

Abstract
Objective
20-Hydroxyeicosatetraenoic acid (20-HETE), a ω-hydroxylation product of arachidonic acid catalyzed by cytochrome P450 4A (CYP4A), may play a role in cardiovascular system. It is well known that CYP450 ω-hydroxylase inhibitors markedly reduced the cardiac ischemia reperfusion injury. However, the direct effect of 20-HETE on cardiomyocytes are still poorly investigated. Here, we studied the effect of 20-HETE on cardiomyocyte apoptosis and the apoptosis-associated signaling pathways.
Methods and Results
The cardiomyocyte apoptosis was measured by FITC-annexin V/propidium iodide (PI) double staining cytometry, indicating that the percentage of early apoptotic cells increased from 15.6±2.6% to 25.5%±2.5% in control and 20-HETE-treated cells respectively. The mitochondrial membrane potential (ΔΨm) was measured by detecting the ratio of JC-1 green/red emission intensity. A significant decrease in the ratio was observed after treatment with 20-HETE for 24 h in comparison with control group, suggesting the disruptive effect of 20-HETE on mitochondrial ΔΨm. In addition, 20-HETE stimulated caspase-3 activity and Bax mRNA expression in cardiomyocytes. In contrast, the Bcl-2 mRNA levels were significantly decreased by 20-HETE treatment.
Conclusion
These results demonstrate that 20-HETE induces cardiomyocyte apoptosis by activation of several intrinsic apoptotic pathways. The 20-HETE-induced apoptosis could contribute to the CYP450 ω-hydroxylase-dependent cardiac injure during cardiac ischemia-reperfusion.
Keywords: 20-HETE, Cardiomyocytes, Apoptosis, Mitochondria
Introduction
Arachidonic acid (AA) cleaved from membrane phospholipids sources can be metabolized by Cytochrome P-450 (CYP450) ω-hydroxylase into 20-hydroxyeicosatetraenoic acid (20-HETE), which is a potent constrictor of renal, cerebral and mesenteric arterioles and small coronary arteries and plays an important role in the regulation of vascular tone in these vascular beds 1-3. Several isoforms of CYP4A and CYP4F families have been reported to catalyze arachidonic acid to 20-HETE in extrahepatic tissues, including the heart 1, 4, 5. Changes of the levels of 20-HETE have been recently reported after subarachnoid hemorrhage in rats and ischemia-reperfusion injury in canine myocardium 6, 7. Thus, 20-HETE may be a key important mediator in the regulation of cardiovascular systems. Recently, it is reported that 20-HETE production is enhanced in the heart following ischaemia-reperfusion (I/R) injury and in rat models of diabetes 7, 8. Conversely, inhibitors of CYP ω-hydroxylase markedly reduce infarct size in I/R canine hearts and exogenous 20-HETE administration significantly increases infarct size 7. These results clearly demonstrate that 20-HETE is involved in exacerbated myocardial injury in I/R hearts. This is supported by a recent study in kidney showing that 20-HETE overexpression significantly exacerbate the cellular damage that is associated with renal I/R injury and that the programmed cell death is mediated by activation of caspase-3 9. However, the exact molecular mechanisms mediating the action of 20-HETE in cardiac I/R injury are still not clear.
It is well known that apoptosis plays a key role in myocardial I/R injury 10, 11. Apoptosis, a programmed cell death process, is initiated by the activation of cell-surface receptors (the extrinsic pathway) or by permeability changes of mitochondria (the intrinsic pathway). Accumulated evidences indicate that mitochondria are pivotal to amplify apoptotic signals 12. The Bcl-2 family of proteins plays important roles in the intrinsic pathways by determining the release of cytochrome c and other pro-apoptotic factors from the mitochondria into the cytoplasm, where they initiate death pathways 13, 14. Bcl-2 family regulatory proteins include two classes: the anti-apoptotic members (Bcl-2 and Bcl-xL) protect cells against some forms of apoptosis, whereas the pro-apoptotic members (Bax and Bak) trigger programmed cell death 12, 15. Ultimately, caspase-3 is activated, leading to apoptotic chromatin condensation and DNA fragmentation, as typical events associated with the execution and completion of apoptosis 16. Previous studies have shown that apoptosis contributes to cardiomyocyte cell death in both human and animal models of ischemia/reperfusion injury and in the pathogenesis of heart failure 17, 18. However, beside a large amount of evidences showing CYP450 ω-hydroxylase and 20-HETE are involved in myocardial I/R injury, the interaction between this metabolite of CYP450 ω-hydroxylase and cardiomyocyte apoptosis has not been directly investigated in cardiomyocytes.
Thus, the objectives of the present study are to determine the effect of 20-HETE on apoptosis of cardiomyocytes and to evaluate the possible mechanisms involved in the process.
