

Abstract
Two new indole alkaloids, hainanerectamines A (1) and B (2), and one new β-carboline alkaloids, hainanerectamines C (4), along with five known related alkaloids (3, 5–8), have been isolated from the Hainan marine sponge Hyrtios erecta. The structures of new compounds 1, 2 and 4 were determined by detailed analysis of their 1D and 2D NMR spectra and by comparison of their spectroscopic data with those of related model compounds. Compounds 2–4 exhibited moderate inhibitory activity against Aurora A, a member of serine/threonine kinase family involving in the regulation of cell division and a new target in cancer treatment, with IC50 values of 24.5, 13.6, and 18.6 μg/mL, respectively.
Keywords: sponge, Hyrtios erecta, Thorectidae, hainanerectamines, indole, β-carboline, Aurora A
1. Introduction
Marine sponges of the genus Hyrtios (Family Thorectidae, Order Dictyoceratida) have proven to be a rich source of bioactive secondary metabolites, including terpenoids [1,2,3,4,5], macrolides [6,7,8,9,10], and tryptamine-derived alkaloids [11,12,13]. The most important metabolites of the genus Hyrtios discovered to date include the powerful anticancer agents, spongistatins 1–3 [8,9,10].
In our previous paper [14], we reported the isolation of several sesterterpenes including five new ones, hyrtiosins A–E, and two well-known sesterterpenes hyrtiosal [15,16] and scalaradial [17,18,19] from the title sponge. Interestingly, in the course of this study, we had discovered that hyrtiosal could inhibit not only PTP1B (protein tyrosine phosphatase 1B) [20], a promising target in the treatment of type-II diabetes and obesity [21], but also HIV-1 integrase [22]. While scalaradial was found for the first time to be a secretory phospholipase A2 inhibitor [23,24]. These findings stimulated our interest to search for more structurally unique and biologically active substances from the sponge. Recently, we re-collected this sponge off the Lingshui Bay of Hainan Province, China, and chemically investigated it. On separation of the Et2O- and n-BuOH-soluble fractions of an acetone extract of this animal, eight Dragendorff positive compounds, of which three are new, named hainanerectamines A–C (1, 2, 4), along with five known related indole alkaloids (3, 5–8) (Chart 1), were isolated. This paper describes the isolation and structure elucidation of these new compounds.
Chart 1.

Structures of compounds 1–9.
2. Results and Discussion
The usual workup of the Et2O- and n-BuOH-soluble materials of the acetone extract of the sponge yielded the new alkaloids 1, 2, 4, and five known related analogues 3, 5–8, as well as previously reported sesterterpenes [14]. The known alkaloids except for 3 were readily identified as 5-hydroxyindole-3-carbaldehyde (5) [11], 5-hydroxyindole-3-carboxylic acid (6) [25], 5-hydroxy-3-(2-hydroxyethyl)-indole (7) [13], and 5-hydroxytryptophan (8) [25], respectively, by analysis of their NMR data and by comparison with the data reported in the literature. While the identification of alkaloid 3 as hyrtioreticulin B was not straight forward, it’s a matter to be discussed herein. The NMR data of new compounds 1, 2, 4 are strongly reminiscent of those of the co-occurring 3, 5–8. In particular, interpretation of their NMR spectra [1H, 13C NMR (Table 1), 1H-1H COSY, HMQC, and HMBC] indicated the presence of a common 3,5-disubstituted indole moiety in 1 and 2, and a 1,3,6-trisubstituted tetrahydro-β-carboline moiety in 4, respectively. In fact, new compounds 1 and 2 differ from 5–8 only by the substituents at C-3 of the indole ring, while 4 differs from 3 only by the substituents at C-1 of β-carboline system.
Table 1.
NMR Data of Compounds 1 a and 2 b.
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC Mult | δH (J in Hz) | δC Mult | δH (J in Hz) | |
| 1 | - | - | - | - |
| 2 | 136.0, CH | 8.19, s | 125.6, CH | 7.05, s |
| 3 | 114.8, qC | - | 105.3, qC | - |
| 3a | 129.1, qC | - | 130.8, qC | - |
| 4 | 107.8, CH | 7.65, d (2.4) | 104.0, CH | 6.58, d (2.4) |
| 5 | 155.0, qC | - | 150.0, qC | - |
| 6 | 114.6, CH | 6.76, dd (8.4, 2.4) | 111.7, CH | 6.65, dd (8.4, 2.4) |
| 7 | 113.9, CH | 7.26, d (8.4) | 111.6, CH | 7.14, d (8.4) |
| 7a | 133.1, qC | - | 126.9, qC | - |
| 8 | 197.5, qC | - | 138.6, qC | - |
| 9 | 72.4, CH | 5.18, dd (8.7, 4.5) | 151.6, CH | 6.80, q (7.3) |
| 10 | 50.4, CH2 | 2.84, dd (16.8, 8.7) | 16.5, CH3 | 1.94, d (7.3) |
| - | 2.95, dd (16.8, 4.5) | - | - | |
| 11 | 209.7, qC | - | 194.9, CH | 9.52, s |
| 12 | 31.3, CH3 | 2.22, s | - | - |
a NMR data were measured in CD3OD on a Bruker DRX 400 MHz spectrometer; chemical shifts (ppm) are referenced to CD3OD (δ 3.33 ppm, 49.5 ppm). Proton coupling constants (J) in Hz are given in parentheses. The assignments were based on DEPT, 1H-1H COSY, HMQC, and HMBC experiments; b NMR data were measured in CDCl3/CD3OD (1:1) on a Bruker DRX 400 MHz spectrometer; chemical shifts (ppm) are referenced to CD3OD (3.25 ppm, 48.9 ppm). Proton coupling constants (J) in Hz are given in parentheses.
