

Abstract
Background
The molecular characteristics of human diseases are often represented by a list of genes termed “signature genes”. A significant challenge facing this approach is that of reproducibility: signatures developed on a set of patients may fail to perform well on different sets of patients. As diseases are resulted from perturbed cellular functions, irrespective of the particular genes that contribute to the function, it may be more appropriate to characterize diseases based on these perturbed cellular functions.
Methods
We proposed a profile-based approach to characterize a disease using a binary vector whose elements indicate whether a given function is perturbed based on the enrichment analysis of expression data between normal and tumor tissues. Using breast cancer and its four primary clinically relevant subtypes as examples, this approach is evaluated based on the reproducibility, accuracy and resolution of the resulting pathway profiles.
Results
Pathway profiles for breast cancer and its subtypes are constructed based on data obtained from microarray and RNA-Seq data sets provided by The Cancer Genome Atlas (TCGA), and an additional microarray data set provided by The European Genome-phenome Archive (EGA). An average reproducibility of 68% is achieved between different data sets (TCGA microarray vs. EGA microarray data) and 67% average reproducibility is achieved between different technologies (TCGA microarray vs. TCGA RNA-Seq data). Among the enriched pathways, 74% of them are known to be associated with breast cancer or other cancers. About 40% of the identified pathways are enriched in all four subtypes, with 4, 2, 4, and 7 pathways enriched only in luminal A, luminal B, triple-negative, and HER2+ subtypes, respectively. Comparison of profiles between subtypes, as well as other diseases, shows that luminal A and luminal B subtypes are more similar to the HER2+ subtype than to the triple-negative subtype, and subtypes of breast cancer are more likely to be closer to each other than to other diseases.
Conclusions
Our results demonstrate that pathway profiles can successfully characterize both common and distinct functional characteristics of four subtypes of breast cancer and other related diseases, with acceptable reproducibility, high accuracy and reasonable resolution.
Keywords: Signature genes, Pathway, Pathway profile, Enrichment analysis, Breast cancer
Background
Diseases are abnormal conditions of the human body resulted from significant nonlethal malfunctions that affect the human physiological system. Traditionally, diseases are characterized by pathology and observation of clinical phenotypes. Although these methods have proved successful in many applications, they lack the sensitivity to detect diseases before the appearance of symptoms and also have a limited ability to distinguish complex disease classes [1,2] which may present confusing or overlapping symptoms.
With the development of genomic technology, a promising approach to overcome limitations of the traditional method is to identify a set of genes as a genetic signature whose combined expression pattern is the uniquely characteristic of a given phenotype [3,4]. In the last decade, some gene signatures have been developed for cancers [5-9] and other diseases [10,11], indicating that the use of these signatures can assist in defining disease, predicting disease recurrence, aiding disease diagnosis and guiding treatment decision.
There are still obvious obstacles that prevent the application of gene signatures in clinical practice [12,13]. One major problem is the low reproducibility between signature genes. The overlaps of gene signatures derived from different data sets of the same disease are generally very few compared to the total number of signature genes [14], while stable gene signatures are crucial to the robustness of predictors [15]. The reasons for this discrepancy may include different cohorts of patients, different statistical methods, and different experimental technologies involved in identifying the signature genes [16]. It has been further suggested that a large number of samples are required to achieve a robust gene signature [14,15]. However, another reason for this discrepancy may lie in the observation that diseases are directly resulted from perturbed cellular functions which are generally carried out by groups of genes in the forms of complexes, modules or pathways [17]. Therefore, it is reasonable to assume that any gene whose change of expression leads to the perturbed molecular function may be a potential signature gene. This assumption is partially evidenced by the fact that both gene signatures developed in [18] and [19] can capture cell proliferation related biological processes and pathways [20], and that dysregulations of functionally related genes result in similar clinical phenotypes [21]. This assumption may also explain why sophisticated methods can rarely find much better gene signatures than simple methods [22]. From this perspective, it may be more appropriate to characterize diseases at the functional level.
Pathway-based methods have been extensively applied to analyze large-scale genome-wide data with varied purposes and applications [23-29]. Some of them classify tumor samples based on pathway-level measurements [30]; many of them, such as PWEA [31] and GSEA [32] identify perturbed pathways between two distinct phenotypes (e.g., tumor vs. normal) using expression data [28]. These enrichment methods often require a significant number of samples to achieve a statistically robust analysis. With appropriate stability and reliability, the resulting pathways of these methods may serve as reference pathways to be compared against pathways identified by sample-based analysis [33,34] in clinical applications such as disease diagnosis and personalized medicine.
In this study, we report a new approach to characterize diseases at the functional level, with our aim being to both consolidate redundant gene lists and to generate a list of pathways which is both accurate and reproducible. For a given disease, a pathway profile is generated based on the enrichment analysis of differential gene expression data between normal and tumor tissues: a binary vector whose elements indicate whether a given function (represented by a KEGG pathway) is perturbed. Using breast cancer and four clinically-relevant subdivisions (luminal A, luminal B, triple-negative and HER2+) as examples, we examine the new approach from three perspectives: to determine whether the pathway profile can be reproduced from the data generated by different technologies (Microarray vs. RNA-Seq), as well as from separate cohorts (The Cancer Genome Atlas (TCGA) vs. The European Genome-phenome Archive (EGA)), to determine whether the resulting pathways are associated with the functional perturbation resulted from the breast cancer and its subtypes, and finally to determine whether the pathway profile can distinguish different subtypes of breast cancer as well as distinguish breast cancer from other diseases. Our results indicate that the new approach achieves 68% average reproducibility between different data sets (TCGA microarray vs. EGA microarray data) and 67% average reproducibility between different technologies (TCGA microarray vs. TCGA RNA-Seq data). Among the enriched pathways, 74% of them are known to be associated with breast cancer or other cancers by extensive literature search. Approximately 40% of the pathways are enriched in all four subtypes and there are 4, 2, 4, and 7 pathways enriched only in the luminal A, luminal B, triple-negative, and HER2+ subtypes, respectively, implying that pathway profiles not only reveal shared mechanisms in the four subtypes but also outline the subtype-specific operations that may potentially be used as signature pathways to distinguish them. Comparison of profiles between subtypes, as well as other diseases including ovarian cancer, glioblastoma multiforme (GBM), and obesity, reveals that the luminal A and luminal B subtypes of breast cancer are closer to each other than to other subtypes, luminal A and luminal B subtypes are closer to the HER2+ subtype than to the triple-negative subtype, and subtypes of breast cancer are more likely to be closer to each other than to other diseases.