Materials and Methods
Animals and drugs
2-3 days old Wistar rats were obtained from the Experimental Animal Department of Jilin University. All experiments conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All experimental procedures were approved by the North Dakota State University and Northeast Normal University Institutional Animal Care and Use Committees. 20-HETE and HET0016 were purchased from Cayman Chemical (Ann Arbor, USA). 20-HETE was dissolved in ethanol and the solution were bubbled with nitrogen and stored at −80 °C in small volume aliquots, as desribed in the production information sheet provided by manufacturer (Cayman Chemical). During experiment, 20-HETE stock solution was kept on the ice until it was diluted into different working concentrations with the aqueous buffer. The concentration of ethanol in the solution was used as control. The JC-1 probe was provided by Beyotime Institute of Biotechnology (Haimen, China). The annexin V-FITC apoptosis detection Kit was purchased from KeyGen Biotech. Co. Ltd (Nanjing, China). Others were obtained from Sigma-Aldrich Co. (St. Louis, USA).
Cardiomyocyte culture
Primary culture of neonatal rat ventricular myocyte was prepared as described previously 19. Briefly, cells were obtained from ventricular myocytes of 2-3 days old Wistar rats. The hearts were quickly excised and minced by use of fine scissors in D-Hanks solution containing trypsin 0.1 % and digested at 37°C for 5 min. Cells were isolated by 5-min rounds of tissue digestion for 10-12 times. After each incubation, the supernatant was added to an equal volume of DMEM containing 10% fetal bovine serum (FBS). The total cell supernatants were centrifuged at 1,000 rpm for 10 min. Supernatants were discarded and the cell pellets were resuspended in DMEM containing 10% FBS. The cells were plated onto plastic culture dishes for 90 min so that most of the non-myocytes attached to the dish and the myocardiocytes remained in the suspension. The myocardocytes were harvested and seeded onto 100-mm culture dishes at 1×105 cells/cm2 or onto 96-well dishes at density of 2×104/100μL. The cardiomyocytes were cultured in an incubator filled with a humidified atmosphere of 5% CO2 at 37°C.
MTT viability assay
After neonatal rat cardiocmyocytes were cultured in 96-well dishes for 48h, the medium was changed to serum-free medium for another 24h and followed by incubation with 20-HETE (0.5 nM, 1 nM, 10 nM, 50 nM) and ethanol (vehicle) for additional 24h. The concentration of ethanol in the medium was less than 0.1% (v/v). After treatment, the cells were incubated for 4 h in a medium containing 0.5% 3-[4,5-dimethylthiazol- 2-yl]-2, 5-diphenyltetrazolium bromide (MTT), which was reduced to formazan dye in a functional mitochondria of viable cells 20. At the end of incubation, the MTT-containing medium was removed and 1.5 mL of Me2SO3 was added in each well and incubated for 10 min at 37 °C. The amount of formazan produced was detected by measurement of the absorbance at 490 nm. Results are expressed as percentage of control values.
Annexin V and propidium iodide staining of the cells
The apoptosis of cardiomyocyte was measured using a FITC-annexin V detection kit by exactly following the instruction provided by manufacturer with a minor modulation. Briefly, neonatal rat cardiomyocytes were grown in culture Flasks for 48h in DMEM containing 10% FBS. The medium was then changed to serum-free medium and continued to culture for additional 24h. After starvation, the cells were treated with 20-HETE (1-50 nM), ethanol (vehicle control), arachidonic acid (AA, 1 μM) or AA plus a CYP ω-hydroxylase inhibitor, HET0016 (10 μM) for 24 h. The 20-HETE doses (1-50 nM) used in the current study were chosen based on previous in vivo studies demonstrating that the plasma 20-HETE levels were elevated up to 2.5 nM and 18 nM after cardiac ischemia/reperfusion in rats and dogs respectively (11, 27). Cells were then washed with PBS and resuspended in a binding buffer. The fluorescein isothiocanate (FITC)-conjugated annexin V (5 μL) and propidium iodide (PI) reagent (5 μL) were added and the mixture was incubated at room temperature for 15 min. The samples were immediately measured by FACScan flow cytometer (Beckman, USA).
Measurement of mitochondrial membrane potential (ΔΨm)
The fluorescent probe JC-1 (5, 5′, 6, 6′,-tetrachloro-1, 1′, 3, 3′,-tetraethylbenzimid azoly-carbocyanide iodide) was used to measure the mitochondrial membrane potential ΔΨm of cardiomyocyte. JC-1 is a lipophilic cationic dye that accumulated in the mitochondria depending upon the mitochondrial membrane potential. Its aggregated form emits red fluorescence in mitochondria with higher ΔΨm, but dissociates into monomers and emits green fluorescence when mitochondria loses ΔΨm. Cultured cardiomyocytes were stained with JC-1 (5μg/mL) at 37°C for 20 min and washed twice with PBS. Mitochondrial membrane potentials were monitored using an Olympus laser scanning confocal microscope (Olympus FV1000). JC-1 monomer fluorescence (green) was observed by examination at 488 nm laser excitations and examination of the emissions at 530 nm. JC-1 aggregate fluorescence (red) was observed by examination of the emissions at 590 nm. Sixteen areas in three separated cultured dishes were scanned and the average intensity for each region was quantified in the cardiomyocytes treated under the following conditions: control, vehicle control, or 20-HETE (10 nM). The ratio of JC-1 aggregate to monomer intensity for each region was calculated. An decrease in this ratio was interpreted as decrease of ΔΨm, whereas a increase in the ratio was interpreted as gain in ΔΨm.
