

Abstract
Control over ionic composition and volume of the inner ear luminal fluid endolymph is essential for normal hearing and balance. Mice deficient in either the EphB2 receptor tyrosine kinase or the cognate transmembrane ligand ephrin-B2 (Efnb2) exhibit background strain-specific vestibular behavioral dysfunction and signs of abnormal endolymph homeostasis. Using various loss-of-function mouse models, we found that Efnb2 is required for growth and morphogenesis of the embryonic endolymphatic epithelium, a precursor of the endolymphatic sac (ES) and duct (ED), which mediate endolymph homeostasis. Conditional inactivation of Efnb2 in early-stage embryonic ear tissues disrupted cell proliferation, cell survival, and epithelial folding at the origin of the endolymphatic epithelium. This correlated with apparent absence of an ED, mis-localization of ES ion transport cells relative to inner ear sensory organs, dysplasia of the endolymph fluid space, and abnormally formed otoconia (extracellular calcite protein composites) at later stages of embryonic development. A comparison of Efnb2 and Notch signaling deficient mutant phenotypes indicated that these two signaling systems have distinct and non overlapping roles in ES/ED development. Homozygous deletion of the Efnb2 C terminus caused abnormalities similar to those found in the conditional Efnb2 null homozygote. Analyses of fetal Efnb2 C-terminus deletion heterozygotes found mis-localized ES ion transport cells only in the genetic background exhibiting vestibular dysfunction. We propose that developmental dysplasias described here are a gene dose sensitive cause of the vestibular dysfunction observed in EphB-Efnb2 signaling-deficient mice.
Keywords: Ephrin-B2, EphB2, Eph-ephrin, Foxi1, Notch, Pendrin, Slc26a4, Proton-translocating, ATPase, Dlx5, Gbx2, Signalling, Morphogenesis, Proliferation, Growth, Mouse, Embryo, Cre-mediated gene inactivation, Inner ear, Otocyst, Vestibular, Deafness, Endolymph, Otoconia, Fluid homeostasis, Endolymphatic sac, Endolymphatic duct
Introduction
The inner ear comprises a fluid-filled epithelium within a labyrinthine space of the temporal bone. A prevalent inner ear malformation is the Enlarged Vestibular Aqueduct (EVA; OMIM 600791). EVA is diagnosed by radiological identification of a dilated bony canal for the endolymphatic duct (ED) [1]. The ED is a soft tissue structure joining an absorptive/secretory transport epithelium, the endolymphatic sac (ES) [2], to the rest of the inner ear epithelium. Dilation of ES and ED epithelia is thought to cause the bony malformation pathognomonic for EVA. EVA is associated with a distinctive clinical phenotype involving progressive hearing loss of early onset and, less frequently, vestibular dysfunction [reviewed in 3-5].
Roughly one-half of EVA patients carry one or two mutant alleles of the SLC26A4 gene [3-5], which encodes the protein pendrin, an electroneutral exchanger of anions such as HCO3−, Cl−, and I− [6]. Studies of the ear in targeted mouse mutants that lack functional pendrin have uncovered a pathological cascade involving dilation of the fluid-filled ear epithelium, altered ionic composition of the luminal fluid endolymph, malformation of acellular matrix structures (otoconia), changes in protein expression, metabolic stress, and post-natal death of the ear's primary sensory (hair) cells [7-12]. Conditional genetic and gene replacement strategies in the mouse indicate that these phenotypes result from loss of pendrin at the ES during late embryonic stages [13,14]. Furthermore, mice lacking Foxi1, a forkhead transcription factor that activates Slc26a4 transcription at the ES but not at other ear epithelia, have an inner ear phenotype similar to that of Slc26a4−/− mice [15]. These and other results [16-18] indicate that mammalian ear development requires ion transport at the prenatal ES.
The ES and ED derive from the primitive ear (otic) epithelium, which is initially patterned by extrinsic signals from neighboring embryonic structures. Reverse genetic and embryological studies define a genetic hierarchy for the early stage specification or growth of an ES/ED rudiment from the otic epithelium: rhombomere patterning by Hoxa1 and Mafb/kr positions a hindbrain source of Fibrobast Growth Factor (Fgf3) signaling, which maintains Wnt-dependent expression of the homeobox-containing gene Gbx2 in the otic epithelium [19-26]. Mice lacking any of these genes lack an ES/ED epithelium and have a dilated, dysmorphic inner ear. Gbx2 is required for proper otic expression of the homeobox-containing gene, Dlx5 [26], and mice lacking Dlx5 show moderately impaired growth of the ES/ED [27-30]. These and other [31] results offer insight into early ES/ED growth, but the question of how the endolymphatic epithelium differentiates into a sac and duct with apparently distinct physiological roles has not been addressed.
Eph receptor tyrosine kinases and their transmembrane ephrin-B ligands comprise a group of cell contact mediated signaling molecules [32]. Eph-ephrin signaling is bidirectional (transduction occurs in both receptor- and ligand-bearing cells) [33] and pleiotropic in its developmental roles. Eph-ephrin signaling mediates axon guidance and topographic mapping, cell migration and proliferation, tissue boundary formation and segmentation, and epithelial morphogenesis [reviewed in 34-37]. Eph-Ephrin signaling also influences synaptogenesis and synaptic plasticity through biochemical effects on ligand-gated ion channel activity [reviewed in 38,39] Mice heterozygous for deletion of the ephrin-B2 (Efnb2) C-terminus or homozygous for Ephb2 mutant alleles show background strain-specific, incompletely penetrant vestibular-behavioral dysfunction and altered ionic composition of endolymph fluid [40,41]. Efnb2 C-terminal deletion heterozygotes have mildly elevated auditory brainstem response thresholds [42]. Biochemical evidence indicates that EphB2 and ephrinB C-termini recognize cytoplasmic domains of anion exchangers and aquaporins through PDZ domain containing proteins [40]. Ephb2 and Efnb2 mRNA or gene products are found at sites of inner ear ion transport in late fetal and post-natal mice [40,41,43,44] and in the earlier-stage otic epithelium before differentiation of transport epithelia [40,45].
The question of how EphB-Efnb2 signaling influences inner ear fluid physiology has remained unanswered. Here, we report on the use of Cre-mediated conditional gene inactivation in the mouse to circumvent early embryonic lethality and show that Efnb2 is required for normal proliferation of early-stage inner ear epithelial cells, timely outgrowth and morphogenesis of the endolymphatic epithelium, and proper localization of pendrin+/Foxi1+ ES ion transport cells (mitochondrion-rich cells, or MRCs) relative to sensory organs of the ear. In Efnb2 heterozygote mutants, mis-localization of ion transport cells was found only in a genetic background with reported vestibular dysfunction.
Results
Efnb2 loss-of-function causes dysplasia of the endolymphatic duct and sac
We used the targeted alleles Sox9-IRES-Cre [46], Efnb2-exon1-floxed [47], and Efnb2-tau-lacZ [48] to effectively inactivate Efnb2 in early-stage otic epithelium, peri-otic mesenchyme, and branchial arch mesenchyme, while avoiding Cre-mediated recombination in embryonic arteries and tissues with known or suspected inductive influences on the developing ear (Fig. 1A-D). Efnb2 inactivation was confirmed by immunofluorescence and in situ hybridization with an exon 1-specific probe (Fig. 1E-J). Live isogenic C57BL/6 Sox9-IRES-Cre+;Efnb2LacZ/flox embryos (referred to here as Efnb2 CKO) were recovered at a Mendelian ratio through stage E11.5, and at just over half the expected ratio between E12.5 and E19 (Supporting Table 1). Live Efnb2 CKO embryos were not distinguished from control littermates based on size or external features. As with Efnb2 C-terminal deletion homozygotes [41], Efnb2 CKO neonates were delivered live, but died within 12 hours of birth. Germline and conditional heterozygote littermates showed a normal lifespan and no evidence of deafness or disequilibrium.
Figure 1. Distribution of Sox9-IRES-Cre activity and validation of Efnb2 inactivation in otic tissues.
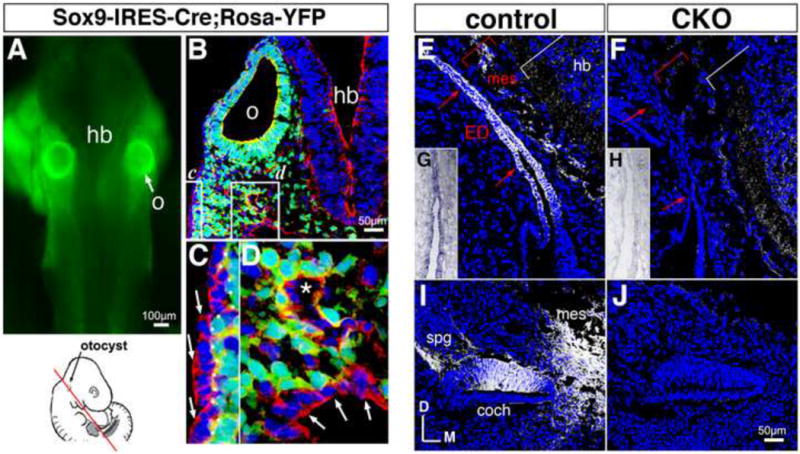
(A-D) Sox9-IRES-Cre+;Gt(ROSA)26Sor(EYFP) embryo at E9.5, showing native reporter fluorescence in a fresh embryo (green, A) and anti-GFP antibody-enhanced reporter signal (green, B-D). Transverse section (B-D) is counter-labeled with rhodamine-phalloidin (red) and DAPI (blue). Reporter signal is absent in definitive ectoderm (arrows in C), pharyngeal endoderm (arrows in D), and endothelium of dorsal aorta (asterisk in D; red cytoplasm). (C,D) correspond to boxed regions in (B). o, otic vesicle; hb, hindbrain. (E,F) Sections through endolymphatic duct of E12.5 Efnb2 (F) and control (E) littermates, immunolabeled for Efnb2 protein (grey). White brackets highlight retention of signal in marginal zone of the mutant hindbrain (hb). ED, endolymphatic duct; mes, periotic mesenchyme. (G,H) Sections through endolymphatic duct of E13.5 Efnb2 CKO (H) and control (G) littermates, hybridized to detect Efnb2 exon 1. (I,J) Sections through cochlear duct of E12.5 Efnb2 CKO (J) and control (I) littermates, immunolabeled for Efnb2 protein (grey), coch, cochlear epithelium; mes, peri-otic mesenchyme spg, spiral ganglion neurites. All controls are Efnb2flox/+. (E-J) are shown to scale. All sections are in transverse plane.
The inner ear epithelial labyrinth is derived from a spheroid otocyst (Fig. 1A,B). The endolymphatic sac (ES) and duct (ED) first appear as an undifferentiated epithelial tube projecting from a dorso-medial region of the otocyst at stage E10.5 (Fig. 2A,B). Efnb2 CKO otocysts failed to form an endolymphatic epithelium on schedule (Fig. 2B′, asterisk; 10/10 otocysts), as determined by hybridization of E10.5-10.75 embryo sections with a Gbx2 antisense probe that marks the undifferentiated endolymphatic epithelium [26]. At more mature stages, Efnb2 CKO inner ears showed fully penetrant hypoplasia and dysmorphism of the ES and ED (n=72; E12.5-E19; Fig. 2C,C′), fully penetrant shortening of the cochlear duct (n = 65; E13.5-E19), and otherwise near-normal size and morphology of the labyrinth through E15.5. A decrease in average luminal width of Efnb2 CKO vestibular canals compared to control (31%; p < 0.002) was first noted at stage E17.5 (Fig. 2E,G).
Figure 2. Anatomical abnormalities of the Efnb2 CKO inner ear.
