcription of the so-called activity-regulated genes, which occurs in response to extracellular stimuli such as growth factors or, in the case of neurons, depolarization that opens ion channels that allow calcium (Ca2+) influx [1, 2]. These extracellular events activate intracellular signal transduction pathways, many of which regulate transcription factors, that in turn cause changes in the expression of their downstream target genes [5]. Activity-regulated genes have been extensively implicated in a variety of neuronal processes, including cell survival, regulation of dendrite morphogenesis, and synaptic plasticity [2, 6].
While many activity-regulated genes have been identified as positive regulators of neuronal structure and function, it is important to note that the upregulation of genes that restrict neuronal growth, synapse development, or synaptic transmission is equally important to maintain neuronal function in an appropriate physiological range in response to increased network activity [7] (Figure 1). For example, the activity-regulated genes Arc and Mef2 limit the strength and formation of excitatory synapses following increased activity, respectively [8–10]. In neurons, calcium-dependent signaling pathways are triggered by neuronal depolarization primarily via calcium entry into a neuron through N-methyl-D-aspartate (NMDA) receptors, or P/Q or L-type voltage-gated calcium channels (L-VGCCs) [11, 12, 13]. Interestingly, expression of some activity-dependent genes is dependent on calcium entry via only one of these sources (e.g. calcium entry through L-VGCCs but not NMDA receptors), suggesting that specific signal transduction pathways are activated in response to particular neuronal stimuli [11, 14]. In general, activity-regulated genes are well-poised to link changes in sensory experience to changes in neuronal structure and function.
Figure 1.
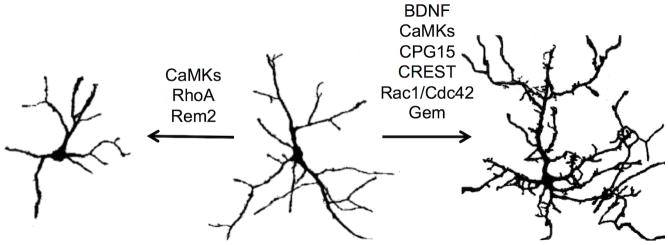
Positive (right) and negative (left) regulators of activity-dependent changes in dendritic morphology. Neuron images are 5 DIV cultured rat cortical neurons transfected with a GFP-expressing plasmid and treated with nifedipine (left), untreated (center), or treated with potassium chloride (right) (for further details, see [14]). While the net effect of increased activity is increased complexity (right), a number of molecules that either enhance or inhibit the dendritic arbor are upregulated and contribute to the net change.
II. Activity-Dependent Regulation of Dendritic Morphology
Structurally, one of the most salient aspects of neurons is their polarized morphology. Neurons are typically comprised of a cell body and an axon, through which they transmit information to other neurons, and a dendritic arbor, where input from other neurons is primarily received [15]. This dendritic arbor is usually highly branched, with the degree of complexity (a term which describes both the length of dendrites and the degree of branching of the arbor) playing a major role in the function of the neuron. The dendritic morphology of a given neuron determines the connections that neuron will make, and neurons with distinct morphologies often serve different functions in neural circuits [16]. For example, pyramidal cells in the mammalian cortex and hippocampus are easily identified by their distinct apical and basal dendritic arborizations. Local interneurons (which have their own distinctive morphologies) and projections from other brain regions will target specific areas of the pyramidal neuron dendritic arbor, soma, or axon initial segment, and the proper integration of these multiple inputs is essential for proper circuit integration and ultimately, function [17].
Dendritic morphology is highly subject to regulation by changes in neuronal activity. In general, the net effect of increased neuronal activity is an enhancement of dendritic complexity [18, 19]. An elegant example of this comes from the optic tectum of Xenopus laevis tadpoles, where increased activity in the form of 4 hours of visual experience leads to an increase in dendritic complexity of tectal projection neurons in vivo; application of the NMDA receptor antagonist 2-amino-5-phosphonopentanoate (APV) suppresses the effect of visual experience on dendritic complexity, firmly demonstrating the role of activity in this process [18].
Much of our understanding of the molecular mechanisms that regulate activity-dependent cellular processes has come from studies using depolarization of neurons in culture with potassium chloride (KCl) [5], which results in an increase in dendritic complexity [14, 19–21]. Although not a perfect mimic of neuronal activity in vivo, KCl-mediated neuronal depolarization causes physiologically-relevant calcium influx into neurons [22–24]. Further, co-treatment of cultures with KCl and either APV or the L-VGCC blocker nifedipine attenuates depolarization-dependent increases in dendritic complexity, suggesting that calcium entry from multiple sources contributes to this net effect [14, 19, 20]. The identification and characterization of molecules that transduce this increase in calcium entry into a corresponding increase in dendritic morphology remains an area of intense research.
Importantly, it has become clear that activity regulates the function of both positive and negative mediators of dendritic complexity; it is the integration of these opposing signals that ultimately results in the proper dendritic morphology [14, 18]. Presumably, the central nervous system evolved such that both positive and negative regulators of dendritic arborization are activated in order to ensure that growth-promoting processes do not proceed unchecked. A similar mechanism maintains homeostasis and allows for synaptic plasticity in the face of changing network activity, providing gain control such that signals are propagated successfully throughout a neuronal network [7]. This type of regulation is also present in many other tissue systems within an organism (e.g. immune system homeostasis) [25, 26], where a balance between positive and negative signal transduction networks achieves the final outcome.
III. Molecules that Regulate Dendritic Morphology
The development of the dendritic arbor is a highly dynamic yet carefully controlled process consisting of an early period of dynamic extension and retraction, followed by subsequent stabilization, pruning, and maturation of the arbor [27]. Studies in both invertebrate and vertebrate model organisms demonstrate that the ultimate morphology of a neuron is regulated by a variety of both intrinsic and extrinsic factors (Figure 2) [28, 29]. Within a given neuron, a number of molecules have been identified that help to shape the dendritic arbor in several different ways. Many genes, including transcription factors such as CUT, Abrupt, NeuroD and CREST, instruct dendritic morphology by initiating changes in gene expression either during development or in response to changes in neuronal activity [1, 28]. Others, such as the Rho GTPases, interact directly with the actin cytoskeleton to effect changes in the cytoskeletal organization of dendrites [30]. In addition, secreted proteins such as neurotrophins (e.g. nerve growth factor (NGF) and BDNF) also play a role in shaping dendritic morphology [31].