Reverse transcription–polymerase chain reaction (RT–PCR)
The neonatal rat cardiomyocytes were cultured in the presence or absence of 20-HETE (10 nM) for 24 h. Total RNA was isolated using the Trizol reagent according to the manufacturer's instructions (Invitrogen, USA). First-strand cDNA was synthesized from total RNA using a reverse transcription kit with the AMV-reverse transcriptase according to the manufacturer's instructions (TaKaRa, China). Reverse transcription was performed at 42°C for 30 min followed by incubation at 94°C for 10 min. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was chosen as a housekeeping gene. The cDNA was amplified by PCR using 5 units of Taq polymerase, 10mmoles of dNTPs and 20 pmoles of each primer. The sequences of the primers used for PCR were as follows: Bcl-2 sense primer: 5′-TCCTTCCAGCCTGAGAGCAACC-3′ and antisense primer: 5′-GACAGCCAGGAGAAATCAAACAGA-3′; Bax sense primer: 5′-TCCAGGATCGAGCAGA-3′ and antisense primer: 5′-AAGTAGAAGAGGGCAACC-3′; GAPDH sense primer: 5′-ATTGCTCTCAATGACAACTT-3′ and antisense primer: 5′-GAACTTTATTGATGGTATTCG-3′. The specific primer yielded expected PCR products of 491, 256 or 277 bp for Bcl-2, Bax or GAPDH genes respectively. The PCR conditions were set as: initial denaturation for 2 min at 94°C; repeated cycles of denaturation for 1 min at 94°C; annealing for 1 min at 56°C (for Bcl-2), 53°C (for Bax) or 50°C (for GAPDH); extension for 1 min at 72°C; final extension for 10 min at 72°C. The amplification was conducted for 40 cycles (Bcl-2), 40 cycles (Bax) or 30 cycles (GAPDH). The synthesized PCR products were separated by electrophoresis on a 2% agarose gel. The gels were stained with ethidium bromide and visualized under UV light.
Assay of caspase-3 activity and detection of apoptosis through Hoechst staining
Caspase-3 activity was measured with a caspase-3 activity assay kit according to the manufacturer's protocol (KeyGEN, China). Briefly, after treatment with 20-HETE (10 nM) or vehicle control for 24h, cell cultures were harvested with a cell scraper in lysis buffer supplied in the kit. The lysate was centrifuged at 4°C for 10 min at 16000 g, and protein was measured with a spectrophotometer. Approximately 50 μg of total protein for each sample was transferred to a 96-well microplate, and incubated with substrate (Ac-DEVD-pNA) in 50μL reaction buffer for 4 h at 37 °C. The absorbance of yellow product (pNA) cleaved from its corresponding precursors was measured at 405 nm using a plate reader.
To measure the cardiomyocyte apoptosis, the cardiomyocytes were stained with Hoechst 33342. After treated with 20-HETE (10 nM) or vehicle control for 24 h, the cells were washed with PBS and then stained with Hoechst 33342 for 30 min at room temperature. The condensed or fragmented apoptotic nuclei were observed under a laser scanning confocal microscope (Olympus FV1000).
Statistics analysis
All data are expressed as means ± SE. Differences between groups were assessed using Student's t test or one-way analysis of variance (ANOVA) for multiple comparisons. P values <0.05 were taken as significant and significance levels are given in the text.
Results
Treatment with 20-HETE decreased cardiomyocyte viability
Blockade of 20-HETE formation with cytochrome P450 ω-hydroxylase inhbitors inhibits cardiomyocyte apoptosis 21. Thus, the first experiment in current study was designed to examine the effect of 20-HETE on the cardiomyocyte viability using MTT assay (Fig. 1). The results shown in Figure 1 demonstrated that treatment of cardiomyocytes with 20-HETE significantly decreased cell viability. The viable cardiomyocytes dropped to 79.1±6.4% (n=3, p<0.05) of control level after treatment with 20-HETE (10 nM). High dose of 20-HETE (50 nM) induced a greater decrease (56.9±7.9%). However, treatment of cells with vehicle (ethanol) alone did not alter the cell viability (Fig.1). The result suggests that treatment of 20-HETE decreased the number of cardiomyocytes in culture dishes. Thus, we next examined whether 20-HETE causes apoptosis of cardiocmyocyte.
Figure 1. Effect of 20-HETE on the survival rate of cultured neonatal rat ventricular myocytes.

Serum deprivated cardiomyocytes were treated with control, vehicle, or 20-HETE (0.5 nM, 1 nM, 10 nM, 50 nM) for 24h. Cell viability was determined using the MTT assay as detailed in the Methods. Values are expressed for each time point as percentage of control. Data are mean±SE obtained from three experiments using independent batches of cells in each group. *P<0.05 compared with control.