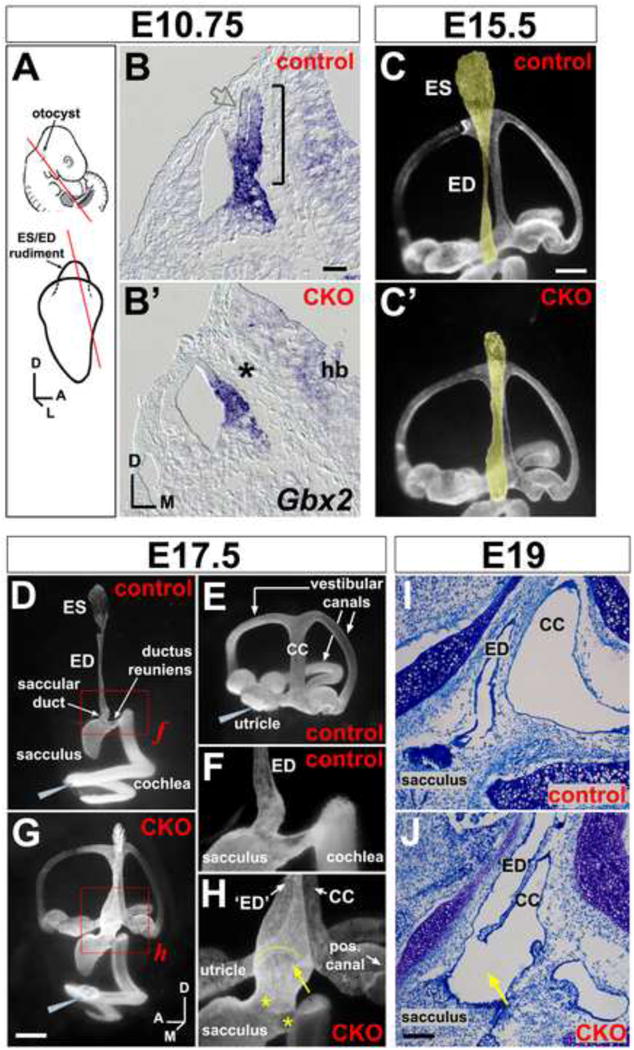
(A) Location of the otocyst at mid-gestation and projection of ES/ED rudiment from the dorso-medial otocyst. Lines show plane of section for (B,B′). (B,B′) E10.5 control (Efnbflox/+) and CKO littermates, hybridized for Gbx2 mRNA. Bracket in (B) highlights the endolymphatic projection; asterisk in (B′) highlights its absence; open grey arrow in (B) highlights weak Gbx2 signal at the endolymphatic epithelial lateral wall. Scale bar = 50 um; hb = hindbrain. (C,C′) Endolymph space of E15.5 control (Cre+;Efnb2flox/+) and CKO ears (medial views), visualized by paint injection. Endolymphatic epithelia are false-shaded yellow. Scale bar = 200 um. (D-H) Endolymph space of E17.5 control ears (Cre+;Efnb2flox/+; D,E), filled from either the cochlea (D) or utricle (E). Efnb2 CKO ear (G), filled with one injection to the cochlea. Grey wedges indicate injection site. (F and H) are magnified views of boxed regions in (D and G). Yellow arrow and arc in (H) highlight abnormal continuity across ED and common crus (CC). Asterisks in (H) highlight dysmorphic saccular duct and ductus reuniens. Scale bar in (G) = 400 microns in (D,E,G) and 150 microns in (F,H). Axes in (G) apply to (C-J). (I,J) Toluidine blue stained saggital sections through the head show abnormal luminal continuity (arrow in J) across endolymphatic epithelium (‘ED’) and common crus (CC) in an E19 Efnb2 CKO ear. (I) shows an Efnb2flox/+ control. Scale bar in (J) = 100 um and applies to (I,J).
By E17.5, the endolymph-filled luminal space of the normal mouse labyrinth is partitioned into distinct cochlear-saccular-ED-ES and utricular-canal compartments [49], as assessed by injection of paint into each of these compartments (Fig. 2D,E). By contrast, a single injection of paint to the E17.5-18.5 Efnb2 CKO cochlea filled the entire endolymph space in 5 of 6 ears (Fig. 2G). In serial sections of E19 Efnb2 CKO ears, we discovered an abnormal communication across the base of the ED and common crus of the vestibular canals (10/14 specimens; E19; Fig. 2H,I,J), which can account for unrestricted filling of the entire mutant labyrinth with one paint injection to the cochlea. In E17.5-18.5 Efnb2 CKO ears, ducts between the cochlea and ED - the ductus reuniens and saccular duct (Fig. 2D,F-H) - were either rudimentary or large in luminal diameter compared to control, and the proximal segment of endolymphatic epithelium was variably dilated (Fig. 2H). All sensory epithelia (auditory: organ of Corti; and vestibular: utricular and saccular maculae, canal cristae) of Efnb2 CKO ears were present and contained supporting cells and innervated hair cells (data not shown). Together, these results indicate that Efnb2 is required for growth and morphogenesis of the ES/ED, as well as for proper partitioning of the endolymph-filled lumen of the fetal inner ear.
Efnb2 loss-of-function affects cell proliferation and survival in the otocyst epithelium
Since outgrowth of the Efnb2 CKO endolymphatic epithelium is developmentally delayed (Fig. 2B,B′), we assayed proliferation and apoptosis in mutant otocysts during the 6-12 hour period preceding normal outgrowth. Stage E10-10.25 Efnb2 CKO otocysts were hypoplastic (Fig. 3A), and the percent reduction in Efnb2 CKOGbx2+ domain area was comparable to that for Efnb2 CKO total otocyst surface area (Table 1), suggesting that loss of Efnb2 has wide spread effects on otocyst growth. Mitotic indices for a sample of E10 otocysts, obtained by subjective counts of phosho Histone-H3- (pH-H3)-positive apical mitotic figures, revealed a 42% decrease in density of M-phase cells in mutant ears compared to those of control ears (Fig. 3B). We obtained comparable absolute and percentage change values for whole otocysts by applying normalized pixel counts (labeling index) to the same samples (Fig. 3C), and this approach was used to characterize regional differences in pH-H3 abundance. At E10, significant decreases of between 42 and 47% in Efnb2 CKO pH-H3 labeling indices compared to control were found at dorso-lateral (DL) and ventral (V) regions (Fig. 3C).
Figure 3. Altered proliferation and apoptosis in Efnb2 CKO otocysts.
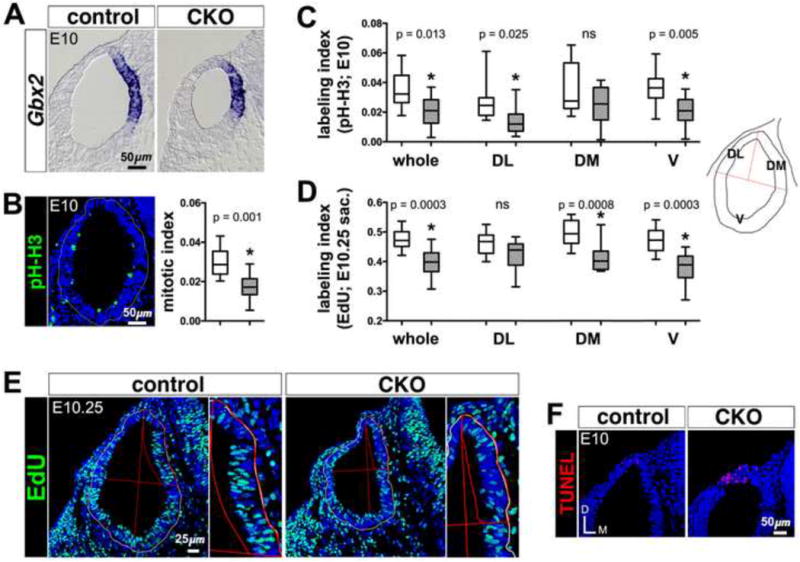
(A) E10 Efnb2flox/+-CKO littermate pair, hybridized for detection of Gbx2 mRNA. (B) phospho-histone-H3 immunolabeling of an E10 control otocyst section. Box and whisker plot summary of mitotic indices for Efnb2flox/+ control (clear box) and CKO (hatched box) whole E10 otocysts (n = 10 otocysts per group), obtained by subjective counts of pH-H3-positive figures. Whiskers on all plots define the sample range. Two-tailed P values on all plots are from unpaired t-tests. (C) Summary of pH-H3 labeling indices for Efnb2flox/+ control (clear boxes) and CKO (hatched boxes) whole otocysts and otocyst regions (n = 10 otocysts per group) at E10, obtained by normalized pixel counts (see Materials and Methods). Non-significance (ns) set at P > 0.05. Schematic at right defines otocyst regions; DL, dorso-lateral; DM, dorso-medial; V, ventral. (D) Summary of EdU labeling indices for Efnb2flox/+ control (clear boxes) and CKO (hatched boxes) whole otocysts and otocyst regions (n = 10 otocysts per group) at E10.25, obtained by normalized pixel counts. (E) EdU incorporation signals from an E10.25 Efnb2flox/+-CKO littermate pair processed in parallel. Images at right are magnified views of the dorso-medial otocyst region. (F) E10 Efnb2flox/+-CKO littermate pair, showing TUNEL signals (red) in the dorsal otocyst. Axes in (F) apply to all photos.
Table 1.
Summarized surface areas (× 103 square microns) for total otocysts and Gbx2-positive domains in Efnb2 CKO and control otocysts at stage E10.
| Control (mean;sd) | Mutant (mean;sd) | % change | |
|---|---|---|---|
| Total otocyst area | 170.0; 19.3 | 136.1; 11.8** | −20.0 |
| Gbx2-positive domain area | 56.4; 9.9 | 41.6; 7.2* | −26.2 |
Values (× 103) refer to surface areas measured at the epithelial basement membrane for regions of interest.
P < 0.01;
P < 0.02, by unpaired 2-way t-tests, n = 6 for each sample.
We next assessed otocyst cell proliferation by injecting pregnant dams with the thymidine analog EdU (10ug/gram weight of mouse) at the onset of E10 (as defined in Materials and Methods: Animals) and again 2 hours later; embryos were collected and fixed six hours after the first injection (E10.25). At E10.25, we found EdU fully incorporated into many interphase or apical M-phase nuclei and partially incorporated (speckling) into a smaller subset of interphase nuclei (Fig. 3E), suggesting that embryos were exposed to EdU for much of the six-hour labeling period. Whole Efnb2 CKO otocysts showed an 18% reduction in EdU labeling index compared to that of controls, providing further support for a slowing of cell-cycle kinetics in the mutant otocyst compared to control (Fig. 3D). By contrast to the E10 pH-H3 regional results, dorso-medial (DM) and ventral (V) E10.25 EdU mutant regional indices were significantly decreased by 17-20% from those of control.
We next analyzed the distribution of apoptotic bodies by TUNEL in a sample of E10 otocysts. We found increased TUNEL in the Efnb2 CKO otocyst compared to control specifically at a previously described [50] dorsal focus of programmed cell death (Fig. 3F). However, TUNEL staining of the Efnb2 CKO endolymphatic epithelium between stages E11.5 and E14.5 was judged to be unchanged from control. Taken together, these results correlate Efnb2 loss-of-function with dysregulated cell proliferation and apoptosis during the 12-hour period of development preceding outgrowth of the normal endolymphatic epithelium.
Efnb2 and cognate Eph receptor gene expression patterns define specific regions of the otocyst epithelium
Since growth defects of the Efnb2 CKO ear were apparent as early as stage E10, we surveyed gene expression of Efnb2 and its cognate receptors in normal C57BL/6 embryos beginning at stage E9. The otocyst forms by invagination of an ectodermal (otic) placode. Efnb2 mRNA expression in the early stage otic epithelium was regionalized and dynamic (Fig. 4C,J,Q). Comparison of Gbx2 mRNA, Efnb2-tau-lacZ, and Efnb2 mRNA signals found Efnb2-lacZ histochemical staining and Efnb2 mRNA signals at the dorsal rim of the otic cup, whereas Gbx2 signal was located medial to the Efnb2-lacZ/Efnb2 dorsal rim signals (Fig. 4A-C; brackets). Regional complementarity of Efnb2-lacZ/Efnb2 and Gbx2 signals was maintained at the dorsal otocyst through the time of otocyst formation at E9.5 (Fig. 4H-J; arrows). However, Efnb2-lacZ and Efnb2 mRNA signals were re-distributed to the dorso-medial otocyst by E10.25, resulting in partial overlap with the Gbx2 domain and marking of the initial outgrowth of endolymphatic epithelium (Fig. 4O-Q). Since fate mapping of chicken embryos places the origin of the endolymphatic epithelium at the dorsal rim of the otic cup [51], our results associate Efnb2 transcription with the earliest known origins of the endolymphatic epithelium. However, we could not reliably detect Efnb2 cell surface protein in the E9-E10.25 otic epithelium using a polyclonal antibody that specifically labels Efnb2 at later stages (Fig. 1E,F,I,J).
Figure 4. Comparative developmental expression of Gbx2, Efnb2, and cognate Eph receptor mRNA.