Methods
Data sources
808 tumor and 106 normal samples of TCGA RNA-Seq data (Illumina HiSeq 2000 RNA Sequencing platform) were downloaded from the TGGA portal on Oct. 2012. 522 tumor samples with available PAM50 classification [35] and 63 normal samples of TCGA microarray data (Agilent G4502A platform) were downloaded on Nov. 2012. 496 tumor and 58 normal samples overlap between the two TCGA data sets (drawn from the same patients). Both the “discovery” and “validation” EGA data sets were also downloaded, which consisted of 997 and 995 tumor samples, respectively, and 144 normal samples (Illumina HT-12 v 3 platform, accession number EGA S00000000083) [36]. The EGA discovery data set was used in our analysis and the EGA validation data set was used to further verify our major results (See discussion in Additional file 1). For brevity, the “discovery” EGA data is referred to as the EGA data set in the remainder of this paper unless otherwise stated. 37 tumor and 8 normal samples of the TCGA ovarian cancer data set (Affymetrix HG-U133A platform, batch 9) were downloaded on Feb. 2013, and 24 tumor and 10 normal samples of TCGA GBM data set (Affymetrix HG-U133A platform, batch 8) were download on Nov. 2013. The obesity data set (Affymetrix HG-U133_Plus_2 platform) of 5 obesity and 6 control samples was downloaded from GEO [GDS3688] (omental adipose from obese, prepubertal children).
No major batch effects were observed for the two TCGA breast cancer data sets [35]. The batch effect of EGA data set has been removed by a linear model [36]. To eliminate batch effects in the ovarian cancer data, only batch 9 of TCGA ovarian cancer data set was used [37]. Similarly, only batch 8 of the TCGA GBM data set was used because the possibility of batch effects within the GBM data set may not be ignored (http://bioinformatics.mdanderson.org/main/TCGABatchEffects:Overview).
A total of 269 human pathways were downloaded from KEGG [38] on Jun. 2013. 175 of these pathways in 30 pathway categories were used in our analysis after excluding all disease pathways and pathways with size either smaller than 16 or larger than 350 (to increase statistical power).
Classification of breast cancer samples
To be consistent with clinical practice, we classified tumor samples into luminal A, luminal B, triple-negative, and HER2+ subtypes using the following steps. First, HER2+ samples were identified based on the test results of immunohistochemistry (IHC) or florescence in situ hybridization (FISH). The rest of the samples were then split into ER+ and ER- samples based on estrogen receptor (ER) status provided by IHC test results. ER+ samples were then further classified into luminal A and luminal B subtypes using PAM50 classification. Finally, triple-negative samples were extracted from the pool of ER- samples according to progesterone receptor (PR) status based on IHC test results. PAM50 classification results alone were used whenever ER or HER2 status was not available in the clinic data. As EGA data does not provide the PR status, instead we used the expression-based classification result for PR status [36]. PAM50 classification results were downloaded from the UCSC Cancer Genomics Browser [39] for TCGA data sets and the supplementary materials in its original publication [36] for the EGA data set. More details on sample classification can be found in Additional files 1 and 2.
Pathway enrichment analysis
We focused on the enrichment of pathways abnormally perturbed in the disease state compared to the normal state in four major subtypes of breast cancer. PWEA [31] was used in this study to carry out pathway enrichment analysis, as a comprehensive study previously indicated that it has a higher sensitivity than other enrichment analysis methods including GSEA [32] with little or no loss of specificity [31,40]. The PWEA results of all data sets are provided in Additional file 3.
Definition of reproducibility
Our measure of reproducibility was applied to both pathway and gene-based profiles. Using pathways as an example, the reproducibility r can be defined as
where N and N’ are the number of enriched pathways for two different data sets, and N c is the number of overlapping of enriched pathways between two data sets.
Pathway profile
For a given disease, a pathway profile p can be defined as:
where N is the total number of pathways used in the analysis and p n (1 ≤ n ≤ N) is equal to either 1 or 0 to indicate whether nth pathway is enriched or not, respectively.
Analysis workflow
The sketch of our approach is depicted in Figure 1. Our approach utilized three main data sets which encompassed two separate patient cohorts and three distinct gene expression measurement platforms. The samples were first split into luminal A, luminal B, triple-negative, and HER2+ subgroups on each platform independently. Perturbed functions (represented by KEGG pathways [38]) were then identified using the PWEA algorithm [31]. Pathway profiles were constructed thereafter as binary vectors with length equal to the number of pathways with each element set to either 1 or 0 to indicate whether the corresponding pathway is enriched. Finally, correlations between breast cancer subtypes and other diseases were calculated by comparing their pathway profiles using hypergeometric statistics. The detail of our results is described below according to three key factors that may impact the performance of our analysis: reproducibility, accuracy and resolution.
Figure 1.

The workflow of pathway profiling. The tumor samples of three data sets are classified into luminal A, luminal B, triple-negative, and HER2+ subtypes by clinical data and PAM50 classification results. We then perform pathway enrichment analysis for each subtype in each data set individually using the PWEA algorithm. After that we evaluate our method based on its reproducibility, accuracy, and resolution. Finally, we build a small disease/subtype network based on the pathway profiles of each of the four subtypes of breast cancer as well as related diseases.
Results
Breast cancer is a complex and heterogeneous disease [41] which consists of four major clinically-relevant subtypes: luminal A, luminal B, triple-negative, and HER2+, which vary in prognosis and require distinct treatments [5,42]. Gene signatures play an important role in the classification of breast cancer subtypes. However, a comparative study of five sets of gene signatures for breast cancer indicated that the overlap between gene signatures is still low [43]. Our goal therefore is to have a stable and accurate characterization of breast cancer subtypes using pathway profiles so that the common perturbed pathways among the four subtypes, as well as specific pathways uniquely perturbed for each subtype, will be appropriately identified.
The numbers of enriched pathways for each subtype in different data sets are shown in Table 1. It is clear that, on average, the luminal A subtype has fewer enriched pathways than the other three subtypes. All of the enriched pathways are identified based on a false discovery rate (FDR) cut-off of 0.1, calculated using the Benjamini-Hochberg method [44].
Table 1.
Number of enriched pathways for each subtype of breast cancer and data set
| Data set | Luminal A | Luminal B | Triple-negative | HER2+ |
|---|---|---|---|---|
| TCGA RNA-Seq |
66 |
84 |
55 |
85 |
| TCGA microarray |
48 |
66 |
65 |
69 |
| EGA microarray | 61 | 87 | 111 | 103 |
Reproducibility
Reproducibility is an essential requirement for almost all published scientific work. We focused on the reproducibility of our method over varying patient cohorts and platforms, with the intention to show that our result can be broadly applied in practice.
The reproducibility (See definition in Methods) of our method was evaluated using the gene expression data generated either from different expression measurement technology (Microarray vs. RNA-Seq), different sample sets (TCGA vs. EGA), or both. Only matched tumor samples (496) and normal samples (58) from each TCGA data set were used when examining the reproducibility between two technologies, and all 914 TCGA RNA-Seq samples (808 tumor and 106 normal) and 585 TCGA microarray samples (522 tumor and 63 normal) were used for the rest of the analysis.