20-HETE induced apoptosis in cultured cardiomyocytes
Flow cytometric analysis with annexin V and PI staining was undertaken to determine the effect of 20-HETE on cardiomyocyte apoptosis. Compared to the control group, treatment of cardiomyocytes with 20-HETE (10 nM) significantly decreased the percentage of living cells and resulted in a significant increase in the percentage of apoptosis cells (Fig. 2). As shown in Fig 2A and B, the vital cells are negative for both annexin V binding and PI uptake (annexin V-/PI-); the early apoptotic cells are positive for FITC-annexin V binding but negative for PI uptake (annexin V+/P-), while the advanced apoptotic/necrotic cells are positive for both FITC-annexin V binding and PI uptake (annexin V+/PI+). The percentage of early apoptotic cells was 15.6±2.6% and 25.5±2.5% in cardiomyocytes treated with vehicle control and 20-HETE (10 nM) respectively (n=3, P<0.05). However, the same amount of ethanol vehicle for 20-HETE altered neither percentage of apoptotic cells nor percentage of living cells. These results demonstrated that treatment of cardiomyocytes with exogenous 20-HETE induced cardiomyocyte apoptosis. In addition, we also examined the effect of arachidonic acid (AA), the substrate for 20-HETE production, on the cardiomyocyte apoptosis. Treatment of cardiomyocytes with AA (1 μM) significant increased the percentage of early apoptotic cells from 14.1±2.9% to 26.8±2.1% (n=3, P<0.05). AA-induced increases in the percentage of apoptotic cells were significantly attenuated by co-treatment with CYP-ω hydroxylase inhibitor, HET0016 (10 μM), by 94.2±4.1% (n=3, P<0.05). These results indicate increased endogenous 20-HETE also increases cardiomyocyte apoptosis. Thus, we next examined the cellular mechanisms underlying 20-HETE-induced apoptosis in cardiomyocytes.
Figure 2. Effect of 20-HETE on apoptosis of cultured neonatal rat ventricular myocytes.
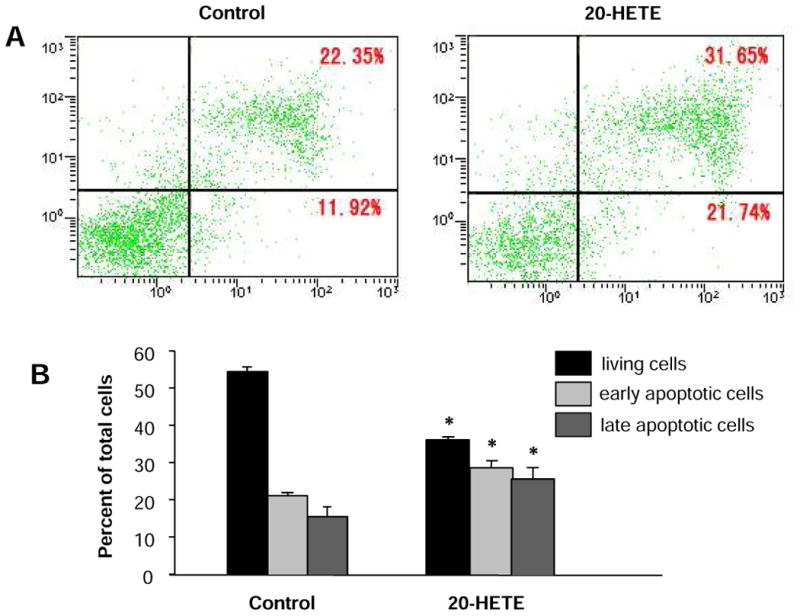
The cell apoptosis was measured by flow cytometry following anexin V and propidium iodide (PI) double staining in cardiomyocytes treated with vehicle control or 20-HETE(10 nM) for 24 h. A. Examples of dot-plots of cells treated with control and 20-HETE. Horizontal axis represents annexin V intensity and vertical axis shows PI staining. The lines divide each plot into four quadrants: lower left quadrant, living cells; lower right quadrant, early apoptotic cells; upper left quadrant, necrotic cells; upper right quadrant, late apoptotic cells. B. Bar graphs summarizing the mean percentage of living cells, ratio of cells in early and late apoptosis as represented in A. Data are expressed as mean±SE obtained from three experiments using independent batches of cells in each group. *P<0.05 significant difference compared to control.
20-HETE resulted in dissipation of mitochondrial membrane potential (ΔΨm)
It is well known that maintenance of intact mitochondrial membrane potential (ΔΨm) is critical for cell survival, and loss of ΔΨm triggers a cascade of reactions, leading to cell apoptosis 22. Thus, the involvement of mitochondria in initiation of apoptosis was evaluated by recording changes in its membrane potential with JC-1 staining in cardiomyocytes treated with 20-HETE (10 nM) or vehicle control. Under control condition, cardiomyocytes were stained with JC-1, emitting red fluorescence with low density of green fluorescence (Fig 3A). This aggregated JC-1 within normal mitochondria was dispersed to the monomeric form after treatment with 20-HETE for 24 h, emitting green fluorescence (Fig. 3A). The ratio of JC-1 green/red fluorescence intensity was significantly decreased in the cardiomyoctyes treated with 20-HETE, as compared with vehicle control (Fig. 3B). In addition, we also examine the time-course of 20-HETE-induced alteration in the ratio of JC-1 green/red fluorescence. The results are shown in Figure 3C, demonstrating that the ΔΨm response to 20-HETE starts in 12 hours, reaches to the peak in 24 hours, and lasts at least 48 hours after treatment. These results demonstrate that 20-HETE has disruptive effect on mitochondrial ΔΨm of cardiomyocytes and this pathway may contribute to the 20-HETE-induced apoptosis observed in above experiments.