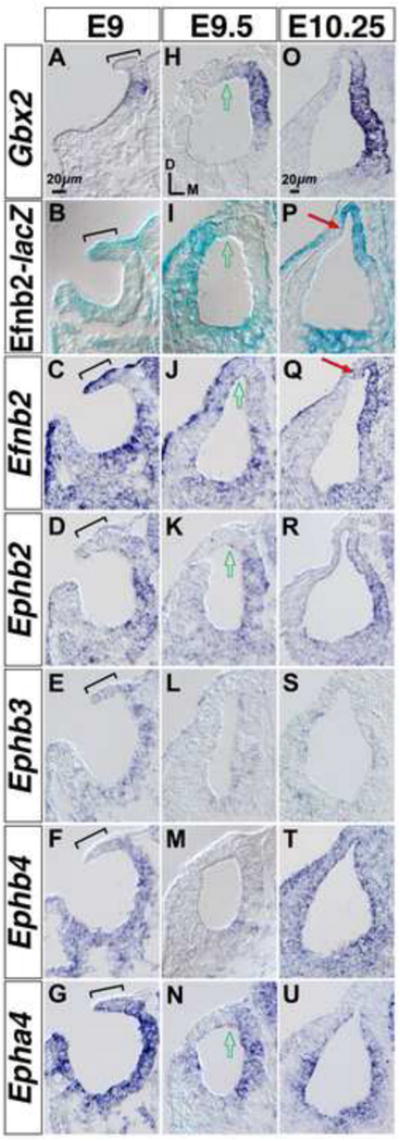
(A-G) Comparative gene expression at the otic cup. Brackets highlight the Efnb2+ dorsal rim of the otic cup. (H-N) Comparative gene expression at the newly formed otocyst. Open arrows highlight the medial-lateral midline of the dorsal otocyst. (O-U) Comparative expression at the later-stage otocyst, which shows first indications of endolymphatic epithelial outgrowth and folding of the dorsal otocyst (red arrows in P,Q). All panels show RNA hybridization signals, except for (B,I,P), which shows Efnb2-lacZ histochemical staining. (A-N) are shown to scale. (O-U) are shown to scale. Axes in (H) apply to all panels.
Cognate Eph receptor mRNA signals were either not detected or were relatively reduced in signal intensity at the Efnb2+ dorsal rim of the otic cup (Fig. 4C-G; brackets). Rough complementarity of Efnb2 and Ephb2/Epha4 signals was maintained dorsally upon formation of an otocyst at E9.5 (Fig. 4I-N). Re-distribution of Efnb2 expression to the dorso-medial otocyst by E10.25 resulted in overlap with the domains of Ephb2, Ephb4, and Epha4 (Fig. 4P-U). Detection of Efnb2 and Eph receptor gene expression within the early-stage otic epithelium is consistent with our findings of otocyst stage growth defects in the Efnb2 CKO.
Efnb2 loss-of-function affects epithelial folding and gene expression at the nascent endolymphatic epithelium
After a delay in outgrowth, the Efnb2 CKO endolymphatic epithelium attained roughly 80% of control length (proximal-distal, measured at medial wall) by E12.5 (Fig. S1). We therefore characterized the mutant structure between stages E10.5 and E12.5 to probe developmental events subsequent to growth initiation. Normally, the dorsal otocyst forms endolymphatic and vertical canal plate epithelia, the latter a rudiment for vestibular canals and the associated common crus. Morphogenic events distinguishing the normal endolymphatic epithelium from vertical canal plate involve folding and invagination of dorsal otic epithelium (Fig. 5A; 4P,Q), and the ventral edge of the invagination fuses with another region of otic epithelium at E16.5 to partition the endolymph-filled lumen (data not shown). Depth of invagination at the E11.5-11.75 Efnb2 CKO otocyst was markedly reduced compared to control (8/8 otocysts; Fig. 5B arrowheads). By E12.5, the depth of mutant invagination was abnormally shallow but comparable to that of control (10/10; Fig. 5H,H′, arrowheads); however, the proximal segment of the invagination (ED lateral wall) was hypocellular and lacked normal low columnar form (Fig. 5C).
Figure 5. Altered morphogenesis and gene expression at the Efnb2 CKO dorsal otic epithelium.
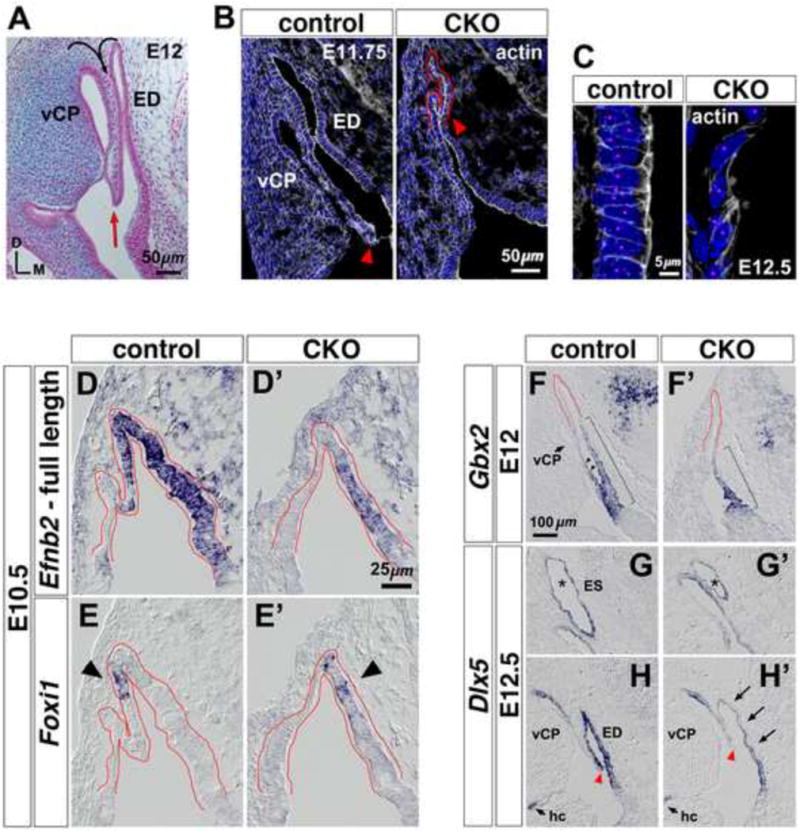
(A) Histology of E12 dorsal otic epithelium in the transverse plane. Curved arrows indicate prior-stage folding and invagination to form the ED and vertical canal plate (vCP). Red arrow highlights ventral edge of invagination. Axes apply to all photos. (B) Sections from an E11.75 Efnb2flox/+-CKO littermate pair, labeled with phalloidin and DAPI. Arrowheads highlight ventral extent of invagination. (C) High power views of proximal segments from the lateral ED wall of an E12.5 Efnb2flox/+-CKO littermate pair. Magenta dots indicate individual nuclei. (D-E′) Efnb2flox/+-CKO littermate pair hybridized to detect Foxi1 or full-length Efnb2. The recombined Efnb2 floxed allele produces non-translated transcript, allowing domains of Efnb2 transcription to be identified in the null mutant (D′). Dorsal otocyst epithelium is outlined in red. Arrowheads in (E,E′) highlight the location of initial Foxi1 mRNA signal. (F-H′) RNA-hybridized sections from Efnb2flox/+-CKO littermate pairs, shown to scale. Brackets in (F,F′) highlight proximal-distal extent of Gbx2 signal. Arrowheads (F) highlight Gbx2 signal at the ED lateral wall. Asterisks in (G,G′) highlight the ES. Arrows in (H′) highlight attenuated Dlx5 mRNA signal. Arrowheads in (H,H′) highlight ventral extent of invagination; fields in (H,H′) are aligned relative to the horizontal crista (hc). vCP, vertical canal plate.
We asked whether deficient morphogenesis correlates with changes in expression of genes required for development of the endolymphatic epithelium. Foxi1 mRNA marks prospective ion-transporting MRCs of the developing endolymphatic sac [15]. Foxi1 mRNA is first detected at the lateral (invaginated) wall of the normal endolymphatic projection at E10.5 [52] and within the domain of Efnb2 expression (Fig. 5D,E). As expected, the E10.5 Efnb2 CKO dorsal otocyst showed no evidence of folding or invagination. Foxi1 mRNA signal appeared without developmental delay within the region of Efnb2 transcription, but was mis-localized to the medial side of the mutant dorsal otocyst (Fig. 5D′,E′).
The homeodomain-containing genes Gbx2 and Dlx5 are required for normal growth of the endolymphatic epithelium [26-30], and expression of Dlx5 at the dorso-medial otocyst requires Gbx2 activity [26]. In wild-type embryos at E12, the Gbx2+ domain formed an intensity gradient across the proximal two-thirds of the endolymphatic epithelial medial wall, and it extended to proximal regions of the lateral (invaginated) ED wall (Fig. 5F; bracket and arrowheads). Thus, Gbx2 appears to mark the early-stage definitive ED. In mutant littermates, the Gbx2+ domain was truncated at the medial wall and did not extend to regions that ultimately form the ED lateral wall (Fig. 5F′). In contrast to Gbx2, normal Dlx5 mRNA signal formed an intensity gradient across the entire proximal-distal length of the endolymphatic epithelium, with intensity decreasing distally (Fig. 5G,H). In stage-matched mutants, Dlx5 mRNA signal intensity was attenuated specifically at the proximal (ductal) segment of epithelium compared to control (Fig. 5G′,H′). RNA hybridization on earlier stage Efnb2 CKO otocysts (E9.5-E10.25) revealed no change in Dlx5 signal intensity compared to control (data not shown). Taken together, deficient folding and invagination of the Efnb2 CKO dorsal otic epithelium correlate with mis-localized initiation of Foxi1, a gene required for ES MRC differentiation [15], and altered expression of Gbx2 and Dlx5, both of which influence endolymphatic epithelial growth [26-30].
Efnb2 loss-of-function affects regionalization of the endolymphatic epithelium into sac and ductal components
Having identified early-stage effects of Efnb2 loss-of-function on gene expression at the endolymphatic epithelium, we asked whether overt differentiation of the ES and ED is affected in the perinatal stage Efnb2 CKO. In wild type embryos between E11.5 and E15.5, Efnb2 mRNA and gene product, together with Ephb2 and Epha4 mRNA signals, were progressively restricted to the ED (proximally), whereas Foxi1 mRNA signal formed a mosaic pattern distally at the ES (Fig. S2). Slc26a4, encoding the anion exchanger pendrin, is activated by the Foxi1 transcription factor and marks ion-transporting MRCs in the fetal endolymphatic sac [15,53,54]. In E19 controls, Slc26a4 signals were confined to the distal third of the endolymphatic epithelium (ES) and a small domain where the ED joins the sacculus (saccular duct [11]), but the mutant epithelium expressed Slc26a4 along its entire proximal-distal length (Fig. 6A,A′). To confirm that dysregulated Slc26a4 signal in the Efnb2 CKO represents ectopic localization of ES MRCs, we double labeled ears from E19 mutant mice and control littermates with antibodies against pendrin and either Foxi1, v-H+-ATPase subunits, or Carbonic anhydrase II (CAII), as the latter proteins are detected specifically in ES MRCs (Fig. 6B, data not shown; [54]). In all cases, distal/ES co-localization profiles were found throughout the entire proximal-distal length of the mutant endolymphatic epithelium (Fig. 6B-D), with the exception that a small proximal rostro-medial region of the mutant endolymphatic epithelium did not display ES markers (Fig. 6C, arrows). Proximal mis-localization of ES markers occurred in mutants as young as stage E12.5 (data not shown). Apical plasma membrane co localization of pendrin and v-H+-ATPase, as well as nuclear localization of Foxi1, was maintained in proximal cells of the mutant epithelium (Fig. 6D), suggesting that these proximal cells are functional acid-base secretory cells (MRCs) of an ES lineage.
Figure 6. Failed proximal-distal (duct-sac) regionalization of the Efnb2 CKO endolymphatic epithelium.
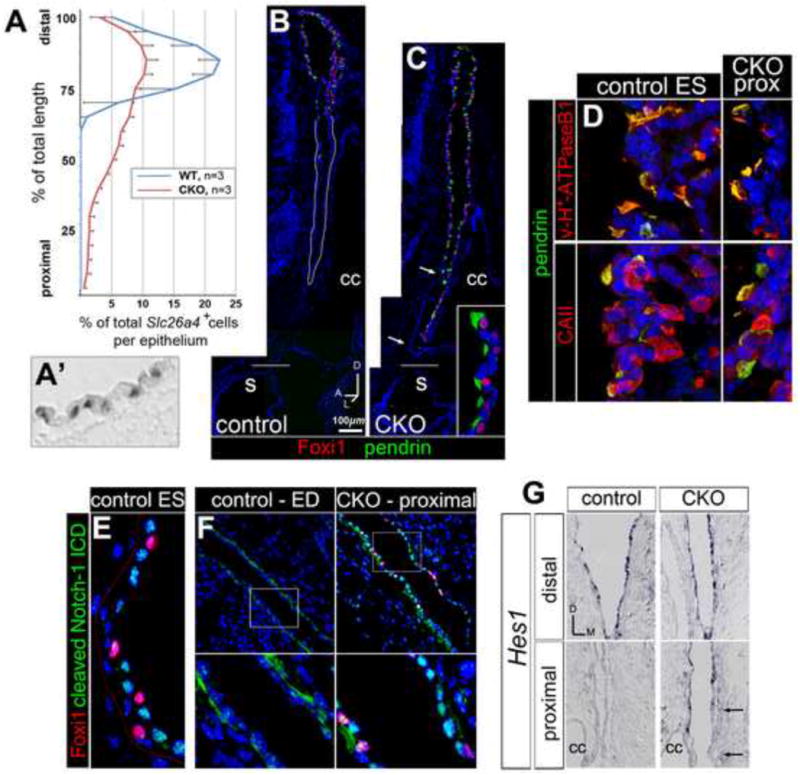
(A) Distributions of Slc26a4 mRNA signals along the proximal-distal length of endolymphatic epithelium for E19 Efnb2 CKO and control littermates. Error bars show standard deviation. (A′) Discrete Slc26a4 signals in a control epithelium. (B,C) Saggital sections through control (Efnb2+/flox, B) and Efnb2 CKO (C) E19 littermate heads, double immunolabeled for Foxi1 and pendrin. Distal/ES is toward the top. Horizontal lines mark level of insertion into the sacculus (s). Arrows in (C) highlight a proximal rostro-medial segment lacking ectopic signal. Inset in C shows proximal mutant cells (lumen is left). cc, common crus. (D) Control (Efnb2+/flox) and Efnb2 CKO E19 littermates, double immunolabeled for pendrin and either B1/B2 subunits of the vesicular proton pump or carbonic anhydrase II. CKO fields are of proximally (prox) located cells (lumen is left). (E,F) Control (Efnb2+/flox) and Efnb2 CKO E14.5 littermates, double immunolabeled for Foxi1 and cleaved Notch1-ICD. Lower panels in F show boxed regions in upper panels. Lower left panel in (F) highlights basal cytoplasmic Notch1-ICD signal in the control duct. (G) Transverse sections of control and CKO E14.5 littermates, hybridized for Hes1. Arrows highlight a proximal medial segment lacking ectopic signal. cc, common crus. Axes in (G) apply to (E-G).