Hainanerectamine A (1) was obtained as an optically active brown amorphous solid (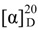 + 4.0 (c 0.05, MeOH)). Its negative ESIMS spectrum displayed a pseudo-molecular ion peak at m/z 246 [M − H]−. The HREIMS experiment established the molecular formula as C13H13NO4 (m/z 247.0835 [M]+, calcd 247.0844), indicating eight degrees of unsaturation. The 1H NMR spectrum of 1 showed downfield signals at δ 8.19 (1H, s, H-2), 7.65 (1H, d, H-4), 7.26 (1H, d, H-7), and 6.76 (1H, dd, H-6) characteristic for a 3,5-disubstituted indole moiety [25]. The 13C NMR spectrum (Table 1) displayed resonances for 13 carbons including six quaternary carbons, five methines, one methylene and one methyl. Substraction of the above elaborated 5-hydroxyl indole moiety from the molecular formula of 1 indicated that the rest part of the molecule (R2), C5H7O2, is composed of two ketons, one oxymethine, one methylene and one methyl. The partial structure 4-hydroxyl-5-oxo-pentan-2-one moiety was readily deduced by the 1H-1H COSY correlations between H-9 and H2-10 and the strong HMBC correlations of H2-10/C-8/C-9 and H3-12/C-10/C-11 (Figure 1), and was further recognized by comparison of the 1H-NMR data of side chain of 1 with those of model compound 9 previously isolated from the sponge Dysidea etberia and Ulosa ruetzler in racemate form [26]. Finally, the location of R2 at C-3 of the indole ring was deduced from the extremely downfield δ value of H-2 (δ 8.19) that implied the presence of a strongly deshielding substituent at C-3. Thus, the planar gross structure of 1 was determined. There is only one chiral center (C-9) in the molecule of 1. To determine its absolute configuration, the modified Mosher’s method [27,28] was applied since hainanerectamine A contains a secondary hydroxyl group at C-9. Thus, compound 1 was esterified separately with (R)- and (S)-MTPA chloride in dry pyridine at room temperature. Unfortunately, we failed to obtain the expected corresponding MTPA esters of 1 due to the decomposition of the molecule during the reaction. Whereas limited amounts of 1 prevent us to try again the Mosher’s reaction. The configuration of hydroxyl group at C-9 of 1 thus remains undefined.
+ 4.0 (c 0.05, MeOH)). Its negative ESIMS spectrum displayed a pseudo-molecular ion peak at m/z 246 [M − H]−. The HREIMS experiment established the molecular formula as C13H13NO4 (m/z 247.0835 [M]+, calcd 247.0844), indicating eight degrees of unsaturation. The 1H NMR spectrum of 1 showed downfield signals at δ 8.19 (1H, s, H-2), 7.65 (1H, d, H-4), 7.26 (1H, d, H-7), and 6.76 (1H, dd, H-6) characteristic for a 3,5-disubstituted indole moiety [25]. The 13C NMR spectrum (Table 1) displayed resonances for 13 carbons including six quaternary carbons, five methines, one methylene and one methyl. Substraction of the above elaborated 5-hydroxyl indole moiety from the molecular formula of 1 indicated that the rest part of the molecule (R2), C5H7O2, is composed of two ketons, one oxymethine, one methylene and one methyl. The partial structure 4-hydroxyl-5-oxo-pentan-2-one moiety was readily deduced by the 1H-1H COSY correlations between H-9 and H2-10 and the strong HMBC correlations of H2-10/C-8/C-9 and H3-12/C-10/C-11 (Figure 1), and was further recognized by comparison of the 1H-NMR data of side chain of 1 with those of model compound 9 previously isolated from the sponge Dysidea etberia and Ulosa ruetzler in racemate form [26]. Finally, the location of R2 at C-3 of the indole ring was deduced from the extremely downfield δ value of H-2 (δ 8.19) that implied the presence of a strongly deshielding substituent at C-3. Thus, the planar gross structure of 1 was determined. There is only one chiral center (C-9) in the molecule of 1. To determine its absolute configuration, the modified Mosher’s method [27,28] was applied since hainanerectamine A contains a secondary hydroxyl group at C-9. Thus, compound 1 was esterified separately with (R)- and (S)-MTPA chloride in dry pyridine at room temperature. Unfortunately, we failed to obtain the expected corresponding MTPA esters of 1 due to the decomposition of the molecule during the reaction. Whereas limited amounts of 1 prevent us to try again the Mosher’s reaction. The configuration of hydroxyl group at C-9 of 1 thus remains undefined.