Figure 2.

Molecular signaling pathways that mediate activity-dependent changes in the dendritic arbor. Depolarization triggers calcium entry through multiple sources, including L-VGCCs and NMDA receptors. This leads to the activation of molecules (e.g. Rho GTPases) that directly interact with the cytoskeleton, as well as molecules (e.g. CaMKs) that regulate transcription factor activity that in turn causes changes in gene expression in the nucleus. Surprisingly little is known about the downstream targets of these pathways that mediate dendritic morphology.
Interestingly, a number of molecules have been shown to preferentially affect either dendritic length or branching, while others mediate both components of dendritic complexity. The apparent selectivity of some molecules for mediating particular aspects of dendritic morphology adds a further layer of intricacy to the regulation of the dendritic arbor. Below, we focus our discussion on some of the best-studied activity-regulated genes that positively or negatively regulate the dendritic arbor.
a. Rho GTPases
Several members of the Rho family of GTPases (RhoA, Rac1, Cdc42) have been demonstrated to play a role in mediating dendritic morphology. Like most GTPases, Rho GTPases cycle between an active, guanosine triphosphate (GTP)-bound and inactive, guanosine diphosphate (GDP)-bound state with the assistance of GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) [6]. It is through these GAPs and GEFs that the function of Rho GTPases can be linked to neuronal activity. For example, the function of the Rac1 GEFs Kalirin-7 [32] and Tiam1 [33] are mediated by transcriptional upregulation and phosphorylation, respectively, in response to activity; thus, the amount of active Rac1 is tied to the activity-dependent regulation of its GEFs.
The Rho GTPases have been shown to mediate activity-dependent changes in dendritic complexity both in vitro and in vivo. Overexpression of constitutively active (CA) mutants of Rac1 or Cdc42 in rodent hippocampal or cortical neurons or the X. laevis optic tectum leads to an increase in dendritic branching, while overexpression of dominant negative (DN) mutants of Rac1 or Cdc42 causes a decrease [34, 35]. These results suggest that Rac1/Cdc42 are positive regulators of dendritic branching. In contrast, overexpression of a CA RhoA mutant in rodent hippocampal or cortical neurons leads to a decrease in total dendritic length, while expression of DN RhoA in X. laevis tectum leads to an increase [34–36]. These results are consistent with a role for RhoA in limiting dendritic outgrowth. Rho GTPases mediate dendritic complexity by directly interacting with the actin cytoskeleton. For example, the ability of RhoA to signal through its downstream kinase ROKβ is required to mediate the length of dendrites [18, 34]. This interaction ultimately destabilizes actin filaments by leading to the phosphorylation and activation of the actin depolymerizing protein cofilin [37].
Thus, members of the same protein family have been implicated as both positive (Rac1, Cdc42) and negative (RhoA) regulators of the dendritic arbor. Importantly, when the function of either Rac1 or RhoA was inhibited by expression of DN mutants in X. laevis tectal neurons, the visual experience-dependent increase in dendritic complexity was suppressed, suggesting that these positive and negative regulators contribute to activity-dependent effects on dendritic morphology [18].
b. CaMKs
Several isoforms of the CaMK family members, including CaMKIV, CaMKI, and CaMKII, have been implicated as either positive or negative regulators of dendritic complexity [19–21, 38–42]. CaMK function is linked to activity via binding of Ca2+-bound CaM, which is required for their activation [43]. Upon CaM binding, CaMKs go on to phosphorylate and activate a number of downstream targets, including transcription factors [43]. CaMKIV, which is restricted to the nucleus, has been implicated in promoting activity-dependent increases in dendritic length in rodent cortical neurons [19]. The α and γ isoforms of CaMKI, both of which have a predominantly cytoplasmic localization, have also been shown to promote both axonal and dendritic complexity in a neuronal activity-dependent manner [21, 44].
In contrast, the role of CaMKII in mediating dendritic complexity is more complicated. CaMKII has been shown to regulate dendritic morphology in a variety of experimental systems, although results have not been consistent between various studies [38–41]. These inconsistencies are possibly due to the use of different approaches to manipulate CaMKII activity, or differences in the ratio of α to β isoforms in the active CaMKII holoenzyme, affecting enzyme distribution and function. For example, RNAi-mediated knockdown of CaMKIIα in vitro in the rodent cerebellum or pharmacological inhibition of CaMKII in sympathetic neurons inhibits complexity, implicating CaMKII as a positive regulator of dendritic complexity [20, 38]. In contrast, overexpression of CA CaMKIIα mutants in X. laevis tectum or in rodent hippocampal culture leads to a decrease in dendritic length, suggesting that CaMKII is a negative regulator of dendritic arbor growth [40, 41]. A thorough analysis of CaMKIIβ in cultured cerebellar neurons also supports the role of CaMKII as an inhibitor of dendritic arbor complexity [39]. Overall, it is clear that activity-dependent signaling through the CaMK family, much like the Rho GTPase family, both promotes and inhibits dendritic complexity.
c. Transcription Factors: CREB, CREST, and NeuroD
A number of transcription factors (TFs) have been identified as essential regulators of dendritic morphology in response to neuronal activity. The two best-studied pathways are the CBP/CREB and CBP/CREST pathways that lie downstream of CaMKIV [19, 45, 46], and the NeuroD pathway downstream of CaMKII [38]. Both cAMP Response Element Binding (CREB) and Calcium Responsive Transactivator (CREST) bind Calcium Binding Protein (CBP) in order to mediate transcription [19, 47–49]. The expression of a DN CREB in rodent cortical neurons suppressed the activity-and CaMKIV- dependent increase in dendritic length, suggesting that CREB is a downstream target of CaMKIV that acts to promote the lengthening of dendrites [19]. Similarly, CREST knockout mice also show decreased activity-dependent dendritic outgrowth, consistent with the role of CREST as a positive regulator of this process [48].