Since Foxi1 is required for expression of the Notch ligand gene Jagged1 in the ES [15] and Notch signaling [55] regulates Foxi1 ortholog expression in developing aquatic vertebrates [56,57], we asked whether spatial dysregulation of activated Notch signaling correlates with the ectopic expression of Foxi1 in the Efnb2 CKO. We surveyed expression of the known mouse Notch receptor genes in the normal endolymphatic epithelium at E14.5 by RNA in situ hybridization and found strong signal for Notch1 and Notch3. We therefore double labeled Efnb2 CKO and control littermates at E14.5 with antibodies against Foxi1 and the cleaved (Val1744) Notch1 intracellular domain (ICD). Robust nuclear Notch1-ICD signal was present in Foxi1-negative nuclei of the control ES, but not in nuclei of the control ED (Fig. 6E,F). By contrast, we found robust nuclear Notch1-ICD signal throughout the proximal-distal axis of the mutant endolymphatic epithelium, especially in nuclei adjacent to ectopic Foxi1+ nuclei (Fig. 6F, right panels; compare with Fig. 6E). We also surveyed expression of the Notch target genes Hes1, Hes5, Hey1, Hey2, and HeyL in the normal endolymphatic epithelium at E14.5 by in situ hybridization and found signal for Hes1 and HeyL only in the endolymphatic sac, with Hes1 the more abundant of these two signals (data not shown). We therefore assessed Hes1 expression in Efnb2 CKO-control littermate pairs at E14.5 and found ectopic Hes1 mRNA signal in the proximal segment of the mutant epithelium (Fig. 6G). Foxi1-dependent MRC differentiation and Notch pathway activation are thus coordinately mis-localized by loss of Efnb2. These results suggest that the Efnb2 CKO fails to form a differentiated ED epithelium.
Conditional inactivation of Notch signaling affects Foxi1+ cell density but not proximal-distal regionalization of the endolymphatic epithelium
As just shown, most of the Efnb2 CKO endolymphatic epithelium was distalized in its molecular profile, but the total number of DAPI+ nuclei within the mutant Foxi1+ domain was decreased compared to control at stage E14.5 (Table 2). Therefore, loss of Efnb2 causes a complex growth deficiency of both sac- and ductal-type tissues, with a differentially severe effect on formation of proximal/ductal-type tissue. Data from two developmental stages revealed that Efnb2 loss-of-function does not affect the local density of Foxi1+ MRCs (Table 2).
Table 2.
Summarized cell counts for Efnb2 CKO and Rbpj CKO mutant and control samples.
| Strain/Stage | Controla (mean;sd) | Mutant (mean;sd) | % change | |
|---|---|---|---|---|
| Efnb2 CKO | ||||
| E14.5(n = 12)b | total nuclei c | 2719; 363 | 2396; 338 * | −11.9 |
| Foxi1+ nuclei | 797; 208 | 603; 116 * | −14.1 | |
| Foxi1+ densityd | 290; 50 | 255; 51 | ns | |
| E19 (n = 4) | total nuclei | 7645; 1344 | 5842; 525 | ns |
| Foxi1+ nuclei | 1942; 372 | 1548; 69 | ns | |
| Foxi1+ density | 241; 26 | 268; 31 | ns | |
| Rbpj CKO | ||||
| E15.5 (n = 6) | total nuclei | 4313; 332 | 1717; 158 ** | − 60.2 |
| Foxi1+ nuclei | 667; 167 | 575; 96 | Ns | |
| Foxi1+ density | 153; 31 | 335; 46 ** | + 118.4 | |
Controls were Efnb2+/flox or Foxg1-Cre+;Rbpj+/flox
Refers to equally sized control and mutant samples
Refers to total number of DAPI+ nuclei within the domain of Foxi1 protein expression
Number of Foxi1+ nuclei per 1000 DAPI+ nuclei
Differences assessed by unpaired t-tests. Two-tailed values of
P < 0.05 and
P < 0.0001.
Non-significance (ns) was set at P > 0.05.
Notch signaling influences embryonic tissue growth, compartmentalization, and fine-scale patterning (i.e., density) of cells within developing tissues, so we asked whether Notch signaling mediates the effects of Efnb2 on growth or regionalization of the endolymphatic epithelium. We analyzed the developing endolymphatic epithelium of a previously characterized Foxg1-Cre-mediated Rbpj CKO [58], which inactivates canonical Notch signaling in the early otocyst epithelium. By RNA hybridization, the E15.5 Rbpj CKO ES showed decreased intensity and extent of Hes1 signal compared to control (Fig. S3A,A′), suggesting that Notch signaling is disrupted in the developing ES. By immunofluorescence, we found no apparent change in Efnb2 protein expression at Rbpj CKO endolymphatic epithelia (data not shown), suggesting that Efnb2 acts upstream of or in parallel to Notch signaling during development of the ED/ES. Notably, neither proximal-distal ED/ES length nor regionalization of Foxi1 and pendrin epitopes differed between Rbpj CKO and littermate controls at E15.5 (Fig. S3B,B′). However, Foxi1+ cell density in the E15.5 Rbpj CKO ES was increased by over 2-fold compared to control; this correlated with a specific hypoplasia of the ES epithelium and a pronounced decrease in absolute number of Foxi1-negative nuclei within the ES (Fig. S3C,C′; Table 2). Taken together, these results suggest that canonical Notch signaling does not mediate the effects of Efnb2 on global growth and regionalization of the endolymphatic epithelium. However, Notch signaling is required for establishing a normal ratio of Foxi1-positive to Foxi1 negative cells within the developing ES epithelium.
Efnb2 loss-of-function disrupts an early phase of otoconial biogenesis
We next used the Efnb2 CKO to characterize inner ear structural abnormalities that might result from mis-localized ES MRCs. Targeted null homozygosity of Foxi1, Slc26a4/pendrin, or the proton pump subunit gene Atp6v0a4 causes defects in otoconia of the utricle and saccule [7,9,10,13,15,17]. Otoconia are extracellular biominerals with a proteinaceous core and calcitic outer shell, and their biogenesis within the endolymph is sensitive to pH and ionic conditions [59,60]. At E19, both mutant and control otoconia had an inorganic outer shell, as determined by birefringence patterns under polarized light, and core glycoprotein constituents, as determined by toluidine blue staining of thin-sectioned specimens (data not shown). Polarized light microscopy of dry frozen sections indicated that both the utricle and saccule of Efnb2 CKO fetuses contained fewer and larger otoconia than those of controls (data not shown). Analyses of E19 utricles by scanning electron microscopy (SEM) confirmed abnormalities in size, number, and shape of mutant otoconia. Control (Efnb2+/flox) otoconia had the typical barrel-shape and faceted ends, with a fine fibrillar network running between and along the cortical surfaces of otoconia (Fig. 7A,E arrowheads). By contrast, Efnb2 CKO otoconia had either of two major atypical forms, regardless of size (Fig. 7C, red and cyan arrows), and proteinacious fibrils running between mutant otoconia were thicker than those of control by an order of magnitude (Fig. 7D,E arrowheads). Measurements of mutant utricular otoconia imaged by SEM (n = 6) gave average lengths ranging between 8 and 135 microns, whereas average otoconial lengths for Efnb2+/flox control utriculi ranged between 2.61 and 2.76 microns. Average otoconial length and number of otoconia per utricle for the mutant sample showed a strong negative correlation (Fig. 7F). Extrapolating the curve to a sample-wide mean length (2.67 microns) measured for control otoconia provided a reasonable estimate (12,000) for the total number of otoconia in a normal peri-natal mouse utricle.
Figure 7. Abnormal otoconia in the Efnb2 CKO fetal utricle.
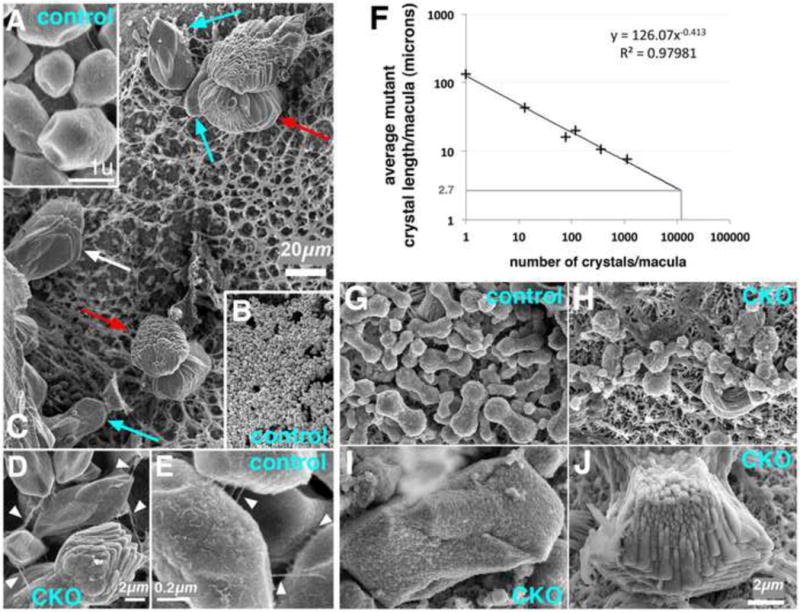
(A-E) Control (A,B,E) and Efnb2 CKO (C,D) utricular otoconia at E19. (B) and (C) are shown to scale. Cyan and red arrows in (C) highlight two prevalent mutant forms; white arrow highlights an intermediate form. White arrowheads in (D, mutant) and (E, control) highlight fibrils between individual otoconia; note 10-plus-fold difference in scale. (F) Logarithmic plot of mean otoconial length vs. total number of otoconia for six Efnb2 CKO utricular maculae at E19. (G-J) Pre-otoconia from utricles of E15.5 control (G) and Efnb2 CKO (H-J) littermates. (G-J) are shown to scale.
We hypothesized that the mass of mutant organic matter available for pre-otoconial formation is similar to normal, but that seeding of organic matter for subsequent mineralization is dysregulated. Seeding occurs in the endolymph space by Ca++- and acidic glycoprotein-mediated aggregation of secreted matrix vesicle-like structures (globular substance) [59,60]. We therefore characterized the morphology of pre-otoconia at E15.5, a stage when the relevant organic constituents are being secreted by vestibular epithelia into the endolymph space [61,62]. SEM of controls revealed stereotypical dumbbell-shaped forms with rough surface morphology (Fig. 7G). By contrast, E15.5 Efnb2 CKO utricles (n = 5) contained either disorganized forms of a similar size to control, or large atypical forms similar in shape to the mutant otoconia at E19 (Fig. 7H-J). These results provide evidence that loss of Efnb2 affects the aggregation of pre-otoconial organic matter, a process that is sensitive to the ionic composition of endolymph.