Figure 3. Effects of 20-HETE on the mitochondrial membrane potential (ΔΨm) of cultured neonatal rat ventricular myocytes.
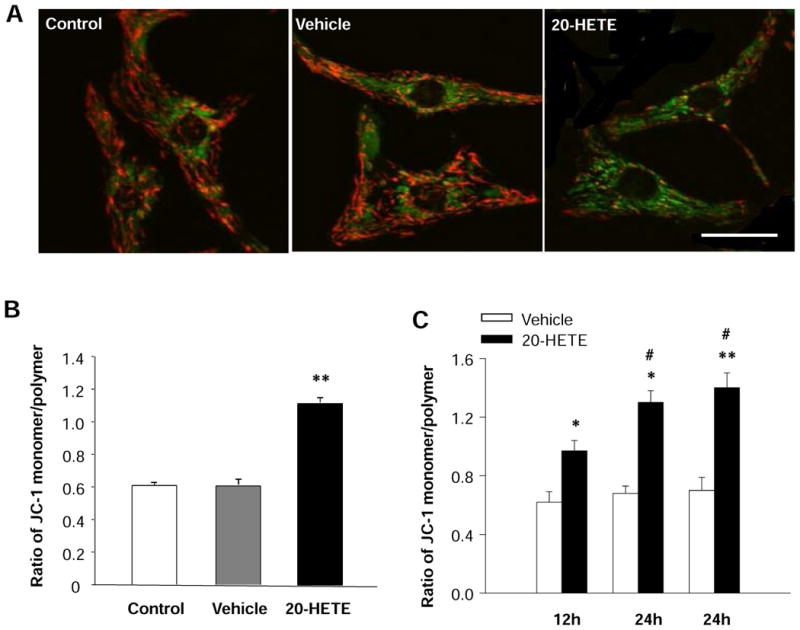
Red fluorescence represents the mitochondrial aggregate form of JC-1, indicating intact mitochondrial membrane potential. Green fluorescence represents the monomeric form of JC-1, indicating dissipation of ΔΨm. A. JC-1 staining of control cardiomyocytes, exhibiting a heterogeneous distribution of mitochondria with low density of green fluorescence and high density of red fluorescence. The green fluorescence intensity was increased and the red fluorescence intensity was decreased by treatment with 20-HETE. Scale bar =20 μm. B. Quantitative analysis of JC-1 fluorescence in cardiomyocyte treated with vehicle control and 20-HETE (10 nM) for 24 h. Treatment of cell with 20-HETE significantly increased the ratio of green to red fluorescence, suggesting a depolarization of ΔΨm. Data are mean±SE obtained from 16 observation fields in three independent experiments in each group. **P<0.01 significant difference compared with control. C. Time-course of the effect of 20-HETE on the ratio of JC-1 monomer/polymer in cardiomyocytes. Data are mean±SE obtained in three independent experiments in each group. *P<0.05 significant difference compared with control. **P<0.01 significant difference compared with control. #P<0.05 compared with treatment with 20-HETE for 12 h.
20-HETE altered the mRNA expression of Bcl-2 and Bax in cardiomyocytes
To examine the intracellular signaling pathways underlying 20-HETE-induced apoptosis, we analyzed the mRNA expression of the Bcl-2 related proteins, Bcl-2 and Bax. The neonatal rat ventricular myocytes were exposed to 10 nM 20-HETE for 24h. Total mRNA was isolated and Bcl-2 or Bax mRNA expression was examined by RT-PCR. Compared with the cells treated with vehicle control, the expression of Bax mRNA was significantly increased in the cardiomyocytes treated with 20-HETE (Fig. 4A and B). In contrast, the Bcl-2 mRNA expression was significantly decreased by 45.4±2.7% (n=3, P<0.05) after treatment of 20-HETE, as compared with vehicle control treatment (Fig. 4A and B). In summary, treatment with 20-HETE significantly results in a down-regulation of the Bcl-2 mRNA and up-regulation of the Bax mRNA in cardiomycytes cultured from neonatal rats.
Figure 4. Effect of 20-HETE on Bcl-2 and Bax mRNA expression and on casepase activity in cultured cardiomyocytes.