Overall, about 67%, 71%, 67% and 76% average reproducibility were achieved (Figure 2) for luminal A, luminal B, triple-negative, and HER2+, respectively over different data sets. This result is consistent with a previous study [45] where RNA samples were analyzed separately on 4 different microarray platforms and the percentage of overlapped functional perturbation between any two platforms fell in the range of 57-70%. On the contrary, it has been shown that any pair of gene signatures developed for breast cancer share only a few common genes [43]. For example, there are only 17 overlapping genes in the two signatures sets [14] (with 456 and 231 genes respectively) developed for breast cancer survival-related prediction [6,18]. Similarly, only three common genes have been found [46] in two sets of genetic markers (each has about 70 genes) predicting the metastasis of breast cancer [7,19] with similar accuracy. To make a more intuitive comparison with the gene-based method, we calculated the reproducibility of the top 1500 DEGs between each pair of our data sets. As shown in Figure 2, the reproducibility of each enriched pathway set is clearly much higher than that of the top DEGs for all pairs of data sets. These results further verified that pathway profiling has a much better reproducibility than gene-based methods.
Figure 2.

Reproducibility of enriched pathways (the left side) and top DEGs (the right side) between each pair of data sets for each subtype of breast cancer. The FDR cut-off is set as 0.1 for enriched pathways. The top 1500 genes are used to calculate reproducibility for DEGs.
Reproducibility across technologies
Recently, RNA-Seq has become a powerful alternative to microarrays due to advantages such as high resolution, increased dynamic range, lower background noise, relatively little technical variation, and the ability to profile the entire transcriptome [47,48]. It is therefore expected that the TCGA RNA-Seq data set may provide more reliable results than other two data sets.
We focus on the matched samples between the TCGA RNA-Seq and TCGA microarray data sets to examine the effect of gene expression measurement technology on the reproducibility of our approach. The comparison of enriched pathways generated from the two data sets results in 60%, 73%, 65% and 70% reproducibility for the luminal A, luminal B, triple-negative, and HER2+ subtypes respectively, with about 67% on average. We also found that the reproducibility based on matched samples was slightly smaller than those based on all samples except in the case of luminal B: 65%, 70%, 66%, and 75% respectively, for luminal A, luminal B, triple-negative and HER2+ subtypes as shown as blue bars in the left side of Figure 2. This observation implies that increasing the number of samples may improve reproducibility [14,15]. Meanwhile, only 45%, 53%, 50%, and 52% reproducibility was found for the top 1500 DEGs for the corresponding subtypes with 50% on average. These results indicate that the functional profile-based method presents advantages over the gene-based method and can be used to directly compare results that are generated from data sets produced by different technologies.
Reproducibility across different data sets
We further investigate the reproducibility of our approach across different data sets. Among the three data sets used in the study, there is, as expected, the smallest difference between TCGA RNA-Seq and TCGA microarray data sets because 496 out of 522 samples from the TCGA microarray data are drawn from the same patient pool as the TCGA RNA-Seq data set, whereas the largest difference separated the TCGA RNA-Seq and EGA microarray data (different patients and different technologies). However, the corresponding average reproducibility over four subtypes is almost invariable with 69%, 68%, and 74% for the data set pairs TCGA RNA-Seq vs. TCGA microarray, TCGA microarray vs. EGA microarray, and TCGA RNA-Seq vs. EGA microarray respectively (left side of Figure 2). On the contrary, there is an obvious difference with regards to the reproducibility of the top 1500 DEGs between each dataset pair, and this is shown on the right side of Figure 2. For example, the reproducibility of top DEGs between the two TCGA data sets is always larger than the reproducibility of the other two data set pairs in all four subtypes. From this perspective, pathway profiles would appear to be better suited for comparative studies with different data sets that may be generated by different technologies.
Determination of the appropriate data set
As mentioned at the beginning of the previous section, RNA-Seq data is expected to generate more reliable results than two other data sets [47,48]. However, in general it is challenging to determine which pathway profiles are better due to the lack of a gold standard [40,49]. Here we adopt a simple strategy to address this issue with the assumption that the pathways predicted by multiple data sets should be more reliable than those predicated by only one data set. This assumption is partly evidenced by the observation that the pathways enriched in all three data sets generally have lower p-value while the pathways enriched in only one data set often have higher p-values (more discussion can be found in Additional files 1 and 4). From these perspectives, we first generated a reference pathway profile as the benchmark where a pathway is enriched only if it is enriched in more than one data set. We then calculated the reproducibility of pathway profiles in each data set against the reference pathway profiles for each subtype. As shown in Table 2, the pathway profile resulted from the TCGA RNA-Seq data set has the highest average reproducibility. As a result of these findings, the analyses in the remainder of this paper are performed against the RNA-Seq data set unless otherwise stated.
Table 2.
Reproducibility between pathway profiles (PPs) of each data set and reference pathway profiles (RPPs)
| Luminal A | Luminal B | Triple-negative | HER2+ | Average | |
|---|---|---|---|---|---|
| PPs of TCGA RNA-Seq data set vs. RPPs |
88% |
90% |
79% |
91% |
87% |
| PPs of TCGA microarray data set vs. RPPs |
76% |
80% |
89% |
85% |
83% |
| PPs of EGA microarray data set vs. RPPs | 86% | 86% | 79% | 88% | 85% |
Accuracy
The accuracy of the perturbed pathways identified in our analysis is measured by their biological relevance to breast or other cancers based on survey of relevant literature sources. About 74% of the enriched pathways (enriched in at least one subtype) are known to be associated with breast cancer or cancers such as those shown in Table 3 and 4.
Table 3.
Overlaps of common enriched pathways across three data sets
| Pathway | Pathway category | Reference a |
|---|---|---|
| Glycolysis/Gluconeogenesis |
Carbohydrate metabolism |
[82] |
| Sphingolipid metabolism |
Lipid metabolism |
[83] |
| Purine metabolism |
Nucleotide metabolism |
[84] |
| Pyrimidine metabolism |
Nucleotide metabolism |
[85] |
| Arginine and proline metabolism |
Amino acid metabolism |
[86] |
| Tyrosine metabolism |
Amino acid metabolism |
[87] |
| Phenylalanine metabolism |
Amino acid metabolism |
[88] |
| One carbon pool by folate |
Metabolism of cofactors and vitamins |
[89] |
| Fanconi anemia pathway |
Replication and repair |
[90] |
| PI3K-Akt signaling pathway |
Signal transduction |
[91] |
| Regulation of actin cytoskeleton |
Cell motility |
[92] |
| Focal adhesion |
Cell communication |
[93] |
| Adipocytokine signaling pathway |
Endocrine system |
[94] |
| Progesterone-mediated oocyte maturation |
Endocrine system |
- |
| Axon guidance | Development | [95] |
aReference shows association between a given pathway and breast cancer or other cancers.
Table 4.
HER2+ specific pathways
| Pathway | Pathway category | Reference a |
|---|---|---|
| Tryptophan metabolism |
Amino acid metabolism |
- |
| Terpenoid backbone biosynthesis |
Metabolism of terpenoids and polyketides |
- |
| Drug metabolism - cytochrome P450 |
Xenobiotics biodegradation and metabolism |
- |
| mTOR signaling pathway |
Signal transduction |
[63] |
| Serotonergic synapse |
Nervous system |
- |
| Long-term depression |
Nervous system |
- |
| Circadian rhythm | Environmental adaptation | [64] |
aReference shows association between a given pathway and the HER2+ subtype.