Ectopic MRCs are associated with background strain-specific vestibular dysfunction of Efnb2 heterozygotes
The Efnb2 C-terminal truncation allele (ephrin-B2LacZ mice in [41,63,64]; here referred to as Efnb2 C-del) produces an Efnb2:ß-galactosidase fusion protein that is targeted to the plasma membrane and binds cognate EphB receptors on adjacent cells, but is unable to transduce signals to the cytoplasmic side of the membrane. Both isogenic CD1 and mixed 129/CD1 Efnb2 C-del homozygotes had a shortened and dysmorphic endolymphatic duct and sac, with CD1 strain dysmorphism and hypoplasia the more severe of the two strains. Homozygous mutants of both backgrounds had otoconia that were large in size and few in number compared to those of controls (Fig. S4A). Immunofluorescence analyses of 129/CD1 Efnb2C-del/C-del fetuses at E19 revealed a profile of molecular mis-localization identical to that of the Efnb2 CKO mutant; i.e., MRC markers were found proximally in the endolymphatic epithelium (Fig. S4B-D; data not shown). As in the Efnb2 CKO, 129/CD1 C-del homozygotes failed to partition the endolymph space into distinct cochlear-saccular-ED-ES and utricular-vestibular canal compartments (5/5 ears). As previously shown [41], vestibular canals were abnormally narrow in 129/CD1 Efnb2 C-del homozygotes. These results indicate that homozygous loss of the Efnb2 C-terminus elicits many features of the Efnb2 CKO fetal ear described in preceding sections.
Dravis et al. [41] characterized incompletely penetrant and background strain-specific vestibular-motor dysfunction (circling, head bobbing) in CD1 Efnb2 C-del heterozygotes, and correlated this behavior with an ionic abnormality of the endolymph. To determine whether defective ES/ED development correlates with vestibular dysfunction, we screened for phenotypes identified in this study using three strains of Efnb2 heterozygote mutant fetuses (CD1 Efnb2 C-del; mixed 129/CD1 Efnb2 C-del; C57BL/6 Efnb2tau-lacZ/+), only one of which (CD1 Efnb2 C-del) shows head bobbing and circling [41]. All E19 heterozygote endolymphatic epithelia were mildly/moderately hypoplastic (Fig. 8C,D), with C-del strains more prominently affected than the C57BL/6 Efnb2tau-lacZ/+. No E19 heterozygotes showed evidence of large otoconia in either the utricle or saccule under polarized light microscopy (0/4 fetuses per strain). All E19 heterozygotes of the circling CD1 Efnb2 C-del strain (4/4) showed Foxi1+/pendrin+/v-H+-ATPase+/CAII+ cells in the proximal half of the endolymphatic epithelium (Fig. 8D-G), while 129/CD1 Efnb2 C-del and C57BL/6 Efnb2tau-lacZ/+ heterozygotes did not (0/4 per strain). The mean number of Foxi1+/pendrin+ MRCs in the proximal half of CD1 Efnb2 C-del heterozygote endolymphatic epithelia was 17% that for CD1/129 Efnb2 C-del homozygote endolymphatic epithelia. Normal sub-cellular localization of selected epitomes was retained in mis-localized MRCs of CD1 ephrin-B2 C-del heterozygotes (Fig. 8E,F). Among all heterozygotes analyzed, one of four CD1 Efnb2 C-del ears had an abnormal luminal communication across the base of the ED and common crus. These results associate dysmorphogenesis of the endolymphatic epithelium and MRC mis-localization with the overt vestibular dysfunction of CD1 Efnb2C-del/+ mice.
Figure 8. Mis-localization of Foxi1 and ion transport proteins in Efnb2c-del/+ fetuses of the circling CD1 strain.
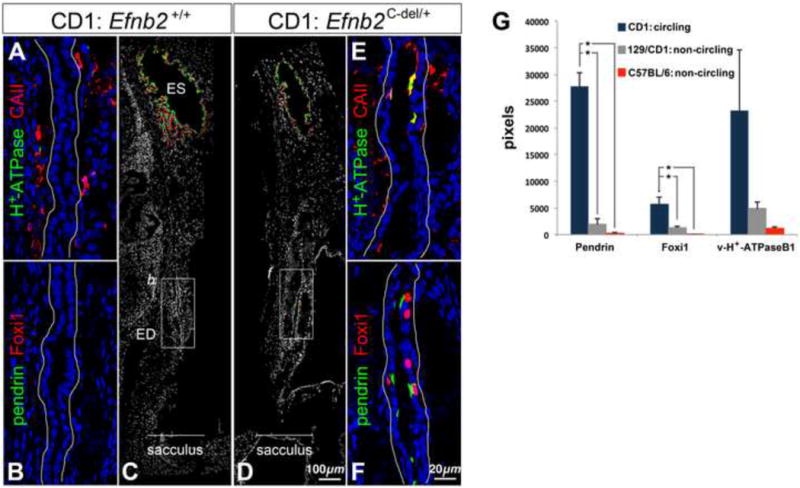
(A-C) Saggital sections of an E19 CD1 wild-type head, double-immunolabeled for v-H+-ATPase and Carbonic anhydrase II (A) or pendrin and Foxi1 (B,C). (B) corresponds to the box in (C). (D-F) Saggital sections of an E19 CD1 Efnb2c-del/+ head, double immunolabeled for pendrin and Foxi1 (D,F) or v-H+-ATPase and Carbonic anhydrase II (E). (F) corresponds to the box in (D). Ductal epithelia (A,B,E,F) are outlined in white. (G) Sums of all positive pixels (means and standard deviations; n = 4 per genotype) for epitopes of interest from the proximal half of the ED in heterozygote fetuses at E19. Asterisks show significant differences (P < 0.05) by Tukey's post-hoc multiple comparison tests.
Discussion
Efnb2 is required for multiple steps in ES/ED development
Growth and morphogenesis of the endolymphatic epithelium are poorly understood. We have demonstrated that conditional Efnb2 loss-of-function disrupts: 1) initiation of endolymphatic epithelial outgrowth; 2) epithelial folding and invagination of the dorsal otocyst; and 3) global growth of the endolymphatic epithelium (Fig. 9). On a more fundamental level, we find that Efnb2 loss-of-function dysregulates otocyst epithelial cell proliferation (globally) and cell survival (focally) during the 12 hours of development preceding normal initiation of endolymphatic epithelial outgrowth (E10-E10.5). Subsequent to formation of an endolymphatic epithelium, Efnb2 loss-of-function correlates with: 1) apparent absence of the ED; 2) mis-localized MRCs; 3) mildly reduced numbers of MRCs compared to control; and 4) failed partitioning of the endolymph fluid space into cochlear-saccular-ED-ES and utricular-canal compartments.
Figure 9. Summary of otic epithelial molecular expression and developmental anatomy in normal and Efnb2 CKO embryos.
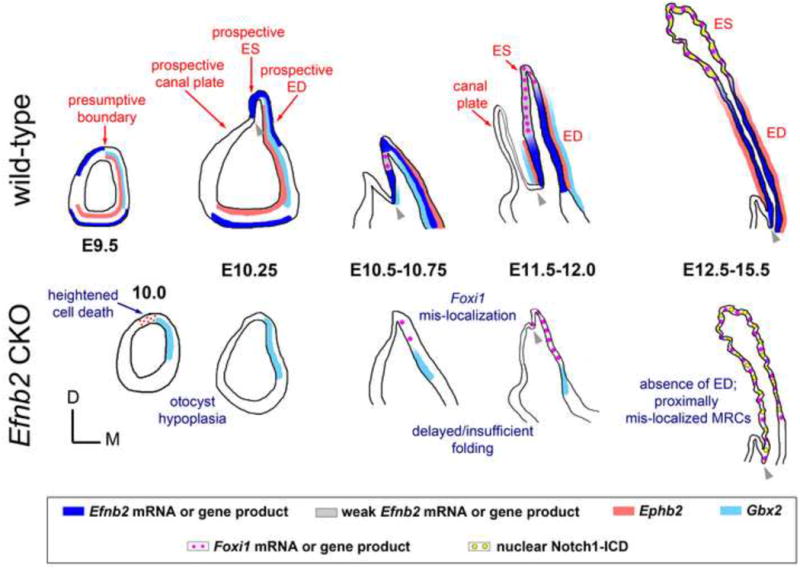
Graphics schematize the entire otocyst (early stages) or dorsal portion of the otic epithelium as seen in transverse section through embryos at specified stages. Colored bars or dots represent gene and/or protein expression, as depicted in the boxed key. Red annotations highlight features of normal development. Blue annotations highlight abnormal features of the mutant. Grey arrowheads highlight the edge of epithelial fold distinguishing endolymphatic epithelium from vertical canal plate or common crus (after stage E12.5). Graphics do not imply loss of Ephb2 signal in the Efnb2 CKO. See Discussion for details.
EphrinB-EphB interactions control progenitor cell proliferation in the intestinal stem cell niche [65,66], adult hippocampus [67], and embryonic palatal shelves [68] through canonical signal transduction pathways such as Abl-cyclin D1 or ERK/MAP kinase. Here, using both pulsed EdU incorporation and an antibody against phosphorylated Histone-H3, we found that loss of Efnb2 reduces total otocyst epithelial proliferation by roughly 20 to 45% at stages E10 (p-Histone-H3) and E10.25 (fixation for EdU analysis). A regional analysis of proliferation in the dorsal otocyst was complicated by differing results from the two techniques at E10 and E10.25; additional controlled studies are therefore necessary to test whether Efnb2 loss-of-function affects proliferation focally and dynamically in the dorsal otocyst. It is intriguing that, in the Efnb2 CKO, reduced proliferation at the E10 dorso-lateral otocyst might be followed by reduced proliferation at the E10.25 dorso-medial otocyst. This spatio-temporal pattern resembles the observed re-distribution of Efnb2-lacZ and Efnb2 mRNA signals from dorso-lateral (E9.5) to dorso-medial (E10.25) regions, and the normal endolymphatic epithelium first emerges from the dorso-medial otocyst at E10.25. A caveat to the hypothesis that close-range Efnb2 signaling drives proliferation in the early otic epithelium is our inability to reliably localize Efnb2 protein to epithelial or periotic mesenchymal cell surfaces during early otocyst stages, despite clear evidence of Efnb2 mRNA expression in both tissue layers.
Efnb2 loss-of-function affects epithelial folding/invagination of the dorsal otocyst and global growth of the endolymphatic epithelium relative to the other major dorsal structure of the late-stage otocyst, the vertical canal plate. At early stages of normal development, we found complementary expression of Efnb2 and cognate Eph receptor mRNAs in the dorsal otic cup and early otocyst (E9.5). During organogenesis, epithelial folding and differential growth can emerge from the more fundamental processes of boundary formation and compartmentalization [69,70], and Eph-ephrin signaling is a well-characterized mediator of these latter processes [34,35]. Interestingly, Brigande and colleagues [51] proposed the existence of a lineage boundary that restricts cell intermingling in the chicken dorsal otocyst and fate mapped this boundary to the fold distinguishing endolymphatic epithelium and vertical canal plate (Fig. 9, arrowheads).
By contrast, we found no evidence of strictly complementary Eph-Efnb2 expression along the proximal-distal axis of the definitive endolymphatic epithelium between E11.5 and E15.5. Efnb2/Efnb2 and cognate Eph receptor mRNA expression domains overlapped proximally in the developing ED and showed graded reductions in signal intensity more distally (Fig. S2A,B). We therefore find the conventional model of Eph-ephrin-mediated cell sorting at a boundary an unlikely explanation for regionalization of ES and ED components after outgrowth of the epithelium.
What could account for MRC mis-localization in Efnb2 mutants? We favor two non-exclusive possibilities, the first of which is delayed endolymphatic epithelial outgrowth and folding of the dorsal otocyst epithelium. Although developmental timing of Foxi1 induction is unaffected in the Efnb2 CKO, delayed growth and folding of the dorsal otocyst correlates with ectopic initiation of Foxi1 signal at E10.5. Thus, Efnb2-independent morphogenic events that ultimately form the mutant endolymphatic projection act on an already mis-localized ES (Foxi1+) domain. We also speculate that delayed folding/insufficient invagination of the Efnb2 mutant dorsal otocyst is a pre-condition to failed partitioning of the Efnb2 mutant endolymph space into cochelar-saccular-ED-ES and utricular-canal fluid compartments (Fig. 2J).
A second possible cause of mis-localized MRCs is differentially severe growth retardation of the ED relative to the ES at stages after initial outgrowth, i.e., stages E10.5-E12.5. As stated above, normal Efnb2 and cognate Eph receptor domains overlap specifically at the ED. In the Efnb2 CKO, we found Dlx5 mRNA signal intensity reduced only at the ED, and the ED-specific Gbx2 expression domain was reduced in absolute size compared to control. Loss of either Gbx2 or Dlx5 causes deficient growth of the endolymphatic epithelium [26-30], but whether these genes are required for differential growth of ED and ES components is not yet known.