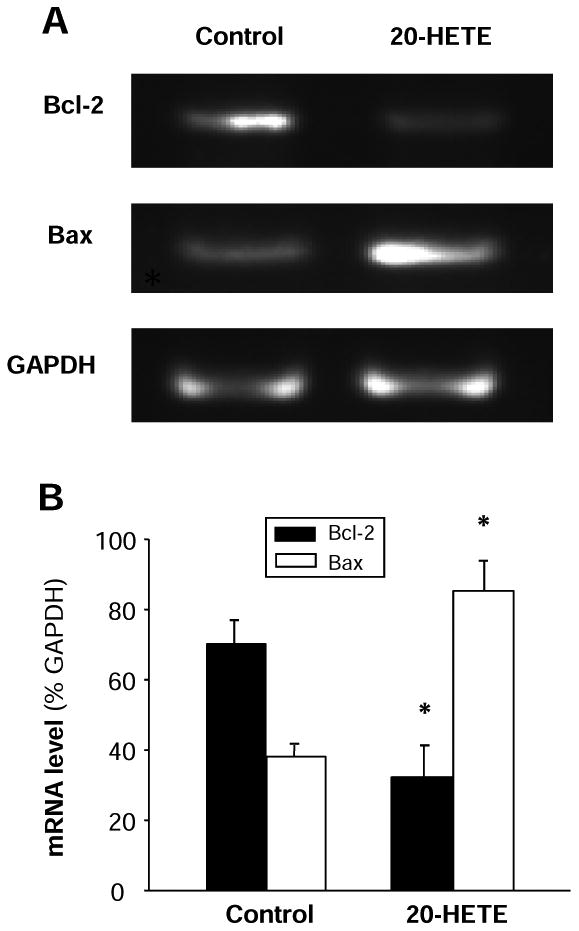
A. Representative amplified PCR products were electrophoresed on a 2% agarose gel and visualized by ethidium bromide staining in cardiomyocytes treated with vehicle control or 20-HETE (10 nM) for 24 h. Treatment of 20-HETE decreased expression of Bcl-2 mRNA and increased Bax mRNA. GAPDH was used as an internal control. B. Bar graphs summarizing the relative density of the bands normalized to GAPDP in the cardiomyocytes under each treatment condition. The mRNA levels of Bcl-2 and Bax were expressed as the percentage of GAPDH mRNA. Data are expressed as mean±SE from three separate experiments using independent batches of cells in each group. *P<0.05 significant difference as compared with control.
20-HETE increased caspase-3 activity and induced nuclear condensation in cardiomyocytes
Caspases are the major class of proteases mediating the apoptosis and caspase-3 is the major form of caspase involved in apoptosis 16. During apoptotic cell death, procaspase-3 is processed into a 16-kDa active subunit, active caspase-3, which is regarded as a signature marker of apoptosis 23. The caspase-3 activity was measured using caspase-3 colorimetric substrate (Ac-DEVD-pNA) in the cells treated with vehicle control and 20-HETE (10 nM) for 24 h. 20-HETE treatment significantly increased caspase-3 like proteases activity by 52.9±12.9% (n=3, P<0.05), as compared with vehicle control (Fig. 5C). However, treatment of cells with vehicle did not alter the activity of caspase-3. This observation demonstrates that 20-HETE stimulates caspase-3 activity, which could contribute to the 20-HETE-induced apoptosis in the cardiomyocytes.
Figure 5. Effect of 20-HETE on caspase-3 activity and nuclear morphology in cardiomyocytes.
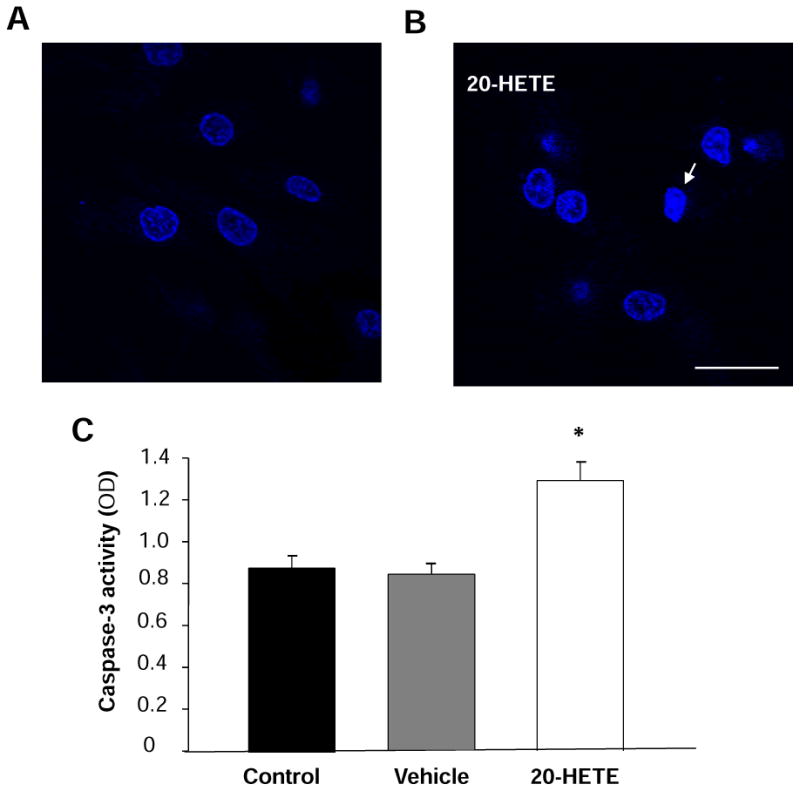
A. Cardiomyocytes were stained with Hoechst 33342, and the nuclei are uniformly weak-stained in the control. B. The chromatin in some nuclei begins to condense after treatment with 20-HETE for 24h. Arrows showed the apoptotic nuclei. Scale bar =20 μm. C. Caspase-3 activity was measured by analyzing the cleavage of colorimetric substrate (Ac-DEVD-pNA) in the cardiomyocytes treated with vehicle control and 20-HETE (10 nM) for 24h. Data are mean±SE obtained in three separated experiments in each group. *P < 0.05 vs. the control group.