Common enriched pathways
As shown in Figure 3, there are 28 common enriched pathways (CEPs) across all four subtypes of breast cancer (See Additional file 5 for the whole list), which indicate the shared features of breast cancer. The CEPs occupy about 42%, 33%, 51%, and 33% of the total enriched pathways in luminal A, luminal B, triple-negative, and HER2+ subtypes, respectively. To test the reliability of CEPs, we first performed the enrichment analysis over all tumor samples without the separation of subtypes, and 86% of these CEPs are enriched in this case. We also checked enrichment analysis results in the TCGA and EGA microarray datasets, where we found that 24 of the total 28 CEPs (86%) are confirmed by at least one other data set and 15 of them are confirmed in both (Table 3). An extensive literature search indicates that 24 CEPs show relationships with either breast cancer or cancers (See Table 3 for details). These results indicate that despite the heterogeneity of breast cancer samples, different subtypes share a relatively high degree of similar molecular mechanisms to support tumor growth and metastatic dissemination. These CEPs might also be used as the “signature pathways” of breast cancer when compared to other diseases.
Figure 3.
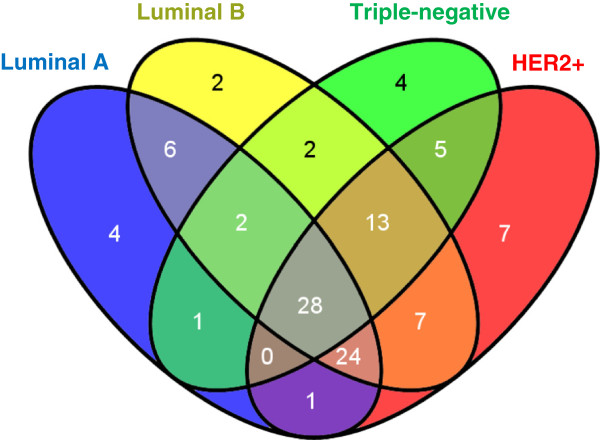
The Venn diagram of pathway enrichment analysis results from the RNA-Seq data. There are 15 regions in the Venn diagram including common enriched pathways and subtype-specific pathways. Common enriched pathways are pathways enriched in all four subtypes of breast cancer. Subtype specific pathways are pathways enriched in only one subtype of breast cancer.
Among 28 CEPs, 9 are relevant to metabolism such as Glycolysis/gluconeogenesis, Purine metabolism and Tyrosine metabolism. This is not surprising, as cancer cells must perturb metabolic pathways to provide energy and building blocks to support aggressive cell growth and proliferation [50]. Thus, these pathways may account for higher biosynthesis of nucleic acids (e.g., pathways in the category of nucleotide metabolism) and proteins (e.g., pathways in the category of amino acid metabolism) and higher energy demands (e.g., pathways in the category of carbohydrate metabolism) of tumors.
We also noted that CEPs consist of some well-known cancer related pathways such as pathways in the categories of replication and repair, signal transduction, cell growth and death. For example, the CEP “Cell cycle” appears to be in agreement with the intuition that inappropriate proliferation is one of the most remarkable characteristics of the cancer cell. This is expected and can be considered as a benchmark result for our method, as several other pathway-based studies in cancer have observed similar results [51-53]. Additionally, the CEPs also contain the PI3K-Akt signaling pathway. It has been shown that many breast cancer tumors harbor mutations in the PI3K-Akt signaling pathway [35]. These mutations are thought to lead to activation of RHEB, which in turn promotes activation of the mTOR gene [54], one downstream effect of which is vastly increased protein production and much larger cancer cells. The notch signaling pathway, on the other hand, has attracted increasing attention as the potential new therapeutic targets for cancer patients [55], and our results indicate that it may also be applicable to the breast cancer.
Two hormonally-related pathways are observed to be enriched in CEPs as well, including Progesterone-mediated oocyte maturation and the Estrogen signaling pathway. As expected, we observe that Estrogen signaling is significantly upregulated in ER+ tumors (luminal A and luminal B) and significantly downregulated in triple-negative tumors. Though HER2+ tumors are not known to be directly enriched in estrogen-related genes, nevertheless we find that the pathway is still significantly dysregulated in HER2+ tumors. It has been suggested that ER+/HER2+ tumors utilize crosstalk between the MAPK signaling pathway and the ER signaling pathway to evade common anti-estrogenic therapies such as Tamoxifen [56].
Subtype-specific pathways
Subtype-specific pathways are especially interesting, since they are potential candidates for signature pathways for each subtype of breast cancer. As shown in Figure 3, luminal A, luminal B, triple-negative, and HER2+ subtypes have 4, 2, 4, and 7 subtype-specific pathways (about 6%, 2%, 7%, and 8% of their enriched pathways) respectively. These pathways represent the heterogeneity of breast cancer subsets.
Luminal A specific pathways
Luminal A has 4 subtype-specific pathways: Ubiquitin mediated proteolysis, Endocytosis, Carbohydrate digestion and absorption, and Vasopressin-regulated water reabsorption. The enrichment of Ubiquitin mediated proteolysis pathway is evidenced by the recent work [57] where it is found that the luminal A subtype had an increased expression level of cyclin D1 which regulates proteolysis mediated by ubiquitin [58].
There are two interesting pathways that are not enriched in luminal A subtype in comparison to the other three subtypes: the Homologous recombination and the p53 signaling pathways. This observation is consistent among all three data sets and may therefore be used to aid in the distinguishability of the luminal A subtype. This observation is also supported in the literature where it has been reported that a functional defect in homologous recombination is common in triple-negative breast cancer and in a subset of high grade ER and/or HER2 positive breast cancer [59]. Homologous recombination may also be associated with the luminal B subtype through the BRCA2 gene which is known to be involved in error-free DNA repair of double-strand breaks (DSBs) through homologous recombination and BRCA2 mutation carriers have a predilection for developing breast cancers of the luminal B subtype [60]. On the other hand, the luminal A subtype has the lowest mutation frequency among the four subtypes of breast cancer [35], therefore it is not surprising that the p53 signaling pathway is not enriched in the luminal A subtype.
Luminal B specific pathways
The luminal B subtype has only two subtype-specific pathways: Histidine metabolism and Phosphatidylinositol signaling systems, although it has many more enriched pathways than the luminal A subtype. It should also be noted that pathways enriched by the luminal B subtype have much more overlaps with those found enriched in the triple-negative and HER2+ subtypes than in the luminal A subtype (Figure 3).
Since ER status is a very important factor in planning breast cancer treatment, we also outline the pathways that are specific to the ER+ subtype (luminal A and luminal B). Among 6 ER+ specific pathways, four of them have supporting evidence from previous studies: the Primary bile acid biosynthesis, Jak-STAT signaling pathway, Complement and coagulation cascades, and GnRH signaling pathways (More details can be found in Additional file 1).