Our results indicate that Efnb2-dependent growth and morphogenesis of the endolymphatic epithelium are not mediated by Notch signaling, since blockade of Notch signaling by conditional inactivation of Rbpj did not phenocopy the Efnb2 CKO ED/ES. Conversely, Efnb2 is unlikely to mediate decisions involved in generating the two major ES cell types, MRCs and Foxi1−/pendrin−/vH-ATPaseB1− ribosomal-rich cells [71]. For example, the Efnb2 CKO endolymphatic epithelium showed no change in Foxi1+ MRC density compared to control; this differs from the Rbpj CKO phenotype of increased MRC density and reduction in absolute number of non-MRC-type ES cells compared to control. In the Efnb2 CKO, nuclear Foxi1 and Notch1-ICD/Hes1 signals were identically mis-localized across the epithelium, and the normal fine-grained pattern of these signals (from cell to cell) was preserved in the proximal mutant epithelium. Finally, normal sub-cellular localization of pendrin and the vH+-ATPaseB1 subunit was retained in proximal MRCs of Efnb2 mutant ears, suggesting that these are functional ion transport cells.
Efnb2 influences endolymph homeostasis through effects on inner ear development
Previous studies indicate that the ES - and MRCs in particular - mediate endolymph homeostasis. Rodent ES physiology and morphology is altered in response to experimental manipulation of endolymph volume [72,73]. ES MRCs exhibit ultrastructural features similar to those of acid-base transporting renal intercalated cells [71]. Like renal intercalated cells, MRCs require the Foxi1 transcription factor for differentiation [15], and MRCs target both the anion exchanger pendrin and protein subunits of the vesicular proton-translocating ATPase to specific plasmalemmal domains [8,54,74,75]. Targeted null homozygosity of Slc26a4 (encoding pendrin) results in an increased volume and acidification of the endolymph, as well as abnormally large otoconia [7,9,10,11]; these defects are rescued by transgenic replacement of SLC26A4 specifically in ES MRCs [13].
We have correlated mis-localization of Foxi1+/pendrin+/vH+-ATPaseB1+/CAII+ MRCs in the Efnb2 CKO with structural evidence of abnormally aggregated pre-otoconial matter at E15.5 and large otoconia at the time of birth. Previous evidence suggests that a lack of pendrin-mediated HCO3− secretion in Slc26a4−/− mice elevates [Ca++] in vestibular endolymph by inhibiting its uptake through acid-sensitive TRPV5/6 channels [9]. Dysregulation of otoconial formation is also observed in mice lacking Foxi1 [15] or the vesicular proton-translocating ATPase subunit a4 [17]. It is presently unclear why mis-localization and a mild decrease from the normal number of MRCs should cause an otoconial phenotype resembling that associated with MRC absence (Foxi1−/−) or dysfunction (Slc26a4−/−; Atp6v0a4−/−). Loss of an insulated ES luminal microenvironment due to absence of the ED is one possible explanation for this (see below).
Heterozygotes of the CD1 Efnb2 C-del strain exhibit partially penetrant vestibular-behavioral dysfunction from the onset of ambulation [41]. This is attributed to abnormal ionic composition of the vestibular endolymph (low [K+]) and collapsed semi-circular canals [41], the latter a likely secondary consequence of abnormal endolymph. In assessing CD1 Efnb2 C-del heterozygotes and heterozygotes from two other non-circling Efnb2 mutant stains, we found proximal mis-localization of MRCs only in fetuses of the circling CD1 C-del strain. This phenotype was mild compared to that of Efnb2 homozygote mutants, and we did not find a gross otoconial defect in CD1 C-del heterozygotes. Mis-localization of MRCs in CD-1 C-del heterozygotes may therefore exert a more direct endolymph-mediated influence on vestibular hair cell function.
MRC mis-localization in CD1 C-del heterozygotes may in itself be insufficient to cause vestibular behavioral dysfunction: all four fetuses analyzed in this study had mis-localized MRCs, but roughly one-quarter of adult animals display circling/head bobbing [41]. Interestingly, one of four CD1 C-del heterozygotes showed failed partitioning of the endolymph fluid space into distinct cochlear-saccular-ED-ES and utricular-canal compartments. A defect of this sort might disrupt chamber-specific ionic concentrations and convective flow of endolymph during head movement.
Precisely how the ES epithelium controls endolymph homeostasis and whether the ED also transports water or solutes remain unknown. The ED provides a path of communication between endolymph-filled chambers of the inner ear and the ES lumen. However, the narrow lumen of the ED may also retard diffusion of solutes and insulate ES luminal contents from other parts of the ear at states of fluid equilibrium [2]. In normal rodents, ES luminal endolymph ionic concentrations [6], pH [11,76], and protein content [72,77] differ from that of endolymph elsewhere in the ear. The apparent absence of an ED in Efnb2 CKO fetal ears should disrupt these solute gradients and dilute the ES endolymph microenvironment, thereby affecting ES homeostatic activity.
Our findings do not conflict with a previous hypothesis – founded partly on biochemical evidence - that B-ephrins and EphB2 regulate endolymph homeostasis through PDZ protein-mediated macromolecular complex formation with aquaporins or the AE-type anion exchangers [40]. From E17.5 through juvenile post-natal stages, EphB2 and ephrin-B2 are expressed across vestibular dark and transitional cell populations [40,41], which control endolymph production and ionic composition at locations distant from the ES [6]. Results communicated here show that Efnb2 is required for ES/ED growth and morphogenesis from stage E10.5, if not earlier. Ephb2 and Efnb2 mutants may thus be useful in addressing the question of how ES and dark cell epithelia cooperate in normal inner ear development and physiology [78].
Conclusion
By studying conditional inactivation of Efnb2 in the otic epithelium and mesenchyme of the head and branchial arches, we have characterized a possible developmental basis for the endolymph fluid abnormalities associated with Efnb2 mutation. Other tissue specific deletions are needed to determine whether cranial neural crest [79] or periotic mesenchymal expression of Efnb2 is specifically required for ES/ED development. We have also shown that homozygous deletion of the Efnb2 C-terminus (C-del) phenocopies the late fetal stage Efnb2 CKO, but existing evidence argues against a model in which these phenotypes derive solely from loss of signaling through the Efnb2 C-terminus (reverse signaling). Previous analyses of adult mice compound homozygous for kinase-inactive Ephb2 and null Ephb3 alleles indicate that bidirectional EphB-Efnb2 signaling is required for normal endolymph homeostasis and vestibular function [40,41]. However, developmental analyses of compound homozygous Ephb2/Ephb3 mutants have not been conducted. In closing, we have shown that Efnb2 is required for growth and morphogenesis of the endolymphatic epithelium. This enhances the usefulness of Efnb2-EphB signaling-deficient mutants to efforts aimed at understanding congenital disorders of endolymph homeostasis.
Materials and Methods
Animals
C57BL/6 mice carrying the null allele Efnb2-tau-lacZ [48] were bred to C57BL/6 Sox9-IRES-Cre knock-in mice [46] to obtain double heterozygotes. Double heterozygotes were bred to C57BL/6 Efnb2flox/flox mice [47] to obtain Cre+;Efnb2LacZ/flox conditional knock-outs (CKO) and control littermate embryos. CD1 and 129/CD1 strain control and Efnb2 C-terminal deletion mutant embryos, wherein ß-galactosidase is fused in-frame with Efnb2 coding sequence (ephrin-B2LacZ mice in [41,63,64]), were obtained as previously described from established colonies at the UT Southwestern Medical Center. Foxg1-Cre+;Rbpjdel/flox experimental and Foxg1-Cre+;Rbpj+/flox control embryos were obtained as previously described [58] from an established colony at the National Institute on Deafness and Other Communication Disorders/NIH. PCR genotyping was performed as previously described. For all breeding, mice were paired in the afternoon and vaginal plugs identified in the early morning of the following day. Noon on the day of plug identification was designated E0.5. Embryos identified as E10 were harvested between 10p.m. and midnight on the day preceding E10, and fulfilled the criterion that the otocyst showed no morphological evidence of an endolymphatic epithelium. Embryos identified as E10.25 were harvested between 6 and 9a.m. on day 10 and fulfilled the criterion that the otocyst had an identifiable dorsal fold. All animal experiments were carried out in strict accordance with recommendations set forth in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Investigational Animal Care and Use Committees of the National Institutes of Health and the UT Southwestern Medical Center.
Tissue preparation
Pregnant dams were euthanized with an anesthetic concentration of CO2 and cervical dislocation. Embryos harvested for immunofluorescence and in situ hybridization were trimmed and immersion-fixed overnight in cold 4% paraformaldehyde/PBS (pH 7.4). Embryos for sectioning were cryoprotected in 30% sucrose/PBS, frozen in OCT compound (Tissue-Tek), and cut serially at 7-micron thickness on a Leica CM 3050 S cryostat. Stages E9 through E16.5 were cut in the horizontal plane of the embryo. E17.5-E19 heads were cut at the mid-sagittal plane prior to fixation and sectioned in the sagittal plane. Embryos harvested for histological staining were trimmed and immersion-fixed in either 4% paraformaldehyde/PBS or Methacarn solution, dehydrated and cleared by standard methods, embedded in paraffin, and cut serially at 7-micron thickness on a Leica RM2145 microtome. For toluidine blue staining of otoconia, E19 otic capsules were immersed in fixative (2.5% glutaraldehyde (EMS), 4% paraformaldehyde (EMS), 50 mM Hepes buffer, 2 mM CaCl2, 1 mM MgCl2, 140 mM NaCl) for 2 hours at room temperature. After washing, utricles and saccules were dissected free of cartilage, processed for embedding in JB4 medium (Polysciences #00226-1), and cut at 0.5 micron thickness on a Leica RM2265 microtome. For visualization of the endolymph space, whole embryos or fetal heads were immersed in Bodian's fixative, washed overnight in absolute ethanol, and cleared in methyl salicylate. E17.5-18.5 heads were cut mid-sagitally after Bodian fixation. A 0.4% suspension of white correction fluid (BiC) in methyl salicylate (i.e., ‘paint’) was injected through a micropipette into either the mid-turn of the cochlea or the utricle.
Detection procedures
RNA hybridization to tissue sections was performed in slide mailers according to standard methods using digoxygenin-labeled probes in weakly acidic hybridization buffer (pH 4.5), anti-digoxigenin-AP Fab fragments (Roche 11093274910) in TBST buffer, and the NBT/BCIP colorimetric substrate reaction in AP buffer at pH 9.5. EdU incorporation was detected with the Click-iT EdU Alexa Fluor 488 imaging kit (Molecular Probes #C10337) according to manufacturer's instructions; two I.P. administrations of 10ug of EdU per gram body weight of pregnant female (midnight and 1:30a.m. on day E10) were followed by harvesting of embryos at 6-7a.m (E10.25). TUNEL activity was assessed on fixed 7-micron sections using the In Situ Cell Death Detection Kit, TMR red (Roche #12156792910). X-gal staining of lightly fixed intact embryos or dissected otic capsules was performed according to standard methods, followed by post-fixation, embedding in OCT compound, and frozen sectioning of stained material. Paraffin sections were rehydrated and stained with either Toluidine Blue or Alcian Blue/Fast Red.
RNA probes
Cre-mediated recombination at the floxed Efnb2 allele was validated with a 140bp exon 1-specific fragment (NM_010111.5, nt 139-278) PCR cloned from C57BL/6 tail DNA and ligated into pCR4-TOPO vector for in vitro transcription. Wild-type Efnb2 was detected with a ∼1kb probe transcribed from NM_010111.5, nt136-1188. Other cDNAs used were as follows: Foxi1 (NM_023907, full length coding region), Hes1 (NM_008235, 1.4kb coding region plus 5′- and 3′-end fragment), HeyL (NM_013905, full coding region), Notch1 (NM_008714, nt4270-5611) Notch2 (NM_010928, nt3732-5181), Notch3 (NM_008716, nt3605-5072), Notch4 (NM_010929, 1.8kb intracellular domain), Slc26a4 (AF167411.1, nt948-1700), Dlx5 (AF022075, 1.3kb full length α-form), Gbx2 (NM_010262), Epha4 (NM_007936, nt2782-4242), Ephb2 (NM_010142), Ephb3 (NM_010143, nt535-1207), and Ephb4 (BC090839.1, nt988-1947).
Antibodies
The following antibodies were used: goat anti-CAII (C-14, Santa Cruz; 1:200), rabbit anti-pendrin (H-195, Santa Cruz; 1:250); mouse monoclonal detecting pendrin (UIRF#01065, MBL International, 1:100); FITC-conjugated goat anti-GFP (GeneTex #26662; 1:200); goat anti-Efnb2 (Neuromics #GT15026, 1:1000); goat anti-FOXI1 (Abcam #ab20454; 1:500), rabbit anti-V-H+-ATPase B1/B2 (H-180, Santa Cruz; 1:200), rabbit anti-cleaved Notch1 (Val1744) (Cell Signaling #2421; 1:100), goat anti-Epha4 (R&D System #AF641; 1:200), rabbit anti-phospho Histone H3 (Ser10) (Cell Signaling; 1:1000); rabbit anti-MyoVlla (Proteus Biosciences #25-6790; 1:500), rabbit anti-Prox1 (Millipore #AB5475; 1:1000). Cross-adsorbed fluorophore- and biotin-conjugated donkey IgG secondary antibodies (Jackson Immunoresearch) were used at 1:500 and 1:2000, respectively, except for biotin-conjugated donkey anti-mouse IgG, which was used at 1:5000.