Overexpression of DN NeuroD constructs results in a simpler dendritic arbor in cultured rodent cerebellar neurons, implicating it as a positive regulator of dendritic complexity. However, unlike CREB and CREST, which lie downstream of CaMKIV, it is CaMKII-dependent phosphorylation of NeuroD that is required for its activity [38]. As CaMKII is itself primarily considered a negative regulator of dendritic complexity [39–41], this suggests that other pathways impinging on NeuroD regulation are also at work. Taken together, the evidence that activity-dependent regulation of TFs influences dendritic complexity begs the question: what is the identity of genes whose transcription is mediated by these TFs?
d. Bdnf
The secreted neurotrophin BDNF plays a role in many aspects of neuronal development and plasticity [50]. Either application of soluble BDNF to, or overexpression of the Bdnf gene in, rodent cortical neurons leads to an increase in dendritic complexity, suggesting that BDNF can act as a positive regulator of the dendritic arbor in both a cell autonomous and non-autonomous manner [51, 52]. In addition, loss-of-function studies in rodent hippocampus have demonstrated decreased dendritic arborization in response to decreased BDNF levels [53, 54]; however, the majority of these studies have not been performed in the context of altered neuronal activity. In fact, a loss of function analysis specifically of the activity-regulated Bdnf gene product revealed no effect on dendritic morphology [55]. Thus, while BDNF is an activity-regulated gene and enhances dendritic morphology, it remains unclear whether BDNF directly links changes in activity to changes in morphology.
e. Cpg15
Candidate plasticity gene 15 is an extracellular protein that attaches to the plasma membrane via a GPI-linker [56] and was first identified in a forward genetic screen for genes whose messenger ribonucleic acid (mRNA) was upregulated in response to seizure activity in the rodent hippocampus [57]. Further characterization of Cpg15 mRNA identified it as an activity-regulated gene in rat cortex in vivo in response to visual stimulation and in vitro by calcium entry via NMDA receptors and L-VGCCs [58, 59]. Functionally, overexpression of cpg15 in X. laevis tadpole optic tectal neurons leads to an increase in dendritic complexity, identifying it as a positive regulator of dendritic morphology [60]. Subsequently, high expression of Cpg15 was found in the rat barrel and visual cortices following sensory stimulation, supporting a role for CPG15 in activity-dependent structural remodeling [61, 62]. Interestingly, and consistent with these results, Cpg15 knockout mice show a delay in the maturation of the dendritic arbor, suggesting that CPG15 supports proper dendritic arbor development in developing circuits [63].
Importantly, both Bdnf and Cpg15 are upregulated in a CREB-dependent manner [59, 64]. The identity of additional CREB targets, as well as NeuroD and CREST target genes, remains largely unknown. Moreover, the few targets of activity-dependent TFs that have been described are positive regulators of dendritic complexity. While the reason for this is unknown, it is possible that positive and negative regulators act in temporally distinct phases of arbor development (i.e. growth vs. retraction), and experimental design has favored the study of the growth-promoting phase. Alternatively, negative regulators may be active during the same temporal window as positive regulators, but their contribution is masked due to experimental designs favoring the identification of positive regulators. Further identification of negative regulators of morphology whose transcription is upregulated in response to activity promises to shed light on this issue.
IV. RGK Proteins: Not Your Typical GTPases
In recent years, members of the Rad, Rem, Rem2, Gem/Kir (RGK) subfamily of small, Ras-like GTPases have emerged as attractive candidates to mediate changes in cellular structure and function in response to external stimuli. This subfamily is structurally and enzymatically unique amongst Ras-like GTPases in several ways, and we refer the reader elsewhere for a detailed review of this topic [65]. The preponderance of the evidence suggests that these proteins may be regulated by mechanisms other than the canonical cycling between GTP and GDP bound states, and no associated GAPs and GEFs have been identified to date [66–68].
Indeed, a unique feature of the RGK family is their ability to be regulated at the transcriptional level by extracellular stimuli [65]. For example, both Rem2 [14] and Gem [69] mRNA are upregulated in neurons following KCl treatment. In addition to this regulation at the transcriptional level, there is evidence of post-translational regulation of RGK proteins as well [65]. The residues on which this phosphorylation occurs are highly conserved amongst the RGK proteins, suggesting a conservation of function as well [65]. RGK proteins also contain multiple putative nuclear localization signals (NLSs) that are conserved across the family [70, 71]. Thus, nuclear localization of RGK proteins is another critical mechanism by which the function of these proteins is regulated [42].
Traditionally, research into RGK protein function has been restricted to studies of two cellular processes: regulation of the actin cytoskeleton [72–77] and inhibition of VGCC function (Box 1). However, the vast majority of these studies were performed via overexpression of the RGK protein of interest (rather than examining functions attributed to RGK proteins at endogenous levels of expression), and/or carried out in heterologous cell types (rather than the cell types in which the RGK protein is endogenously expressed). Thus, the discovery of many of the endogenous functions of RGK family members awaits further research.
Box 1. RGK Proteins as Endogenous Regulators of VGCCs.
Initially, an interaction between RGK proteins and VGCCs was discovered when a yeast two-hybrid screen identified Gem as a binding partner of the accessory calcium channel subunit CaVβ [96]. Subsequently, RGK proteins were identified as potent inhibitors of calcium influx through several types of VGCCs [65, 73, 96–100].
Controversy remains as to the mechanism through which overexpression of RGK proteins inhibit VGCCs in these studies. Each of the four RGK proteins, when co-expressed with L-VGCCs in heterologous cells, bind and sequester newly synthesized channels, reducing their surface expression as evidenced by immunocytochemistry [72, 96, 101, 102]. However, when overexpressed in cardiac myocytes, peripheral neurons, or pancreatic islet cells, the RGK proteins instead decrease the gating kinetics of VGCCs, suggesting another mechanism to account for RGK-mediated VGCC inhibition [97, 100, 103–105]. It remains a possibility that channel- or cell-type specific mechanisms may explain the discrepancies in these studies.
A caveat of this debate is, of course, that VGCC current inhibition may not be a relevant function of the RGK proteins due to a lack of loss-of-function studies of RGK family members with respect to calcium flux. The exception to this statement comes from patch-clamp recordings in cardiac myocytes using mice in which the Rad or Rem genes were constitutively deleted [106, 107]. These experiments demonstrated increased calcium currents in myocytes obtained from Rad knockout animals, representing the first loss-of-function evidence that endogenous expression of an RGK protein inhibits VGCCs [106].