Triple-negative specific pathways
The triple-negative subtype has 4 subtype-specific pathways: Alanine, aspartate and Glutamate metabolism, Lysine degradation, Vascular smooth muscle contraction, and Glutamatergic synapse. Two of these have supporting evidence in previous studies. Metabotropic glutamate receptor-1 (GRM1) has been reported as an oncogene in the progression of triple negative breast cancer [61], whose alteration may affect the pathway Alanine, aspartate and glutamate metabolism. It has been observed that the amino acid metabolism is also a major source of energy and carbon for tumor cell growth and survival in invasive breast cancer such as the triple-negative subtype [62]. Thus, the perturbation of the Lysine degradation pathway is very likely resulted from the changes in the energy metabolism of tumor cells.
HER2+ specific pathway
The HER2+ subtype has the largest number of subtype-specific pathways among the four subtypes as shown in Table 4, which may be due to the important role of HER2 in promoting cell growth and proliferation. Among 7 HER2+ specific pathways, mTOR signaling pathway and Circadian rhythm have supporting evidence from previous studies. For example, it was pointed out that constitutively activating HER2 and EGFR stimulated many of the same intracellular signaling proteins and pathways as wild type receptors, including the mTOR pathway [63]. Additionally, the deregulated expression of the circadian related genes PER1, PER2 and PER3 in breast cancers has been studied [64]. It was found that methylation of the PER gene promoters has a strong correlation with c-erbB2 expression.
Resolution
The resolution of our approach aims to test whether the pathway profiles have enough detail not only to distinguish the different diseases but also to correctly assess the correlations between them based on the perturbed functions. To achieve this, we evaluate the disease correlation by calculating the hypergeometric probability of corresponding pathway profiles: a method that has been successfully applied to calculate the correlation between phylogenetic profiles in our previous study [65]. We further generated pathway profiles for ovarian cancer (TCGA, Affymetrix HG-U133A platform), GBM (TCGA, Affymetrix HG-U133A platform) and obesity (GEO, GDS3688, HG-U133_Plus_2 platform) in order to compare breast cancer against other diseases.
The resulting profiles are drawn in Figure 4A using Gitools [66] with 11, 85, and 14 enriched pathways for ovarian cancer, GBM, and obesity respectively according to PWEA results (See Additional file 6). Figure 4B shows the correlations between seven diseases/subtypes as a small network drawn by VisANT [67-71] where 4 subtypes are encapsulated by the metanode [72-74] of breast cancer and the edge thickness is roughly proportional to correlation strength. With a 0.05 p-value cutoff, a total of 17 disease pairs exhibit significant correlation with varied strength (See Additional file 7). As expected, most of the correlations between four subtypes are stronger than those between subtypes and the other three diseases. Among the four subtypes, luminal A and luminal B subtypes are closer to each other than to the other two subtypes. On the other hand, the luminal A and luminal B subtypes are closer to the HER2+ subtype than to the triple-negative subtype, which may not be surprising because the HER2+ subtype is simply characterized by copy number variation in the HER2 amplicon, while it has been suspected that ER+ (luminal A and luminal B) and ER- subtypes may have differing cells of origin [75]. Similarly, both triple-negative and HER2+ subtypes have stronger correlation to ovarian cancer than luminal A and luminal B subtypes have, most likely because of the effect of P53 gene [35]. Meanwhile, the fact that luminal B, triple-negative, and HER2+ subtypes are more aggressive tumors than the luminal A subtype is reflected by the observation that both triple-negative and HER2+ subtypes are closer to the luminal B subtype than the luminal A subtype. We also find the luminal A subtype is closer to obesity than to ovarian cancer, indicating the potential connection between breast cancer and obesity through estrogen [76]. Interestingly, our results show that GBM is significantly correlated with luminal A, luminal B, HER2+ breast cancer and obesity. It was pointed out that sex hormones are important in the growth of breast cancer and are also important in the development of GBM [77]. Furthermore, the anti-estrogen drug Tamoxifen has been found to be effective in decreasing glioblastoma cell proliferation [78]. On the other hand HER2 is the most frequently expressed tyrosine kinase receptors in GBM cells [79]. The association between GBM and obesity may be explained by the important role of leptin in both of these two diseases. The current model suggests that obesity in human is due to a desensitization to leptin while within gliomas, there is a correlation between tumor grade and tumor expression of leptin and its receptor [80].
Figure 4.

Evaluate disease correlations by using pathway profiles. (A) Pathway profiles for luminal A, luminal B, HER2+, and triple-negative subtypes of breast cancer, as well as ovarian cancer, glioblastoma multiforme (GBM), and obesity. Each red rectangle represents an enriched pathway. From right to left, KEGG pathways are sorted according to pathway categories and have the same order as the FDR values of enriched pathways in these 7 diseases/subtypes (Additional file 6). Due to limitation of space, here only pathway categories are labeled. This figure roughly shows that luminal A and luminal B subtypes are closer to the HER2+ subtype than the triple-negative subtype. (B) Disease/subtype correlation networks based on pathway profile. Each node represents one disease and subtypes are embedded in a metanode representing breast cancer [72-74]. The width of each edge is roughly proportional to the correlation between the disease/subtype pair. Here only the significant correlations (p-value < 0.05) are displayed (See Additional file 7). The network is drawn by the VisANT system [67-71].
Discussion
All the enriched pathways in this study are identified based on a FDR cut-off 0.1. This cut-off was optimized to achieve reasonable reproducibility (Figure 5) while maintaining adequate coverage and accuracy. More detail on the FDR cut-off is addressed in Additional file 1. Another important factor that needed to be taken into account is the coverage of KEGG pathway genes with the corresponding gene expression measurement technology. All three data sets used in this study have a good coverage of KEGG pathway genes (TCGA RNA-Seq, TCGA microarray and EGA microarray data sets contain about 20360, 17814 and 17621 genes, corresponding to the 97%, 91% and 90% coverage of 5584 total KEGG pathways genes, respectively), which however may not hold for the large sets of microarray array data available in GEO databases. More discussion of the pathway coverage can also be found in Additional file 1.
Figure 5.
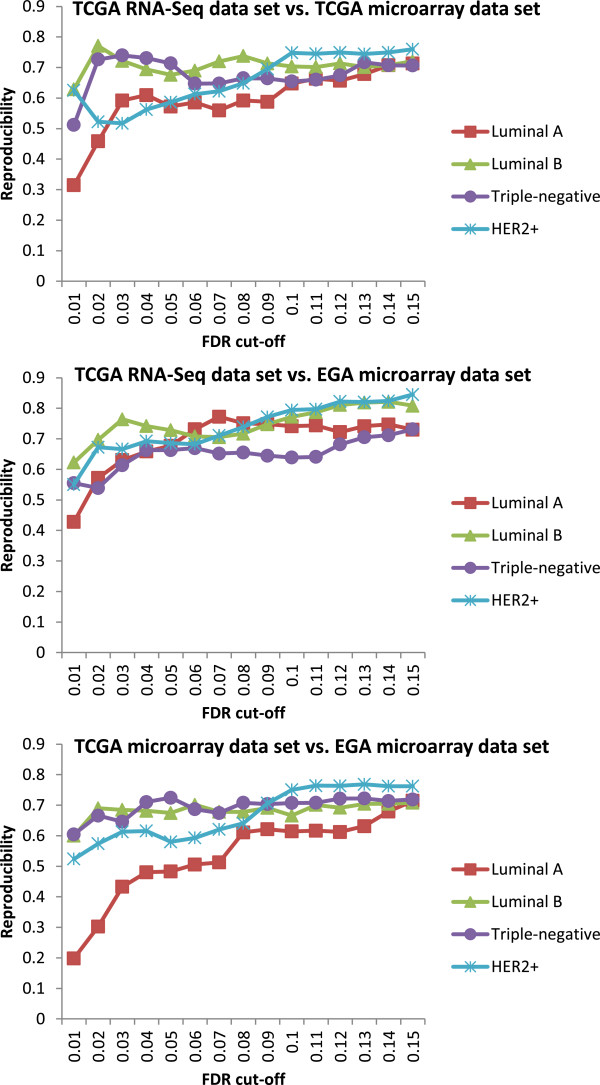
Reproducibility of enriched pathways between different data sets/platforms vs. FDR cut-off for each subtype of breast cancer. In general, the reproducibility increases quickly at the beginning and gradually becomes flat as FDR cut-off increases.