Immunofluorescence on Tissue Sections
Immunofluorescence on fixed tissue cut to 7-micron thick sections involved standard PBS washes, PBS + 0.1% TritonX-100 + 10% donkey serum blocking solution (30 minutes, room temperature), and overnight incubations of primary antibodies at 4°C in PBS + 0.1% TritonX-100 + 1% donkey serum, with the following modifications. CAII/v-H+-ATPaseB1-B2, CAII/mouse anti-pendrin and v-H+-ATPaseB1-B2/mouse anti-pendrin double labeling was initiated with a 5-minute incubation of sections in 1% SDS for antigen retrieval, followed by copious washing. Goat anti-Efnb2 (raised against the extracellular domain) was applied to tissues for 1 hour at room temperature with detergent-free blocking and diluent buffers, followed by biotin-conjugated secondaries and TSA Fluorescence Systems tyramide signal amplification (Perkin Elmer #NEL701). Tyramide substrate reactions for all antibodies lasted 16 minutes. FOXI1/rabbit anti-pendrin and FOXI1/Notch1-ICD double labeling and phospho-HistoneH3 single labeling were initiated with antigen unmasking by boiling in citrate buffer (pH 6) plus 0.05% Tween-20 for 20 minutes, followed by continued incubation in the same solution as it cooled for another 20 minutes at room temperature. Rabbit anti-pendrin, anti-cleaved Notch1-ICD, and anti-phospho Histone H3 signals were amplified using a biotin-conjugated secondary (1:2000) and the Fluorescein ASA system as described above. All mouse monoclonal antibody applications were preceded by incubation with unconjugated monovalent Fab fragment donkey anti-mouse IgG (Jackson Immunoresearch # 715-007-003; 1:50) for 1 hour at room temperature, a quick PBS rinse, 15-minute post-fixation in 4% paraformaldehyde, and 5×5 minute washes in PBS. Slides were mounted using Prolong Gold anti-fade with DAPI (Invitrogen #P36931) and #1.5 glass coverslips, and imaged with a Zeiss LSM-780 confocal laser-scanning microscope.
Quantitative Fluorescence Confocal Laser Scanning Microscopy
Measurements of epitope abundance (for pH-H3 and EdU labeling indices; Foxi1+ nuclei, DAPI+ nuclei) were obtained by summing automated pixel counts from fluorescence images sampling serial section sets. A mutant-control littermate pair was serially sectioned on the same day. Each slide for a littermate pair contained mutant and control sections, distributed in equal proportions across the minimum number of slides needed to uniformly sample entire domains of interest from every second 7-micron serial section. Mutant and control sections were randomized in their placement from slide to slide. Within an imaging session, mutant and control sections were captured in alternation. The total number of fields sampling an entire domain of interest varied according to stage (mean ± sd = 17+/−2 fields/ear for pH-H3 or EdU at E10-10.25; 22+/−5 for Foxi1 at E14.5-15.5; 32+/−8 for Foxi1 at E19). Data were collected with a Plan-NEOFLUAR 40×/1.3 oil objective (for anti-FOXI1 and pH-H3) or a Plan-APOCHROMAT 20×/0.8 dry objective (for Alexa-488-EdU) on a Zeiss Oberver.Z1 and LSM-780 confocal laser-scanning microscope controlled by ZEN 2010b software. A 561 DPSS laser excited Rhodamine Red-X (Jackson Immunoresearch) (anti-FOXI1) and a 488 Argon laser excited fluorescein (tyramide amplified anti pH-H3) or Alexa 488 (EdU). GaAsP photodetectors were used. Integration mode line-scan acquisition parameters included a frame size of 2048 × 2048 pixels, pixel dwell time of 1.58 microseconds, and pixel averaging of 4 at 16 bit depth. Each field was acquired as a single optical plane with pinhole size set to 1AU for the 488nm line. The optical plane for each field was chosen objectively and without reference to the epitopes of interest by maximizing the number of DAPI pixels in the field during fine focusing (collimator adjusted at the beginning of imaging session). Laser power and gain settings were unchanged during imaging of any one slide. Small changes in Master Gain were made between slides if necessary. Other settings were largely unchanged across biological replicates for a particular epitope or epitope pair.
Fluorescence Image Data Analyses
Thresholding and Bezier curves were used to select signal from non-signal in the raw Zeiss LSM file format (Zen 2010b). Thresholding was held constant across all specimens processed in parallel (within-batch) and, if necessary, modified slightly across processing batches. Each processing batch comprised an equal number of experimental and control specimens. For pH-H3 and EdU signals, automated counts of total pixels for the channel of interest and DAPI were summed across fields for each specimen. To obtain a normalized index of pH-H3 or EdU labeling for each specimen, the summed pH-H3 or EdU signal for a specimen was divided by the specimen's summed DAPI pixel count. Normalized values were grouped by genotype and subjected to two-tailed unpaired t-tests. To assess absolute numbers of Foxi1+ nuclei in the endolymphatic epithelium, automated pixel counts of nuclear Foxi1 signal (colocalizing with DAPI) were summed across fields for each specimen. The sum of nuclear Foxi1+ pixels for a specimen was then divided by a conversion factor estimating the average number of Foxi1+ pixels per positive nucleus. The Foxi1 conversion factor (2520 pixels/nucleus) was obtained using the freehand selection tool in ImageJ to sample more than 1200 DAPI+ nuclei from control and mutant specimens colocalizing with 25 or more Foxi+ pixels. These same freehand selections provided a conversion factor for the number of DAPI+ pixels per nucleus (3048 pixels/nucleus). Absolute numbers of nuclei and Foxi1 nuclear densities were grouped by genotype and subjected to two-tailed unpaired t-tests.
Scanning Electron Microscopy
Fetal heads were cut mid-sagittally and immersed in fixative (2.5% glutaraldehyde (EMS), 4% paraformaldehyde (EMS), 50 mM Hepes buffer, 2 mM CaCl2, 1 mM MgCl2,140 mM NaCl) for 2 hours at room temperature. Shortly after immersion, the brain was removed and a small volume of fixative was injected into the ear with a needle. After washing, utricles with anterior and lateral ampullae attached were dissected free of the otic capsule, subjected to osmium tetroxide/tannic acid post-fixation (OTOT, each step for 1 hour with 3 × 5 minute washes with distilled water between each step), ethanol dehydration, and critical point drying from liquid CO2. Specimens were mounted on stubs and the utricular roof was lifted with a tungsten needle to expose the macula. Preparations were sputter-coated with platinum and viewed on a Hitachi S-4800 field-emission scanning electron microscope at 5kV acceleration voltage.
Measurements from scanning electron and light micrographs
Measurements of otoconial lengths, vestibular canal diameters, endolymphatic epithelial lengths, Gbx2+ domain areas, and total otocyst surface areas were made in ImageJ. Spatial distributions of Slc26a4 signals and endolymphatic epithelial lengths were obtained from automatically aligned images of serial sections (Autoaligner, BitplaneAG). The endolymphatic epithelial length for a specimen was obtained by averaging the three longest lengths measured from a serial section set. For E11.5-12.5 specimens, the Gbx2 expression domain ventral border (at the medial wall of the otocyst) was used to mark the proximal extent of the endolymphatic epithelium. Morphological criteria were used for these measurements at later stages.
Statistics
Two-tailed, unpaired t-tests were used to assess differences in pH-H3 and EdU labeling indices, absolute counts of DAPI+ or Foxi1+ nuclei, and Foxi1+ nuclear densities, with statistical significance set at the 95% confidence level. Repeated measures one-way ANOVAs with Tukey's multiple comparison post-test were applied to summed pixel data for Efnb2 C-del and null heterozygotes.
Figure Preparation
Fluorescence confocal microscopic images were optimized uniformly by applying a 0.45 gamma setting prior to export from Zen 2010. Further brightness/contrast optimization of confocal microscopic images, and brightness/contrast optimization of scanning electron and light micrographs was carried out in Photoshop after flattening all mutant-control photo comparisons to a single layer. The Unsharp mask and Hue/Saturation tools (Photoshop) were also applied uniformly to mutant and control DIC images of RNA-hybridized tissue.
Supplementary Material
Acknowledgments
We thank Thomas M. Coate, Lisa Cunningham, Andrew J. Griffith, and Matthew W. Kelley for insightful comments on the manuscript. Weise Chang and Matthew W. Kelley kindly provided pregnant Rbpjflox/flox female mice for experiments described herein. Olivia Bermingham-McDonogh, Angelika Doetzlhofer, Andrew K. Groves, and Ryoichiro Kageyama provided cDNA plasmids.
FINANCIAL DISCLOSURE: This work was supported by the Intramural Program of NIDCD (to D.K.W) and an NIH/NIMH grant MH066332 (to M.H).
Non standard abbreviations
- ES
endolymphatic sac
- ED
endolymphatic duct
- MRCs
mitochondrion rich cells
References
- 1.Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88:723–728. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- 2.Salt AN. Regulation of endolymphatic fluid volume. Ann NY Acad Sci. 2001;942:306–312. doi: 10.1111/j.1749-6632.2001.tb03755.x. [DOI] [PubMed] [Google Scholar]
- 3.Griffith AJ, Wangemann P. Hearing loss associated with enlargement of the vestibular aqueduct: Mechansitic insights from clinical phenotypes, genotypes, and mouse models. Hear Res. 2011;281:11–17. doi: 10.1016/j.heares.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T, Choi BY, King KA, Zalewski CK, Muskett J, et al. SLC26A4 genotypes and phenotypes associated with enlargement of the vestibular aqueduct. Cell Physiol Biochem. 2011;28:545–552. doi: 10.1159/000335119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dror AA, Brownstein Z, Avraham KB. Integration of human and mouse genetics reveals pendrin function in hearing and deafness. Cell Physiol Biochem. 2011;28:535–544. doi: 10.1159/000335163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am J Physiol Cell Physiol. 2007;293:C1187–1208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- 7.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, et al. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- 8.Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, et al. Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol. 2003;4:394–404. doi: 10.1007/s10162-002-3052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, et al. Lack of pendrin HCO3- transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am J Physiol Renal Physiol. 2007;292:F1314–1321. doi: 10.1152/ajprenal.00432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dror AA, Politi Y, Shahin H, Lensz DR, Dossena S. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J Biol Chem. 2010;285:21724–21735. doi: 10.1074/jbc.M110.120188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS One. 2011;6:el7949. doi: 10.1371/journal.pone.0017949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh R, Wangemann P. Free radical stress-mediated loss of Kcnj10 protein epression in stria vasularis contributes to deafness in Pendred sysndrome mouse model. Am J Physiol Renal Physiol. 2008;294:F139–F148. doi: 10.1152/ajprenal.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Sanneman JD, Harbidge DG, Zhao F, Ito T, et al. SLC26A4 targeted to the endolymphatic sac rescues hearing and balance in Slc26a4 mutant mice. PLoS Genet. 2013;9(7):e1003641. doi: 10.1371/journal.pgen.1003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi BY, Kim HM, Ito T, Lee KY, Li X, et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest. 2011;121:4516–4525. doi: 10.1172/JCI59353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, et al. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130:2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- 16.Lorente-Canovas B, Ingham N, Norgett EE, Golder ZJ, Karet Frankl FE, et al. Mice deficient in the H+− ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis Model Mech. 2012;6(2):434–442. doi: 10.1242/dmm.010645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norgett EE, Golder ZJ, Lorente-Canovas B, Ingham N, Steel KP, et al. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc Natl Acad Sci U S A. 2012;109(34):13775–13780. doi: 10.1073/pnas.1204257109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, et al. Mutations in the gene encoding B1 subunit of the H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet. 1999;21:84–90. doi: 10.1038/5022. [DOI] [PubMed] [Google Scholar]
- 19.Pasqualetti M, Neun R, Davenne M, Rijli FM. Retinoic acid rescues inner ear defects in Hoxa1 deficient mice. Nat Genet. 2001;29:34–39. doi: 10.1038/ng702. [DOI] [PubMed] [Google Scholar]