In neurons, an intriguing possibility is that the Ca2+-dependent upregulation of Rem2 functions in a negative feedback loop to shut off Ca2+ entry into the cell and ultimately, its own expression. Such a mechanism was previously suggested for Rem2 regulation of Ca2+-mediated insulin secretion from pancreatic β-cell [97]. However, results implicating endogenous RGK proteins in VGCC inhibition in neurons have been equivocal. A recent study identified the Gem/VGCC interaction as essential for mediating activity-dependent changes in dendritic morphology, but did not find a role for the interaction of Gem with the channel in mediating calcium currents [69]. In addition, the Rem2/VGCC interaction is dispensable for the role of Rem2 in mediating dendritic morphology [79], and loss-of-function studies using either a pool of shRNAs [108] or two individual shRNAs [109] targeting Rem2 and transfected into cultured neurons failed to reveal a role for Rem2 in inhibiting VGCC-mediated calcium currents in neurons. Thus, a definitive conclusion as to whether an endogenous function of RGK proteins, particularly Rem2, is regulation of VGCC function awaits further experiments.
V. RGK Proteins in the Nervous System
Rem2 is the most highly expressed RGK protein in the central nervous system, and has been studied in both cortical and hippocampal neurons [14, 78, 79]. Gem is also detectable at lower levels in these same brain regions [69]. Importantly, the expression of both Rem2 and Gem is upregulated in response to KCl-mediated depolarization of rodent neuron cultures [14, 69, 80], consistent with the extracellular stimulus-dependent regulation of gene expression that is a hallmark of the RGK family.
a. Rem2
Recent studies revealed that Rem2 functions during multiple stages of neuronal development. RNA interference (RNAi) to decrease Rem2 expression in human embryonic stem cells (hESCs) leads to cellular arrest and apoptosis, suggesting a role for Rem2 in the maintaining hESC survival [81]. Moreover, Rem2 overexpression drove induced pluripotent stem cells (iPSCs) towards an ectodermal cell fate [81]. This role for Rem2 in neuronal precursor proliferation and neuronal development was confirmed in a later experiment in zebrafish embryos [82].
What is the function of Rem2 in post-mitotic neurons? An RNAi-based screen in primary hippocampal cultured neurons to identify new molecules required for the proper development of synapses identified Rem2 as positive regulator of excitatory and inhibitory synapse development [80]. A follow up study described Rem2 as a positive regulator of dendritic spine development, and simultaneously identified Rem2 as a negative regulator of dendritic complexity [79]. In addition, an interaction between Rem2 and 14-3-3 proteins was required for Rem2 regulation of both excitatory synapse development and dendritic morphology, while an interaction with CaM was required only for Rem2 to mediate dendritic morphology [79]. This finding suggests that Rem2 regulates dendritic complexity and synapse development via distinct signaling pathways (Box 2), thus distinguishing these two neuronal functions of Rem2 at the molecular level. Interestingly, an interaction between Rem2 and the beta subunit of VGCCs was found to be dispensable for both functions of Rem2 [79].
Box 2. The Synaptotrophic Hypothesis.
A key feature of the synaptotrophic hypothesis is that synapse formation and dendritic outgrowth are concurrent; synapses mark branch points along the dendritic arbor, and the maturation of a stable synapse causes the stabilization of a newly-formed dendritic branch at that point [110, 111]. This implies that synapse number and dendritic arbor complexity should trend the same way: more synapses leads to more branches. However, it is clear that synapse development and dendritic arbor complexity do not always coincide. For example, Rem2 promotes synapse development while inhibiting dendritic complexity [79], as does postsynaptic ephrinB3 [112]. One plausible explanation is that this may represent a homeostatic mechanism by which neurons attempt to keep the same number of synapses regardless of arbor complexity. In support of this idea, loss-of-function of the RNA-binding protein Hermes in X. laevis retinal granule cells results in decreased axonal arborization but increased presynaptic puncta, suggesting that synaptogenesis is upregulated in these neurons to compensate for a simpler arbor [113]. Similarly, inhibition of synaptic AMPA receptors in the X. laevis optic tectum leads to a decrease in the number of dendritic branches (supporting the synaptotrophic hypothesis), but the length of individual branches is increased, as if neurons are searching for synaptic partners [114].
In light of these findings, it appears that the synaptotrophic hypothesis needs to be revised in order to account for the highly dynamic nature of both synapses and dendritic branches. For example, perhaps the increase in branching that is observed despite fewer excitatory synapses with Rem2 knockdown [79] is the result of increased transient branches that extend and remain for a number of days before retracting in the absence of a mature synapse to stabilize them. Live imaging studies of dendritic morphology dynamics over time in the context of, for example, Rem2 manipulation will influence our interpretation of the synaptotrophic hypothesis in the future.
Rem2 is widely expressed throughout the soma, dendrites, and axon in cultured hippocampal neurons [79, 80]. Recent work has further characterized the subcellular localization of Rem2 in response to activity. A yellow fluorescent protein (YFP)-tagged Rem2 cDNA was found to redistribute from a diffuse to punctate expression pattern following activation of the NMDA receptor in rat hippocampal neurons, and to co-associate with green fluorescent protein (GFP)-tagged CaMKII following this redistribution [83]. A subsequent epistatic analysis of Rem2 and several CaMK family members provided a functional role for this Rem2/CaMKII interaction. Rem2 was found to lie downstream of CaMKII, upstream of CaMKIV, and parallel to CaMKIα in a signaling cascade that regulates the dendritic arbor [42]. Moreover, Rem2 was identified as a novel CaMKII substrate, and phosphorylation of Rem2 at S241 and S308 leads to enhanced nuclear localization of Rem2 and further, phosphorylation of Rem2 at these sites is required for its ability to mediate dendritic complexity [42]. An intriguing hypothesis has emerged from this work: Rem2 functions in the nucleus to regulate the expression of genes that mediate dendritic complexity perhaps by inhibiting CaMKIV-dependent regulation of the CREB transcription factor [19]. Interestingly, a recent study described a role for the RGK protein Rad in regulating gene transcription in the nucleus by inhibiting the activity of NFκB [84]. Thus, a thorough investigation of the nuclear function of Rem2 has important implications for our understanding of an activity-dependent program of gene expression that regulates dendritic complexity.