The comparison of the reproducibility resulted from the pathway profiles and top DEGs are in general conservative. From a hypergeometric statistics perspective, a larger number of genes resulted from the different analyses will achieve better reproducibility. We therefore used the top 1500 DEGs in the reproducibility comparison instead of known signature genes available in the literature because the number of the latter is generally much smaller than 1500. In addition, we also performed the comparison using the top 6000 DEGs and the enriched pathways still achieves better reproducibility (See details in Additional file 1). The robustness of our approach is further verified by using the EGA validation data set [36]. There is little change in the reproducibility between two TCGA data sets and the EGA data set when substituting the EGA discovery set with the validation set (in Additional file 1 Figure S5 where more discussion of the EGA validation set can also be found).
Despite the improvement over classical gene-based methods in characterizing breast cancer and related diseases, this approach has great potential to be further enhanced. First, it depends on the prior knowledge of pathways which are far from complete, and identifying new pathways is a difficult, time-consuming, and labor intensive work. An alternative approach to partially ameliorate this limit is to replace pathways with functional modules that may be computationally identified [81]. Second, current pathway profiles are represented as a simply binary vector that may be improved by incorporating some additional information such as the corresponding statistical significance of the enriched pathway to make each profile more quantitative. For a simple approach, one may directly set elements of pathway profile as the p-values or enrichment scores of given pathways.
Conclusion
Reproducibility is one of the main challenges for the identification of gene signatures. Besides technical factors, the disparity may result from the fact that diseases are directly caused by the perturbations of the molecular function that are generally carried out by a set of genes in the form of modules or pathways. Therefore it may be more appropriate to characterize diseases at the functional level than at the gene level. Following this perspective, we developed a novel approach to characterize diseases using the pathway profiles and evaluated the approach based on profiles’ reproducibility, accuracy and resolution. Using four subtypes of breast cancer as an example, the results of this new approach are promising with 70% average reproducibility, 74% average accuracy (e.g., references in Table 3[82-95]) and reasonable resolution to identify the correlations between not only different diseases, but also their subtypes.
Abbreviations
TCGA: The Cancer Genome Atlas; EGA: The European Genome-phenome Archive; GBM: Glioblastoma multiforme; DEGs: Differentially expressed genes; FDR: False discovery rate; PPs: Pathway profiles; RPPs: Reference pathway profiles; CEP: Common enriched pathway.
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
FT and YW designed the method and experiments. FT carried out the computation and both FT and YW performed the analysis. FT drafted the manuscript. MS normalized TCGA breast cancer RNA-Seq data, provided constructive advice, and revised the manuscript. ZH supervised the project, gave suggestions, and revised the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Supplementary Material
Supplementary materials on determination of ER, PR, and HER2 status, differentially expressed genes, determination of FDR cut-off, coverage of KEGG pathway genes, comparison between consistent and inconsistent pathways, EGA validation data set, and ER+ specific pathways.
Lists of samples for three data sets and four subtypes of breast cancer.
PWEA results of all used data sets.
Lists of consistent and inconsistent pathways across three data sets for four subtypes of breast cancer.
Lists of CEPs and subtype specific pathways.
FDR of enriched pathways for pathway profiles shown in Figure 4A.
p -values for subtype/disease correlations shown in Figure 4B.
Contributor Information
Feng Tian, Email: fengtian@bu.edu.
Yajie Wang, Email: tiantanwyj@aliyun.com.
Michael Seiler, Email: miseiler@gmail.com.
Zhenjun Hu, Email: zhenjun.hu@gmail.com.
Acknowledgements
This work is supported by the National Institutes of Health (R01GM103502-05).
References
- Loscalzo J, Kohane I, Barabasi AL. Human disease classification in the postgenomic era: a complex systems approach to human pathobiology. Mol Syst Biol. 2007;7:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte AJ. Medicine. The ultimate model organism. Science. 2008;7:325–327. doi: 10.1126/science.1158343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itadani H, Mizuarai S, Kotani H. Can systems biology understand pathway activation? Gene expression signatures as surrogate markers for understanding the complexity of pathway activation. Curr Genomics. 2008;7:349–360. doi: 10.2174/138920208785133235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JG, Campen A, Huang SG, Peng SB, Ye X, Palakal M, Dunker AK, Xia YN, Li SY. Identification of a gene signature in cell cycle pathway for breast cancer prognosis using gene expression profiling data. BMC Med Genet. 2008;7:39. doi: 10.1186/1755-8794-1-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;7:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;7:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van ’t Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;7:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Bandres E, Malumbres R, Cubedo E, Honorato B, Zarate R, Labarga A, Gabisu U, Sola JJ, Garcia-Foncillas J. A gene signature of 8 genes could identify the risk of recurrence and progression in Dukes’ B colon cancer patients. Oncol Rep. 2007;7:1089–1094. [PubMed] [Google Scholar]
- Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, Chan WK, Li HN, Liu CC, Singh S, Chen WJ, Chen JJ, Yang PC. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;7:11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007;7:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- Okahara S, Arimura Y, Yabana T, Kobayashi K, Gotoh A, Motoya S, Imamura A, Endo T, Imai K. Inflammatory gene signature in ulcerative colitis with cDNA macroarray analysis. Aliment Pharmacol Ther. 2005;7:1091–1097. doi: 10.1111/j.1365-2036.2005.02443.x. [DOI] [PubMed] [Google Scholar]
- Koscielny S. Why most gene expression signatures of tumors have not been useful in the clinic. Sci Transl Med. 2010;7:14ps12. doi: 10.1126/scitranslmed.3000313. [DOI] [PubMed] [Google Scholar]
- Dowsett M, Sestak I, Lopez-Knowles E, Sidhu K, Dunbier AK, Cowens JW, Ferree S, Storhoff J, Schaper C, Cuzick J. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol. 2013;7:2783–2790. doi: 10.1200/JCO.2012.46.1558. [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;7:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci U S A. 2006;7:5923–5928. doi: 10.1073/pnas.0601231103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Eklund AC, Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006;7:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;7:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- van’t Veer LJ, Dai HY, van de Vijver MJ, He YDD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;7:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;7:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Drier Y, Domany E. Do two machine-learning based prognostic signatures for breast cancer capture the same biological processes? Plos One. 2011;7(3):e17795. doi: 10.1371/journal.pone.0017795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti M, Brunner HG. The modular nature of genetic diseases. Clin Genet. 2007;7:1–11. doi: 10.1111/j.1399-0004.2006.00708.x. [DOI] [PubMed] [Google Scholar]