- 20.Choo D, Ward J, Reece A, Duo H, Lin Z, et al. Molecular mechanisms underlying inner ear patterning defects in kreisler mutants. Dev Biol. 2006;289:308–317. doi: 10.1016/j.ydbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Riccomagno MM, Takeda S, Epstein DJ. Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev. 2005;19:1612–1623. doi: 10.1101/gad.1303905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK. Wnt signals mediate a fate decision between otic placode and epidermis. Development. 2006;133:865–875. doi: 10.1242/dev.02271. [DOI] [PubMed] [Google Scholar]
- 23.Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, et al. FGF/FGFR-2(lllb) signaling is essential for inner ear morphogenesis. J Neurosci. 2000;20(16):6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mansour SL, Goddard JM, Capecchi MR. Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development. 1993;117:13–28. doi: 10.1242/dev.117.1.13. [DOI] [PubMed] [Google Scholar]
- 25.Hatch EP, Noyes CA, Wang X, Wright TJ, Mansour SL. Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development. 2007;134:3615–3625. doi: 10.1242/dev.006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin Z, Cantos R, Patente M, Wu DK. Gbx2 is required for the morphogenesis of the mouse inner ear: a downstream candidate of hindbrain signaling. Development. 2005;132:2309–2318. doi: 10.1242/dev.01804. [DOI] [PubMed] [Google Scholar]
- 27.Depew MJ, Liu JK, Long JE, Presley R, Meneses JJ, et al. Dlx5 regulates regional development of the branchial arches and sensory capsules. Development. 1999;126:3831–3846. doi: 10.1242/dev.126.17.3831. [DOI] [PubMed] [Google Scholar]
- 28.Acampora D, Merlo G, Laeari L, Zerega B, Postiglione MP, et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less related gene Dlx5. Development. 1999;126:3795–3809. doi: 10.1242/dev.126.17.3795. [DOI] [PubMed] [Google Scholar]
- 29.Merlo GR, Paleari L, Mantero S, Zerega B, Adamska M, et al. The Dlx5 homeobox gene is essential for vestibular morphogenesis in the mouse embryo through a BMP4-mediated pathway. Dev Biol. 2002;248:157–169. doi: 10.1006/dbio.2002.0713. [DOI] [PubMed] [Google Scholar]
- 30.Sajan SA, Rubenstein JA, Warchol ME, Lovett M. Identification of direct downstream targets of Dlx5 during early inner ear development. Hum Mol Genet. 2011;20:1262–1273. doi: 10.1093/hmg/ddq567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerlach-Bank LM, Cleveland AR, Barald KF. DAN directs endolymphatic sac and duct ourgrowth in the avian inner ear. Dev Dyn. 2003;229:219–230. doi: 10.1002/dvdy.10414. [DOI] [PubMed] [Google Scholar]
- 32.Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron. 1996;17(1):9–19. doi: 10.1016/s0896-6273(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 33.Cowan CA, Henkemeyer M. Ephrins in reverse, park and drive. Trends Cell Biol. 2002;12:339–346. doi: 10.1016/s0962-8924(02)02317-6. [DOI] [PubMed] [Google Scholar]
- 34.Klein R. Eph/ephrin signaling during development. Development. 2012;139:4105–4109. doi: 10.1242/dev.074997. [DOI] [PubMed] [Google Scholar]
- 35.Batlle E, Wilkinson DG. Molecular mehanisms of cell segregation and boundary formation in development and tumorigenesis. Cold Spring Harb Perspect Biol. 2012;4:a008227. doi: 10.1101/cshperspect.a008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genander M, Frisen J. Ephrins and Eph receptors in stem cells and cancer. Curr Opin Cell Biol. 2010;22:611–616. doi: 10.1016/j.ceb.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Feldheim DA, O'Leary DM. Visual map development: bidirectional signaling, bifunctional guidance molecules, and competition. Cold Spring Harb Perspect Biol. 2010;2:a001768. doi: 10.1101/cshperspect.a001768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu NJ, Henkemeyer M. Ephrin reverse signaling in axon guidance and synaptogenesis. Semin Cell Dev Biol. 2012;23:58–64. doi: 10.1016/j.semcdb.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein R. Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat Neurosci. 2008;12(1):15–20. doi: 10.1038/nn.2231. [DOI] [PubMed] [Google Scholar]
- 40.Cowan CA, Yokoyama N, Bianchi LM, Henkemeyer M, Fritzsch B. EphB2 guides axons at the midline and is necessary for normal vestibular function. Neuron. 2000;26:417–430. doi: 10.1016/s0896-6273(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 41.Dravis C, Wu T, Chumley MJ, Yokoyama N, Wei S, et al. EphB2 and ephrin-B2 regulate the ionic homeostasis of vestibular endolymph. Hear Res. 2007;223:93–104. doi: 10.1016/j.heares.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Miko IJ, Henkemeyer M, Cramer KS. Auditory brainstem responses are impaired in EphA4 and ephrin-B2 deficient mice. Hear Res. 2008;235:39–46. doi: 10.1016/j.heares.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pickles JO, Claxton C, Van Heumen WRA. Complementary and layered expression of Ephs and ephrins in developint mouse inner ear. J Comp Neurol. 2002;449:207–216. doi: 10.1002/cne.10231. [DOI] [PubMed] [Google Scholar]
- 44.Zhou CQ, Lee J, Henkemeyer MJ, Lee KH. Disruption of EphrinB/Eph B interaction results in abnormal cochlear innervation patterns. Laryngoscope. 2011;121:1541–1547. doi: 10.1002/lary.21861. [DOI] [PubMed] [Google Scholar]
- 45.Henkemeyer M, Marengere LE, McGlade J, Olivier JP, Conlon RA, et al. Immunolocalization of the Nuk receptor tyrosine kinase suggests roles in segmental patterning of the brain and axonogenesis. Oncogene. 1994;9:1001–1014. [PubMed] [Google Scholar]
- 46.Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102:14665–14670. doi: 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerety SS, Anderson DJ. Cardiovascular ephrinB2 function is essential for embryonic angiogenesis. Development. 2002;129:1397–1410. doi: 10.1242/dev.129.6.1397. [DOI] [PubMed] [Google Scholar]
- 48.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 49.Cantos R, Cole LK, Acampora D, Simeone A, Wu DK. Patterning of the mammalian cochlea. Proc Natl Acad Sci U S A. 2000;97:11707–11713. doi: 10.1073/pnas.97.22.11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lang H, Bever MM, Fekete DM. Cell proliferation and cell death in the developing chick inner ear: spatial and temporal patterns. J Comp Neurol. 2000;417:205–220. doi: 10.1002/(sici)1096-9861(20000207)417:2<205::aid-cne6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 51.Brigande JV, Iten LE, Fekete DM. A fate map of chick otic cup closure reveals lineage boundaries in the dorsal otocyst. Dev Biol. 2000;227:256–270. doi: 10.1006/dbio.2000.9914. [DOI] [PubMed] [Google Scholar]
- 52.Ohyama T, Groves AK. Expression of Foxi class genes in early craniofacial development. Dev Dyn. 2004;231(3):640–646. doi: 10.1002/dvdy.20160. [DOI] [PubMed] [Google Scholar]
- 53.Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, et al. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4) Am J Hum Genet. 2007;80:1055–1063. doi: 10.1086/518314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dou H, Xu J, Wang Z, Smith AN, Soleimani M, et al. Co-expression of pendrin, vacuolar H+-ATPase alpha4-subunit and carbonic anhydrase II in epithelial cells of the murine endolymphatic sac. J Histochem Cytochem. 2004;52:1377–1384. doi: 10.1177/002215540405201014. [DOI] [PubMed] [Google Scholar]
- 55.Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 56.Janicke M, Carney TJ, Hammerschmidt M. Foxi3 transcription factors and Notch signaling control the formation of skin ionocytes from epidermal precursors of the zebrafish embryo. Dev Biol. 2007;307:258–271. doi: 10.1016/j.ydbio.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 57.Quigley IK, Stubbs JL, Kintner C. Specification of ion transport cells in the Xenopus larval skin. Development. 2011;138:705–714. doi: 10.1242/dev.055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamamoto N, Chang W, Kelley MW. Rbpj regulates development of prosensory cells in the mammalian inner ear. Dev Biol. 2011;353(2):367–379. doi: 10.1016/j.ydbio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 59.Hughes I, Thalmann I, Thalmann R, Ornitz DM. Mixing model systems: using zebrafish and mouse inner ear mutants and other organ systems to unravel the mystery of otoconial development. Brain Res. 2006;1091:58–74. doi: 10.1016/j.brainres.2006.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thalmann R, Ignatova E, Kachar B, Ornitz DM, Thalmann I. Development and maintenance of otoconia: biochemical considerations. Ann N Y Acad Sci. 2001;942:162–178. doi: 10.1111/j.1749-6632.2001.tb03743.x. [DOI] [PubMed] [Google Scholar]
- 61.Nakahara H, Bevelander G. An electron microscope study of crystal calcium carbonate formation in the mouse otolith. Anat Rec. 1979;193:233–242. doi: 10.1002/ar.1091930205. [DOI] [PubMed] [Google Scholar]
- 62.Anniko M. Development of otoconia. Am J Otolaryngol. 1980;1:400–410. doi: 10.1016/s0196-0709(80)80021-4. [DOI] [PubMed] [Google Scholar]
- 63.Cowan CA, Yokoyama N, Saxena A, Chumley MJ, Silvany RE, et al. Ephrin-B2 reverse signaling is required for axon pathfinding and cardiac valve formation but not early vascular development. Dev Biol. 2004;271:263–271. doi: 10.1016/j.ydbio.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 64.Dravis C, Yokoyama N, Chumley MJ, Cowan CA, Silvany RE, et al. Bidirectional signaling mediated by ephrin-B2 and EphB2 controls urorectal development. Dev Biol. 2004;271:272–290. doi: 10.1016/j.ydbio.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 65.Holmberg J, Genander M, Halford MM, Anneren C, Sondell M, et al. EphB receptors coordinate migration and proliferation in the intestinal stem cell niche. Cell. 2006;125:1151–1163. doi: 10.1016/j.cell.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 66.Genander M, Halford MM, Xu NJ, Eriksson M, Yu Z. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 2009;139:679–692. doi: 10.1016/j.cell.2009.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chumley MJ, Catchpole T, Silvany RE, Kernie SG, Henkemeyer M. EphB receptors regulate stem/projenitor cell proliferation, migration, and polarity during hippocampal neurogenesis. J Neurosci. 2007;27(49):13481–13490. doi: 10.1523/JNEUROSCI.4158-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bush JO, Soriano P. Ephrin-B1 forward signaling regulates craniofacial morphogenesis by controlling cell proliferation across Eph-ephrin boundaries. Genes Dev. 2010;24:2068–2080. doi: 10.1101/gad.1963210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taylor J, Adler PN. Cell rearrangement and cell division during the tissue level morphogenesis of evaginating Drosophila imaginal discs. Dev Biol. 2008;313:739–751. doi: 10.1016/j.ydbio.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simpson P, Morata G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev Biol. 1981;85:299–308. doi: 10.1016/0012-1606(81)90261-x. [DOI] [PubMed] [Google Scholar]
- 71.Peters TA, Tonnaer EL, Kuijpers W, Cremers CW, Curfs JH. Differences in endolymphatic sac mitochondria-rich cells indicate specific functions. Laryngoscope. 2002;112(3):534–541. doi: 10.1097/00005537-200203000-00023. [DOI] [PubMed] [Google Scholar]
- 72.Rask-Andersen H, DeMott JE, Bagger-Sjoback D, Salt AN. Morphological changes of the endolymphatic sac induced by microinjection of artificial endolymph into the cochlea. Hear Res. 1999;138:81–90. doi: 10.1016/s0378-5955(99)00153-7. [DOI] [PubMed] [Google Scholar]
- 73.Salt AN, DeMott JE. Ionic and potential changes of the endolymphatic sac induced by endolymph volume changes. Hear Res. 2000;149:46–54. doi: 10.1016/s0378-5955(00)00160-x. [DOI] [PubMed] [Google Scholar]
- 74.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, et al. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest. 2004;113(11):1560–1570. doi: 10.1172/JCI20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vidarsson H, Westergren R, Heglind M, Blomqvist SR, Enerback S. The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney, and epididymis. PLoS One. 2009;4(2):e4471. doi: 10.1371/journal.pone.0004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsujikawa S, Yamashita T, Amano H, Kumazawa T, Vosteen KH. Acidity in the endolymphatic sac fluid of guinea pigs. ORLJ Otorhinolaryngol Relat Spec. 1992;54:198–200. doi: 10.1159/000276298. [DOI] [PubMed] [Google Scholar]
- 77.Rask-Andersen H, Danckwardt-Lilliestrom N, Linthicum FH, House WF. Ultrastructural evidence of a merocrine secretion in the human endolymphatic sac. Ann Otol Rhinol Laryngol. 1991;100:148–156. doi: 10.1177/000348949110000211. [DOI] [PubMed] [Google Scholar]
- 78.Kim HM, Wangemann P. Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLOS One. 2010;5(11):el4041. doi: 10.1371/journal.pone.0014041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adams RH, Diella F, Hennig S, Helmbacher F, Deutsch U, Klein R. The cytoplasmic domain of the ligand ephrinB2 is required for vascular morphogenesis but not cranial neural crest migration. Cell. 2001;104:57–69. doi: 10.1016/s0092-8674(01)00191-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.