Given that Rem2 is both activity-regulated and inhibits dendritic complexity, Rem2 is a likely candidate to regulate changes in the dendritic arbor in response to changes in neuronal activity. As discussed above, the X. laevis visual system is a rich experimental paradigm with which to probe the function of activity-dependent genes such as Rem2, as dendritic complexity of optic tectum neurons in tadpoles exposed to a visual stimulus is enhanced in an activity-dependent manner [18]. This paradigm allows a distinction to be made between the function of a gene that is constitutively expressed versus the function of a gene that is expressed specifically in response to increased neuronal activity, because it is possible to simultaneously manipulate gene expression and sensory experience. This approach was used to demonstrate that Rem2 is a critical regulator of activity-dependent dendritic complexity in vivo, as dialing Rem2 expression up or down, in the context of increased sensory experience, decreases or increases dendritic complexity, respectively [14, 42]. While it is well-known that the net effect of increasing activity is an increase in dendritic arbor complexity [18], this study of Rem2 brings to light a previously underappreciated fact about how neurons achieve the proper morphology in response to changes in sensory experience. Rem2 manipulation demonstrates that both positive and negative regulators of the dendritic arbor are upregulated by activity, and neurons must balance these opposing signals such that the net effect results in the proper dendritic morphology.
Intriguingly, Ghiretti et al. (2014) used pharmacological manipulation to show that Rem2 mRNA expression is activated by calcium influx primarily through L-VGCCs and not through NMDARs. In addition, this study demonstrated that calcium influx through L-type VGCCs activates parallel signaling pathways(s) to Rem2 that function to positively regulate dendritic arborization [14]. Thus, Rem2 and opposing, parallel signaling pathway(s) that promote complexity, appear to be upregulated only during a more global and sustained neuronal depolarization that is required to open L-VGCCs. While the effects of Rem2 on dendritic complexity are primarily dependent on L-VGCCs, calcium entry through NMDA receptors was also pharmacologically implicated in activity-dependent dendritic morphology, independent of the role of Rem2 in this process [14]. Thus, it is likely that there are multiple molecular pathways that both positively and negatively regulate dendritic complexity. Some, such as Rem2, respond only to L-VGCCs, while other molecules may be selectively regulated by NMDA receptors, and still others may be influenced by both modes of calcium entry. It is clear that a complete understanding of activity-dependent changes in structural plasticity will rely on full understanding of these molecular regulators.
b. Gem
A recent study implicated Gem in activity-dependent regulation of dendritic morphology using a knock-in mouse model of the autism spectrum disorder Timothy syndrome [69]. A point mutation (G406R) in the Cav1.2 subunit that is genetically linked to the human disorder suppressed KCl-induced dendritic outgrowth in cortical neuron cultures [69]. Interestingly, Gem knockdown led to a comparable suppression of KCl-induced dendritic outgrowth, while Gem overexpression was able to rescue the Timothy syndrome phenotype, suggesting that Gem is a positive regulator of activity-dependent increases in dendritic outgrowth downstream of L-VGCCs [69]. The authors went on to identify the downstream consequences of the Timothy syndrome mutation: an increase in RhoA activation leading to dendritic retraction. They show that Gem does not bind the mutated Cav1.2 as well as the wild-type channel. As a consequence, Gem is not properly sequestered at the membrane with the Cav1.2 subunit, and is thus unable to inhibit Rho activity [69]. Interestingly, neither the Timothy syndrome mutation nor the Gem/L-VGCC interaction altered calcium currents in these neurons, suggesting the effect on dendritic morphology is not the result of Gem-mediated changes in L-VGCC currents [69]. In fact, it appears that the importance of the Gem/VGCC interaction lies in Gem regulation of Rho signaling pathways. These results again suggest that inhibition of calcium currents may not be a relevant function of endogenous RGKs in neurons (Box 1).
VII. Conclusions
Structural plasticity of the dendritic arbor is critical to maintaining neuronal circuit function in the face of changes in the environment, such as modulation of sensory experience, hormones, and temperature [85]. Disorders of the central nervous system-including epilepsy, mental retardation, autism spectrum disorders, and drug addiction-are characterized by pronounced changes in neuronal architecture, connectivity, and morphology in many different brain regions [86–89]. Moreover, aberrant structural plasticity is a key component of a number of neurological disorders that have a relatively late onset of symptoms (e.g. Rett and Angelman Syndrome), suggesting that experience-dependent processes in the nervous system are profoundly affected in these individuals [90]. In addition, it has been demonstrated that addiction to stimulants such as cocaine causes changes in neuronal structure, and further, these structural changes may explain the persistent features of drug addiction, including drug cravings and relapse [91, 92]. As changes in sensory experience concurrently activate signaling pathways that both promote and inhibit dendritic complexity, a shift in the balance of these signals is likely a contributing factor to these neurological disorders.
Activity-regulated genes are essential mediators of a variety of neuronal processes, including dendritic outgrowth and synaptic plasticity, and these genes are well-poised to rapidly translate changes in neuronal activity into cell autonomous structural changes in individual neurons [2, 93, 94]. To date, the majority of research has focused on activity-regulated genes that act as positive regulators of dendritic morphology [55, 60, 95]. In addition, while a number of loss-of-function studies have demonstrated a role for these genes in mediating dendritic arborization [53, 63], these studies have not been performed in the context of altered neuronal activity. Thus, a true appreciation of the function of activity-regulated genes in mediating structural plasticity can only be discovered by co-manipulation of network activity and gene function [14, 55].
Members of the RGK family of atypical Ras-like GTPases have emerged in recent years as regulators of activity-dependent changes in dendritic morphology. For the past decade, the RGK proteins have been studied in non-neuronal cell types as regulators of VGCCs and the actin cytoskeleton. In recent years, a number of groups have begun to characterize the role of RGK proteins in neurons, and their role in mediating the dendritic arbor in particular. While Gem appears to be a positive regulator of dendritic complexity, Rem2 is among a small group of negative regulators of dendritic complexity to be described [14, 69]. Thus, RGK proteins have emerged as important players in mediating neuronal structural plasticity in response to changing network activity, and ongoing work promises to further elucidate the regulation and function of RGK proteins in the nervous system (Box 3).
Box 3. Open Questions- RGK Proteins in the Nervous System.