- Gonen M. Statistical aspects of gene signatures and molecular targets. Gastrointest Cancer Res. 2009;7:S19–S21. [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Baurley JW, Brown EE, Figueiredo JC, Goldstein A, Hazra A, Wilson RT, Rothman N. Approaches to complex pathways in molecular epidemiology: summary of a Special Conference of the American Association for Cancer Research. Cancer Res. 2008;7:10028–10030. doi: 10.1158/0008-5472.CAN-08-1690. [DOI] [PubMed] [Google Scholar]
- Chin L, Hahn WC, Getz G, Meyerson M. Making sense of cancer genomic data. Genes Dev. 2011;7:534–555. doi: 10.1101/gad.2017311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;7:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A. A module map showing conditional activity of expression modules in cancer. Nat Genet. 2004;7:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang QL, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Marks JR, Dressman HK, West M, Nevins JR. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;7:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Emmert-Streib F, Glazko GV. Pathway Analysis of expression data: deciphering functional building blocks of complex diseases. Plos Comput Biol. 2011;7:e1002053. doi: 10.1371/journal.pcbi.1002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary MP, Bader GD, Sander C. Pathway information for systems biology. FEBS Letters. 2005;7:1815–1820. doi: 10.1016/j.febslet.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kim S, Kon M, DeLisi C. Pathway-based classification of cancer subtypes. Biol Direct. 2012;7:21. doi: 10.1186/1745-6150-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung JH, Whitfield TW, Yang TH, Hu Z, Weng Z, DeLisi C. Identification of functional modules that correlate with phenotypic difference: the influence of network topology. Genome Biol. 2010;7:R23. doi: 10.1186/gb-2010-11-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;7:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu JC, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010;7:i237–i245. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proc Natl Acad Sci U S A. 2013;7:6388–6393. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK, Ally A, Balasundaram M, Butterfield YSN, Carlsen R, Carter C, Chu A, Chuah E, Chun HJE, Coope RJN, Dhalla N, Guin R, Hirst C, Hirst M, Holt RA, Lee D, Li HYI, Mayo M, Moore RA, Mungall AJ. et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;7:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen-Dale AL, Brenton JD, Tavare S, Caldas C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;7:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, Huang M, Huntsman DG, Iacocca M, Imielinski M, Kalloger S, Karlan BY, Levine DA, Mills GB, Morrison C, Mutch D, Olvera N, Orsulic S, Park K, Petrelli N, Rabeno B, Rader JS, Sikic BI. et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;7:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;7:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Craft B, Swatloski T, Goldman M, Ma S, Haussler D, Zhu J. Exploring TCGA Pan-Cancer data at the UCSC Cancer Genomics Browser. Sci Rep. 2013;7:2652. doi: 10.1038/srep02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung JH, Yang TH, Hu ZJ, Weng ZP, DeLisi C. Gene set enrichment analysis: performance evaluation and usage guidelines. Brief Bioinform. 2012;7:281–291. doi: 10.1093/bib/bbr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexe G, Dalgin GS, Scanfeld D, Tamayo P, Mesirov JP, DeLisi C, Harris L, Barnard N, Martel M, Levine AJ, Ganesan S, Bhanot G. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res. 2007;7:10669–10676. doi: 10.1158/0008-5472.CAN-07-0539. [DOI] [PubMed] [Google Scholar]
- Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;7:698–707. doi: 10.1016/j.ctrv.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DSA, Nobel AB, van’t Veer LJ, Perou CM. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;7:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Ser B-Methodological. 1995;7:289–300. [Google Scholar]
- Li ZG, Su ZQ, Wen ZN, Shi LM, Chen T. Microarray platform consistency is revealed by biologically functional analysis of gene expression profiles. BMC Bioinformatics. 2009;7(Suppl 11):S12. doi: 10.1186/1471-2105-10-S11-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol. 2007;7:140. doi: 10.1038/msb4100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;7:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;7:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Sirota M, Butte AJ. Ten Years of pathway analysis: current approaches and outstanding challenges. Plos Comput Biol. 2012;7:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012;7:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernadez V, Hartmann E, Ott G, Campo E, Rosenwald A. Pathogenesis of mantle-cell lymphoma: all oncogenic roads lead to dysregulation of cell cycle and DNA damage response pathways. J Clin Oncol. 2005;7:6364–6369. doi: 10.1200/JCO.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH. et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;7:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham L, Christadore L, Schaus S, Kolaczyk ED. Network-based prediction for sources of transcriptional dysregulation using latent pathway identification analysis. Proc Natl Acad Sci U S A. 2011;7:13347–13352. doi: 10.1073/pnas.1100891108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;7:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Al-Hussaini H, Subramanyam D, Reedijk M, Sridhar SS. Notch signaling pathway as a therapeutic target in breast cancer. Mol Cancer Ther. 2011;7:9–15. doi: 10.1158/1535-7163.MCT-10-0677. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005;7:865s–870s. [PubMed] [Google Scholar]
- Yu ZR, Wang LP, Wang CG, Ju XM, Wang M, Chen K, Loro E, Li ZP, Zhang YZ, Wu KM, Casimiro MC, Gormley M, Ertel A, Fortina P, Chen YH, Tozeren A, Liu ZM, Pestell RG. Cyclin D1 induction of Dicer governs microRNA processing and expression in breast cancer. Nat Commun. 2013;7:2812. doi: 10.1038/ncomms3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontano LL, Aggarwal P, Barbash O, Brown EJ, Bassing CH, Diehl JA. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol. 2008;7:7245–7258. doi: 10.1128/MCB.01085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis JS, Dowsett M, Ashworth A, Turner NC. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res. 2010;7:6159–6168. doi: 10.1158/1078-0432.CCR-10-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson OA, Jonasson JG, Olafsdottir K, Bjarnason H, Th Johannsson O, Bodvarsdottir SK, Valgeirsdottir S, Eyfjord JE. Genomic and phenotypic analysis of BRCA2 mutated breast cancers reveals co-occurring changes linked to progression. Breast Cancer Res. 2011;7:R95. doi: 10.1186/bcr3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda M, Speyer C, Gorski D. Metabotropic glutamate receptor-1 is oncogenic in triple-negative breast cancer. Ann Surg Oncol. 2012;7:23–23. [Google Scholar]