Cardiomyoctyes treated with 20-HETE or vehicle control were stained with Hoechst 33342 to examine nuclear condensation, a major apoptotic character. 20-HETE treatment significantly increased the number of cells with abnormal nucleus as compared with vehicle control (Fig. 5B).
Dissicusion
The present study presents the first evidence that 20-HETE directly induces cardiomyocyte apoptosis via stimulation of several intracellular signaling pathways in primary cultured cardiomyocytes from neonatal rats. This conclusion is supported by the following observations: 1) treatment of cardiomyotyes with 20-HETE significantly decreases the number of living cardiomyocyte and increases the number of both early and late apoptotic cells; 2) 20-HETE treatment significantly decreases the mitochondrial membrane potential in cardiomyocytes; 3) decreased Bcl-2 expression and increased Bax mRNA expression are observed in 20-HETE-treated cells; 4) the activity of caspase-3 is enhanced in the cardiomyocytes treated with 20-HETE. These results demonstrate that 20-HETE directly stimulates apoptosis in cardiomyocytes via mitochondria-dependent pathways. These action of 20-HETE may contribute to CYP ω-hydroxylase-mediated myocardial injury during ischemia-reperfusion. However, our study has the limitations that cultured neonatal rat cardiomyocytes have some different pathological and physiological properties compared with the adult heart. However, the model allows us to examine the direct effects of 20-HETE on the apoptosis of cardiomyocytes with independence of hemodynamic, neural, or locally derived factors.
Several studies reported that the levels of 20-HETE are elevated in the coronary venous plasma of the cardiac ischemia reperfusion injury and that the formation of 20-HETE is also increased in the hearts of diabetic animals 7, 8. In Langendorff isolated perfused rat hearts, administration of nonspecific CYP enzyme inhibitors, such as chloramphenicol, cimetidine, and sulfaphenazole, prevents ischemia/reperfusion-induced myocardial damage, an effect that is associated with reduced ROS production 24. These results are confirmed in an ischemia-reperfusion injury rat model evoked by coronary artery ligation 25. More interestingly, the production of 20-HETE is enhanced during ischemia-reperfusion injury 7. Specific CYP ω-hydroxylase inhibitors, such as 17-octadecanoic acid (17-ODYA) and DDMS, markedly inhibit 20-HETE production and produce a profound reduction in myocardial infarct size 5, 7. Conversely, exogenous 20-HETE administration significantly increased infarct size 7. Thus, it is proposed that 20-HETE production is linked to myocardial injury induced by ischemia-reperfusion. However, the effect of 20-HETE on cardiomyocytes are still poorly understood. In this study, we evaluated the effect of 20-HETE on the apoptosis in cultured cardiomyocytes using flow cytometry, caspase-3 activity assay, and Bax mRNA expression measurement. We firstly presented a direct evidence that 20-HETE induces neonatal rat cardiomyocytes apoptosis by mitochondria-dependent intrinsic apoptotic pathways.
In this study, a significant reduction of mitochondrial potentials was observed after treatment with 20-HETE, suggesting that mitochondrial membrane damage may be involved in 20-HETE-induced apoptosis in cardiomyocytes. Mitochondrial damage and dysfunction has been shown to trigger apoptosis with enhanced expression of the pro-apoptotic protein Bax 26. In other hands, Bcl-2, an anti-apoptotic protein, located in the outer mitochondrial wall controls mitochondrial permeability. In current study, we observed 20-HETE induces a decreased Bcl-2 mRNA expression and an increased Bax mRNA expression, suggesting that both Bcl-2 and Bax are involved in the 20-HETE-induced mitochondrial membrane damage.
These findings indicate that 20-HETE induces cardiomyocytes apoptosis by activating the intrinsic mitochondrial-mediated apoptotic pathways. Mitochondria dysfunction plays a key role in the activation of the caspase cascade 27, which is a crucial mechanism for inducing apoptotic death signals. Caspases participate in a cascade that is triggered in response to pro-apoptotic signals and culminates in the cleavage of a set of proteins, resulting in disassembly of the cell. Caspase-3 is the primary activator of apoptotic DNA fragmentation 22. In the current study we measured caspase-3 activity by analyzing the cleavage of colorimetric substrate (Ac-DEVD-pNA) indicating that 20-HETE significantly stimulates caspase-3 like proteases. These results demonstrate that multiple pathways may be involved in 20-HETE-induced apoptosis in myocardial cells.
In conclusion, our results demonstrate that 20-HETE induces apoptosis in neonatal rat cardiomyocytes, and such an effect is likely to be mediated through multiple intrinsic apoptotic pathways involving mitochondria, Bcl-2, Bax, caspase-3. 20-HETE induced apoptosis in myocardial cells could contribute to the CYPω-hydroxylase-mediated cardiac damage during ischemia-reperfusion.
Acknowledgments
The work was supported by the American Heart Association (0635050N) and by the National Natural Science Foundation of China (30870910). Funding for the NDSU Core Biology Facility used in this study was made possible by NIH Grant Number 2P20 RR015566 from the National Center for Research Resources.