Both Rem2 [14] and Gem [69] are upregulated at the transcriptional level in response to increased neuronal activity. Rem2 is primarily upregulated by calcium entry through L-VGCCs, while the mode of calcium entry that regulates Gem expression remains to be determined. In addition, the Rem2 and Gem promoter element(s) that confer activity-dependent expression to these genes remain to be identified. Comprehensive characterization of the Rem2 and Gem promoters via transcriptional reporter assays will aid in characterizing the relationship between neuronal activity and RGK mRNA expression in neurons, and provide new insight into the selectivity of different modes of calcium entry that regulate the expression of particular genes.
Gem has been implicated as a positive regulator of activity-dependent dendritic morphology [69]. However, the upstream signaling pathway that regulates the interaction between Gem and VGCCs has not been described. As key phosphorylation sites are conserved in both Rem2 and Gem [42], epistatic analyses similar to those carried out for Rem2 and the CaMKs should provide insight into whether Gem function is regulated by phosphorylation [42], and could reveal how Gem signals to promote dendritic outgrowth.
Rem2 is the first RGK protein for which a nuclear function has been described [42]. CaMKII-dependent phosphorylation leads to enhanced nuclear localization of Rem2, where it is hypothesized that Rem2 inhibits the transcription of genes that promote dendritic complexity [42]. The characterization of the conserved Rem2 NLS will provide insight into the manner in which Rem2 is localized in the nucleus. Further, the identification of downstream targets of Rem2 signaling is required to elucidate the nuclear function of Rem2.
Highlights.
Dendritic morphology is dynamic and regulated in part by neuronal activity.
Positive & negative molecular regulators contribute to activity-mediated morphology.
The identification of molecular regulators of morphology is a major research area.
RGK proteins have recently emerged as activity-dependent regulators of morphology.
Acknowledgments
The authors thank Katelyn Kenny, Dr. William Harris, Dr. Michael T. Marr 2nd, Dr. Anna R. Moore, and Dr. Stephen D. Van Hooser for helpful scientific discussions pertaining to this topic. Research in the Paradis lab is funded in part by National Institute of Health grants R01NS065856 from the National Institute on Neurological Disorders and Stroke (S.P.) and by F31DA032181 from the National Institute on Drug Abuse (A.E.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loebrich S, Nedivi E. The function of activity-regulated genes in the nervous system. Physiol Rev. 2009;89(4):1079–103. doi: 10.1152/physrev.00013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leslie JH, Nedivi E. Activity-regulated genes as mediators of neural circuit plasticity. Prog Neurobiol. 2011;94(3):223–37. doi: 10.1016/j.pneurobio.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hubel DH, Wiesel TN, Stryker MP. Orientation columns in macaque monkey visual cortex demonstrated by the 2-deoxyglucose autoradiographic technique. Nature. 1977;269(5626):328–30. doi: 10.1038/269328a0. [DOI] [PubMed] [Google Scholar]
- 4.Hooks BM, Chen C. Critical periods in the visual system: changing views for a model of experience-dependent plasticity. Neuron. 2007;56(2):312–26. doi: 10.1016/j.neuron.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Lyons MR, West AE. Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol. 2011;94(3):259–95. doi: 10.1016/j.pneurobio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saneyoshi T, Fortin DA, Soderling TR. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr Opin Neurobiol. 2010;20(1):108–15. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5(2):97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- 8.Flavell SW, et al. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311(5763):1008–12. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 9.Rial Verde EM, et al. Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron. 2006;52(3):461–74. doi: 10.1016/j.neuron.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepherd JD, et al. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52(3):475–84. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260(5105):181–6. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263(5153):1618–23. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 13.Sutton KG, et al. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature. 1999;401(6755):800–4. doi: 10.1038/44586. [DOI] [PubMed] [Google Scholar]
- 14.Ghiretti AE, et al. Rem2 is an activity-dependent negative regulator of dendritic complexity in vivo. J Neurosci. 2014;34(2):392–407. doi: 10.1523/JNEUROSCI.1328-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solecki DJ, et al. Neuronal polarity in CNS development. Genes Dev. 2006;20 (19):2639–47. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- 16.Jan YN, Jan LY. Dendrites. Genes Dev. 2001;15(20):2627–41. doi: 10.1101/gad.916501. [DOI] [PubMed] [Google Scholar]
- 17.Rudy B, et al. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71(1):45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sin WC, et al. Dendrite growth increased by visual activity requires NMDA receptor and Rho GTPases. Nature. 2002;419(6906):475–80. doi: 10.1038/nature00987. [DOI] [PubMed] [Google Scholar]
- 19.Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34(6):999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- 20.Vaillant AR, et al. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34(6):985–98. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- 21.Wayman GA, et al. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt- 2. Neuron. 2006;50(6):897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Dolmetsch RE, et al. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294(5541):333–9. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 23.Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87(7):1203–14. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 24.Mermelstein PG, et al. Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci U S A. 2001;98(26):15342–7. doi: 10.1073/pnas.211563998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, et al. Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death. Cell. 1994;78(5):739–50. doi: 10.1016/s0092-8674(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 26.Singer AL, Koretzky GA. Control of T cell function by positive and negative regulators. Science. 2002;296(5573):1639–40. doi: 10.1126/science.1071551. [DOI] [PubMed] [Google Scholar]
- 27.Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11(1):118–26. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- 28.Jan YN, Jan LY. Branching out: mechanisms of dendritic arborization. Nat Rev Neurosci. 2010;11(5):316–28. doi: 10.1038/nrn2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao FB. Molecular and cellular mechanisms of dendritic morphogenesis. Curr Opin Neurobiol. 2007;17(5):525–32. doi: 10.1016/j.conb.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19(1):1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 31.McAllister AK. Neurotrophins and neuronal differentiation in the central nervous system. Cell Mol Life Sci. 2001;58(8):1054–60. doi: 10.1007/PL00000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penzes P, et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37 (2):263–74. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- 33.Tolias KF, et al. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45 (4):525–38. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 34.Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20(14):5329–38. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Threadgill R, Bobb K, Ghosh A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron. 1997;19(3):625–34. doi: 10.1016/s0896-6273(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 36.Li Z, Van Aelst L, Cline HT. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci. 2000;3(3):217–25. doi: 10.1038/72920. [DOI] [PubMed] [Google Scholar]