- Budczies J, Denkert C, Muller BM, Brockmoller SF, Klauschen F, Gyorffy B, Dietel M, Richter-Ehrenstein C, Marten U, Salek RM, Griffin JL, Hilvo M, Oresic M, Wohlgemuth G, Fiehn O. Remodeling of central metabolism in invasive breast cancer compared to normal breast tissue - a GC-TOFMS based metabolomics study. BMC Genomics. 2012;7:334. doi: 10.1186/1471-2164-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;7:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ, Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis. 2005;7:1241–1246. doi: 10.1093/carcin/bgi075. [DOI] [PubMed] [Google Scholar]
- Wu J, Hu Z, DeLisi C. Gene annotation and network inference by phylogenetic profiling. BMC Bioinformatics. 2006;7:80. doi: 10.1186/1471-2105-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Llamas C, Lopez-Bigas N. Gitools: analysis and visualisation of genomic data using interactive heat-maps. Plos One. 2011;7:e19541. doi: 10.1371/journal.pone.0019541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, DeLisi C. Analyzing networks with VisANT. Curr Protoc Bioinformatics. 2004;7 doi: 10.1002/0471250953.bi0808s08. Chapter 8:Unit 8.8.1-24. [DOI] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, DeLisi C. VisANT: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;7:17. doi: 10.1186/1471-2105-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Ng DM, Yamada T, Chen C, Kawashima S, Mellor J, Linghu B, Kanehisa M, Stuart JM, DeLisi C. VisANT 3.0: new modules for pathway visualization, editing, prediction and construction. Nucleic Acids Res. 2007;7:W625–W632. doi: 10.1093/nar/gkm295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Snitkin ES, DeLisi C. VisANT: an integrative framework for networks in systems biology. Brief Bioinform. 2008;7:317–325. doi: 10.1093/bib/bbn020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Chang YC, Wang Y, Huang CL, Liu Y, Tian F, Granger B, Delisi C. VisANT 4.0: Integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res. 2013;7:W225–W231. doi: 10.1093/nar/gkt401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, Yamada T, Holloway D, Delisi C. VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res. 2005;7:W352–W357. doi: 10.1093/nar/gki431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, Kanehisa M, Stuart JM, DeLisi C. Towards zoomable multidimensional maps of the cell. Nat Biotechnol. 2007;7:547–554. doi: 10.1038/nbt1304. [DOI] [PubMed] [Google Scholar]
- Hu Z, Hung JH, Wang Y, Chang YC, Huang CL, Huyck M, DeLisi C. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res. 2009;7:W115–W121. doi: 10.1093/nar/gkp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;7:3155–3163. doi: 10.1172/JCI33295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary MP, Grossmann ME. Minireview: obesity and breast cancer: the estrogen connection. Endocrinology. 2009;7:2537–2542. doi: 10.1210/en.2009-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirilli M, Salvati M, Bistazzoni S, Frati A, Brogna C, Giangaspero F, Frati R, Santoro A. Glioblastoma multiforme and breast cancer: report on 11 cases and clinico-pathological remarks. Tumori. 2005;7:256–260. doi: 10.1177/030089160509100309. [DOI] [PubMed] [Google Scholar]
- Leslie KK, Keefe D, Powell S, Naftolin F. Estrogen receptors are identified in the glioblastoma cell line U138MG. J Soc Gynecol Investig. 1994;7:238–244. doi: 10.1177/107155769400100311. [DOI] [PubMed] [Google Scholar]
- Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, Berthou C, Dam Hieu P. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J Neurooncol. 2007;7:281–287. doi: 10.1007/s11060-007-9424-1. [DOI] [PubMed] [Google Scholar]
- Lawrence JE, Cook NJ, Rovin RA, Winn RJ. Leptin promotes glioblastoma. Neurol Res Int. 2012;7:870807. doi: 10.1155/2012/870807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shamir R. Identification of functional modules using network topology and high-throughput data. BMC Syst Biol. 2007;7:8. doi: 10.1186/1752-0509-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;7:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grosch S, Geisslinger G, Holtrich U, Karn T, Kaufmann M. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;7:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Schramm G, Surmann EM, Wiesberg S, Oswald M, Reinelt G, Eils R, Konig R. Analyzing the regulation of metabolic pathways in human breast cancer. BMC Med Genomics. 2010;7:39. doi: 10.1186/1755-8794-3-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miecznikowski JC, Wang D, Liu S, Sucheston L, Gold D. Comparative survival analysis of breast cancer microarray studies identifies important prognostic genetic pathways. BMC Cancer. 2010;7:573. doi: 10.1186/1471-2407-10-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Pervin S, Karimi A, Cederbaum S, Chaudhuri G. Arginase activity in human breast cancer cell lines: N (omega)-hydroxy-L-arginine selectively inhibits cell proliferation and induces apoptosis in MDA-MB-468 cells. Cancer Res. 2000;7:3305–3312. [PubMed] [Google Scholar]
- Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie JX, Ge QY, Gu TL, Polakiewicz RD, Roesel JL, Chen GZ, Boggon TJ, Lonial S, Fu HA, Khuri FR, Kang SM, Chen J. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell. 2011;7:864–877. doi: 10.1016/j.molcel.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong MY, McDunn J, Kakar SS. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PLoS One. 2011;7:e19963. doi: 10.1371/journal.pone.0019963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Gammon MD, Chan W, Palomeque C, Wetmur JG, Kabat GC, Teitelbaum SL, Britton JA, Terry MB, Neugut AI, Santella RM. One-carbon metabolism, MTHFR polymorphisms, and risk of breast cancer. Cancer Res. 2005;7:1606–1614. doi: 10.1158/0008-5472.CAN-04-2630. [DOI] [PubMed] [Google Scholar]
- Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC Biochem. 2007;7(Suppl 1):S10. doi: 10.1186/1471-2091-8-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;7:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim BiophysActa. 2007;7:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya V, Beviglia L, Xu LH, Earp HS 3rd, Craven R, Cance W. Dual inhibition of focal adhesion kinase and epidermal growth factor receptor pathways cooperatively induces death receptor-mediated apoptosis in human breast cancer cells. J Biol Chem. 2002;7:38978–38987. doi: 10.1074/jbc.M205002200. [DOI] [PubMed] [Google Scholar]
- Housa D, Housova J, Vernerova Z, Haluzik M. Adipocytokines and cancer. Physiol Res. 2006;7:233–244. doi: 10.33549/physiolres.930848. [DOI] [PubMed] [Google Scholar]
- Chedotal A, Kerjan G, Moreau-Fauvarque C. The brain within the tumor: new roles for axon guidance molecules in cancers. Cell Death Differ. 2005;7:1044–1056. doi: 10.1038/sj.cdd.4401707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials on determination of ER, PR, and HER2 status, differentially expressed genes, determination of FDR cut-off, coverage of KEGG pathway genes, comparison between consistent and inconsistent pathways, EGA validation data set, and ER+ specific pathways.
Lists of samples for three data sets and four subtypes of breast cancer.
PWEA results of all used data sets.
Lists of consistent and inconsistent pathways across three data sets for four subtypes of breast cancer.
Lists of CEPs and subtype specific pathways.
FDR of enriched pathways for pathway profiles shown in Figure 4A.
p -values for subtype/disease correlations shown in Figure 4B.