References
- 1.Sun CW, Alonso-Galicia M, Taheri MR, Falck JR, Harder DR, Roman RJ. Nitric oxide-20-hydroxyeicosatetraenoic acid interaction in the regulation of K+ channel activity and vascular tone in renal arterioles. Circ Res. 1998;83:1069–1079. doi: 10.1161/01.res.83.11.1069. [DOI] [PubMed] [Google Scholar]
- 2.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 3.Larsen BT, Miura H, Hatoum OA, Campbell WB, Hammock BD, Zeldin DC, Falck JR, Gutterman DD. Epoxyeicosatrienoic and dihydroxyeicosatrienoic acids dilate human coronary arterioles via BK(Ca) channels: implications for soluble epoxide hydrolase inhibition. Am J Physiol Heart Circ Physiol. 2006;290:H491–H 499. doi: 10.1152/ajpheart.00927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML. Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol Regul Integr Comp Physiol. 1999;276:R1691–R1700. doi: 10.1152/ajpregu.1999.276.6.R1691. [DOI] [PubMed] [Google Scholar]
- 5.Gross ER, Nithipatikom K, Hsu AK, Peart JN, Falck JR, Campbell WB, Gross GJ. Cytochrome P450 omega-hydroxylase inhibition reduces infarct size during reperfusion via the sarcolemmal KATP channel. J Mol Cell Cardiol. 2004;37:1245–1249. doi: 10.1016/j.yjmcc.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Kehl F, Cambj-Sapunar L, Maier KG, Miyata N, Kametani S, Okamoto H, Hudetz AG, Schulte ML, Zagorac D, Harder DR, Roman RJ. 20-HETE contributes to the acute fall in cerebral blood flow after subarachnoid hemorrhage in the rat. Am J Physiol. 2002;282:H1556–H1565. doi: 10.1152/ajpheart.00924.2001. [DOI] [PubMed] [Google Scholar]
- 7.Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ. Inhibition of cytochrome P450omega-hydroxylase: a novel endogenous cardioprotective pathway. Circ Res. 2004;95:e65–e71. doi: 10.1161/01.RES.0000146277.62128.6f. [DOI] [PubMed] [Google Scholar]
- 8.Yousif MH, Benter IF, Roman RJ. Cytochrome P450 metabolites of arachidonic acid play a role in the enhanced cardiac dysfunction in diabetic rats following ischaemic reperfusion injury. Auton Autacoid Pharmacol. 2009;29:33–41. doi: 10.1111/j.1474-8673.2009.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilakantan V, Maenpaa C, Jia G, Roman RJ, Park F. 20-HETE-mediated cytotoxicity and apoptosis in ischemic kidney epithelial cells. Am J Physiol Renal Physiol. 2008;294:F562–F570. doi: 10.1152/ajprenal.00387.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kajstura J, Cheng W, Reiss K, Clark WA, Sonnenblick EH, Krajewski S, Reed JC, Olivetti G, Anversa P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab Invest. 1996;74:86–107. [PubMed] [Google Scholar]
- 11.Freude B, Masters TN, Robicsek F, Fokin A, Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S, Schaper J. Apoptosis is initiated by myocardial ischemia and executed during reperfusion. J Mol Cell Cardiol. 2000;32:197–208. doi: 10.1006/jmcc.1999.1066. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 13.Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 14.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 15.Tsujimoto Y, Shimizu S. Bcl-2 family: life-or-death switch. FEBS Lett. 2001;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- 16.Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 17.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 18.Zucchi R, Ghelardoni S, Evangelista S. Biochemical basis of ischemic heart injury and of cardioprotective interventions. Curr Med Chem. 2007;14:1619–1637. doi: 10.2174/092986707780831014. [DOI] [PubMed] [Google Scholar]
- 19.Zeng Q, Zhou Q, Yao F, O'Rourke ST, Sun C. Endothelin-1 regulates cardiac L-type calcium channels via NAD(P)H oxidase-derived superoxide. J Pharmacol Exp Ther. 2008;326:732–738. doi: 10.1124/jpet.108.140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Musser DA, Oseroff AR. The use of tetrazolium salts to determine sites of damage to the mitochondrial electron transport chain in intact cells following in vitro photodynamic therapy with Photofrin II. Photochem Photobiol. 1994;59:621–626. doi: 10.1111/j.1751-1097.1994.tb09666.x. [DOI] [PubMed] [Google Scholar]
- 21.Lv X, Wan J, Yang J, Cheng H, Li Y, Ao Y, Peng R. Cytochrome P450 omega-hydroxylase inhibition reduces cardiomyocyte apoptosis via activation of ERK1/2 signaling in rat myocardial ischemia-reperfusion. Eur J Pharmacol. 2008;596:118–126. doi: 10.1016/j.ejphar.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabó C, Clark RS. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- 23.Chang HY, Yang X. Proteases for cell suicide: functions and regulation of caspases. Microbiol Mol Biol Rev. 2000;64:821–846. doi: 10.1128/mmbr.64.4.821-846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishihara Y, Sekine M, Nakazawa M, Shimamoto N. Suppression of myocardial ischemia-reperfusion injury by inhibitors of cytochrome P450 in rats. Eur J Pharmacol. 2009;611:64–71. doi: 10.1016/j.ejphar.2009.03.069. [DOI] [PubMed] [Google Scholar]
- 26.Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y Acad Sci. 2002;959:93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 27.Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]