- 37.Maekawa M, et al. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285(5429):895–8. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 38.Gaudilliere B, et al. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41(2):229–41. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- 39.Puram SV, et al. A CaMKIIbeta signaling pathway at the centrosome regulates dendrite patterning in the brain. Nat Neurosci. 2011;14(8):973–83. doi: 10.1038/nn.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fink CC, et al. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39(2):283–97. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 41.Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279(5348):222–6. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- 42.Ghiretti AE, et al. CaMKII-Dependent Phosphorylation of the GTPase Rem2 Is Required to Restrict Dendritic Complexity. J Neurosci. 2013;33(15):6504–15. doi: 10.1523/JNEUROSCI.3861-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wayman GA, et al. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron. 2008;59(6):914–31. doi: 10.1016/j.neuron.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takemoto-Kimura S, et al. Differential roles for CaM kinases in mediating excitation-morphogenesis coupling during formation and maturation of neuronal circuits. Eur J Neurosci. 2010;32(2):224–30. doi: 10.1111/j.1460-9568.2010.07353.x. [DOI] [PubMed] [Google Scholar]
- 45.Sun P, et al. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8(21):2527–39. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 46.Impey S, et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34(2):235–44. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 47.Hu SC, Chrivia J, Ghosh A. Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron. 1999;22(4):799–808. doi: 10.1016/s0896-6273(00)80738-2. [DOI] [PubMed] [Google Scholar]
- 48.Aizawa H, et al. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303(5655):197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- 49.Hardingham GE, et al. Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron. 1999;22(4):789–98. doi: 10.1016/s0896-6273(00)80737-0. [DOI] [PubMed] [Google Scholar]
- 50.Cohen-Cory S, et al. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol. 2010;70(5):271–88. doi: 10.1002/dneu.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15(4):791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 52.Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5(11):1177–84. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- 53.Xu B, et al. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26(1):233–45. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 54.McAllister AK, Katz LC, Lo DC. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18(5):767–78. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 55.Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60(4):610–24. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naeve GS, et al. Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A. 1997;94(6):2648–53. doi: 10.1073/pnas.94.6.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nedivi E, et al. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363(6431):718–22. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- 58.Nedivi E, et al. A set of genes expressed in response to light in the adult cerebral cortex and regulated during development. Proc Natl Acad Sci U S A. 1996;93(5):2048–53. doi: 10.1073/pnas.93.5.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujino T, Lee WC, Nedivi E. Regulation of cpg15 by signaling pathways that mediate synaptic plasticity. Mol Cell Neurosci. 2003;24(3):538–54. doi: 10.1016/s1044-7431(03)00230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nedivi E, Wu GY, Cline HT. Promotion of dendritic growth by CPG15, an activity-induced signaling molecule. Science. 1998;281(5384):1863–6. doi: 10.1126/science.281.5384.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee WC, Nedivi E. Extended plasticity of visual cortex in dark-reared animals may result from prolonged expression of cpg15-like genes. J Neurosci. 2002;22 (5):1807–15. doi: 10.1523/JNEUROSCI.22-05-01807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harwell C, et al. Regulation of cpg15 expression during single whisker experience in the barrel cortex of adult mice. J Neurobiol. 2005;65(1):85–96. doi: 10.1002/neu.20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujino T, et al. CPG15 regulates synapse stability in the developing and adult brain. Genes Dev. 2011;25(24):2674–85. doi: 10.1101/gad.176172.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tao X, et al. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20(4):709–26. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 65.Correll RN, et al. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal. 2008;20 (2):292–300. doi: 10.1016/j.cellsig.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sasson Y, et al. RGK Family G-Domain:GTP Analog Complex Structures and Nucleotide-Binding Properties. J Mol Biol. 2011;413(2):372–89. doi: 10.1016/j.jmb.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 67.Reymond P, et al. Structure of the GDP-bound G domain of the RGK protein Rem2. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68(Pt 6):626–31. doi: 10.1107/S1744309112013541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Opatowsky Y, et al. Structure-function studies of the G-domain from human gem, a novel small G-protein. FEBS Lett. 2006;580(25):5959–64. doi: 10.1016/j.febslet.2006.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krey JF, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16(2):201–9. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mahalakshmi RN, et al. Nuclear transport of Kir/Gem requires specific signals and importin alpha5 and is regulated by calmodulin and predicted serine phosphorylations. Traffic. 2007;8(9):1150–63. doi: 10.1111/j.1600-0854.2007.00598.x. [DOI] [PubMed] [Google Scholar]
- 71.Mahalakshmi RN, et al. Nuclear localization of endogenous RGK proteins and modulation of cell shape remodeling by regulated nuclear transport. Traffic. 2007;8(9):1164–78. doi: 10.1111/j.1600-0854.2007.00599.x. [DOI] [PubMed] [Google Scholar]
- 72.Beguin P, et al. Nuclear sequestration of beta-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol. 2006;355(1):34–46. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 73.Ward Y, et al. Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down-regulation of calcium channel activity. Mol Cell Biol. 2004;24(2):651–61. doi: 10.1128/MCB.24.2.651-661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beguin P, et al. Roles of 14-3-3 and calmodulin binding in subcellular localization and function of the small G-protein Rem2. Biochem J. 2005;390(Pt 1):67–75. doi: 10.1042/BJ20050414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beguin P, et al. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J Cell Sci. 2005;118(Pt 9):1923–34. doi: 10.1242/jcs.02321. [DOI] [PubMed] [Google Scholar]
- 76.Pan JY, et al. Ges, A human GTPase of the Rad/Gem/Kir family, promotes endothelial cell sprouting and cytoskeleton reorganization. J Cell Biol. 2000;149(5):1107–16. doi: 10.1083/jcb.149.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelly K. The RGK family: a regulatory tail of small GTP-binding proteins. Trends Cell Biol. 2005;15(12):640–3. doi: 10.1016/j.tcb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 78.Finlin BS, et al. Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras-related GTPases. Biochem J. 2000;347(Pt 1):223–31. [PMC free article] [PubMed] [Google Scholar]
- 79.Ghiretti AE, Paradis S. The GTPase Rem2 regulates synapse development and dendritic morphology. Dev Neurobiol. 2011;71(5):374–89. doi: 10.1002/dneu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paradis S, et al. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron. 2007;53(2):217–32. doi: 10.1016/j.neuron.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edel MJ, et al. Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes Dev. 2010;24(6):561–73. doi: 10.1101/gad.1876710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Edel MJ, et al. Rem2 GTPase controls proliferation and apoptosis of neurons during embryo development. Cell Cycle. 2010;9(17) doi: 10.4161/cc.9.17.12719. [DOI] [PubMed] [Google Scholar]
- 83.Flynn R, et al. Activity-Dependent Subcellular Cotrafficking of the Small GTPase Rem2 and Ca2+/CaM-Dependent Protein Kinase IIalpha. PLoS One. 2012;7(7):e41185. doi: 10.1371/journal.pone.0041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hsiao BY, et al. Rad GTPase inhibits the NFkappaB pathway through interacting with RelA/p65 to impede its DNA binding and target gene transactivation. Cell Signal. 2014;26(7):1437–44. doi: 10.1016/j.cellsig.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 85.McEwen BS. Stress, sex, and neural adaptation to a changing environment: mechanisms of neuronal remodeling. Ann N Y Acad Sci. 2010;1204 (Suppl):E38–59. doi: 10.1111/j.1749-6632.2010.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pickett J, London E. The neuropathology of autism: a review. J Neuropathol Exp Neurol. 2005;64(11):925–35. doi: 10.1097/01.jnen.0000186921.42592.6c. [DOI] [PubMed] [Google Scholar]
- 87.Ben-Ari Y. Epilepsies and neuronal plasticity: for better or for worse? Dialogues Clin Neurosci. 2008;10(1):17–27. doi: 10.31887/DCNS.2008.10.1/ybenari. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blair RE, et al. Epileptogenesis causes an N-methyl-d-aspartate receptor/Ca2+-dependent decrease in Ca2+/calmodulin-dependent protein kinase II activity in a hippocampal neuronal culture model of spontaneous recurrent epileptiform discharges. Eur J Pharmacol. 2008;588(1):64–71. doi: 10.1016/j.ejphar.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fernandez F, Garner CC. Over-inhibition: a model for developmental intellectual disability. Trends Neurosci. 2007;30(10):497–503. doi: 10.1016/j.tins.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 90.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302(5646):826–30. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 91.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 92.Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–32. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- 93.Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–90. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.West AE, Greenberg ME. Neuronal Activity-Regulated Gene Transcription in Synapse Development and Cognitive Function. Cold Spring Harb Perspect Biol. 2011;3(6) doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–69. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beguin P, et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411(6838):701–6. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 97.Finlin BS, et al. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem. 2005;280(51):41864–71. doi: 10.1074/jbc.M414261200. [DOI] [PubMed] [Google Scholar]
- 98.Correll RN, et al. Plasma membrane targeting is essential for Rem-mediated Ca2+ channel inhibition. J Biol Chem. 2007;282(39):28431–40. doi: 10.1074/jbc.M706176200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seu L, Pitt GS. Dose-dependent and isoform-specific modulation of Ca2+ channels by RGK GTPases. J Gen Physiol. 2006;128(5):605–13. doi: 10.1085/jgp.200609631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen H, et al. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J Neurosci. 2005;25(42):9762–72. doi: 10.1523/JNEUROSCI.3111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beguin P, et al. RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+-channel beta-subunits via an uncommon effector binding domain. J Biol Chem. 2007;282(15):11509–20. doi: 10.1074/jbc.M606423200. [DOI] [PubMed] [Google Scholar]
- 102.Sasaki T, et al. Direct inhibition of the interaction between alpha-interaction domain and beta-interaction domain of voltage-dependent Ca2+ channels by Gem. J Biol Chem. 2005;280(10):9308–12. doi: 10.1074/jbc.M413773200. [DOI] [PubMed] [Google Scholar]
- 103.Murata M, et al. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res. 2004;95(4):398–405. doi: 10.1161/01.RES.0000138449.85324.c5. [DOI] [PubMed] [Google Scholar]
- 104.Yang T, Puckerin A, Colecraft HM. Distinct RGK GTPases Differentially Use alpha(1)- and Auxiliary beta-Binding-Dependent Mechanisms to Inhibit Ca(V)1.2/Ca(V)2.2 Channels. PLoS One. 2012;7(5):e37079. doi: 10.1371/journal.pone.0037079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leyris JP, et al. RGK GTPase-dependent CaV2.1 Ca2+ channel inhibition is independent of CaVbeta-subunit-induced current potentiation. FASEB J. 2009;23(8):2627–38. doi: 10.1096/fj.08-122135. [DOI] [PubMed] [Google Scholar]
- 106.Manning JR, et al. Rad GTPase deletion increases L-type calcium channel current leading to increased cardiac contraction. J Am Heart Assoc. 2013;2(6):e000459. doi: 10.1161/JAHA.113.000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Magyar J, et al. Rem-GTPase regulates cardiac myocyte L-type calcium current. Channels (Austin) 2012;6(3):166–73. doi: 10.4161/chan.20192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang HG, Wang C, Pitt GS. Rem2-Targeted shRNAs Reduce Frequency of Miniature Excitatory Postsynaptic Currents without Altering Voltage-Gated Ca Currents. PLoS One. 2011;6(9):e25741. doi: 10.1371/journal.pone.0025741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moore AR, Ghiretti AE, Paradis S. A loss-of-function analysis reveals that endogenous rem2 promotes functional glutamatergic synapse formation and restricts dendritic complexity. PLoS One. 2013;8(8):e74751. doi: 10.1371/journal.pone.0074751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vaughn JE. Fine structure of synaptogenesis in the vertebrate central nervous system. Synapse. 1989;3(3):255–85. doi: 10.1002/syn.890030312. [DOI] [PubMed] [Google Scholar]
- 111.Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586(6):1509–17. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu NJ, et al. A dual shaping mechanism for postsynaptic ephrin-B3 as a receptor that sculpts dendrites and synapses. Nat Neurosci. 2013:2011. doi: 10.1038/nn.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hornberg H, et al. RNA-Binding Protein Hermes/RBPMS Inversely Affects Synapse Density and Axon Arbor Formation in Retinal Ganglion Cells In Vivo. J Neurosci. 2013;33(25):10384–95. doi: 10.1523/JNEUROSCI.5858-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Haas K, Li J, Cline HT. AMPA receptors regulate experience-dependent dendritic arbor growth in vivo. Proc Natl Acad Sci U S A. 2006;103(32):12127–31. doi: 10.1073/pnas.0602670103. [DOI] [PMC free article] [PubMed] [Google Scholar]