Figure 1.
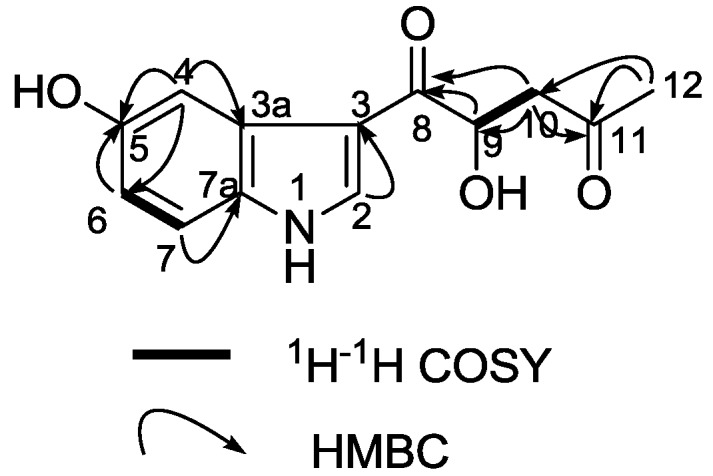
Key 2D NMR correlations of compound 1.
Hainanerectamine B (2), a brown amorphous solid, showed the molecular ion peak at m/z 201 [M]+ in the EIMS, and the molecular formula, C12H11NO2, was established by HREIMS (m/z 201.0787, calcd 201.0790). The IR spectrum showed the absorptions indicated the presence of OH and/or NH (3390 cm−1) group(s) and conjugated aldehyde (2925 and 1629 cm−1). The 1H and 13C NMR spectra of 2 clearly showed the presence of an aldehyde group (δH 9.52, 1H, s; δC 194.9, d) and suggested the presence of the same 3,5-disubstituted indole nucleus as 1. To account for the molecular formula, the side chain (R2) at C-3 has to be a butenal group (C4H5O). Furthermore, the splitting pattern of H-9 (6.80, q, J = 7.3 Hz), H3-10 (1.94, d, J = 7.3 Hz) and H-11 (9.52, s) suggested that the olefin at ∆8 is conjugated with the aldehyde. The distinct NOE cross peak between H-9 and H-11 indicated the E nature of the double bond. Finally, a series of long-range C-H correlations (Figure 2) between H-2 (δ 7.05, s)/C-3 (δ 105.3) and C-8 (δ 138.6), H-4 (δ 6.58, d, J = 2.4 Hz)/C-3a (δ 130.8), C-5 (δ 150.0) and C-6 (δ 111.6), H-9/C-2 (δ 125.6), C-3 and C-11 (δ 194.9), H3-10/C-3, C-8 and C-9 (δ 151.6), H-11/C-3 and C-8 clearly confirmed the position of the 2-butenal and hydroxyl groups to be on C-3 and C-5, respectively. Thus, the structure of compound 2 was unambiguously assigned.
Figure 2.
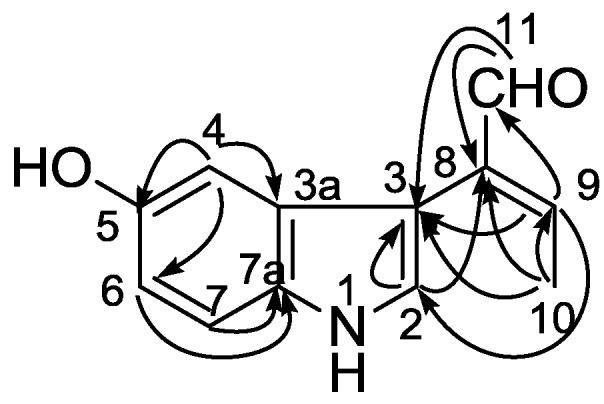
Key HMBC correlations of compound 2.
Both compounds 3 and 4 were isolated from the n-BuOH fraction implying that they are more polar than co-occurring indole alkaloids. Moreover, 3 and 4 showed other common spectroscopic properties: they appeared as strong greenish fluorescence spots on TLC plates under UV light (λ = 366 nm); their UV spectra displayed strong absorption bands at λmax 275 nm (log ε 3.4) and 358 nm (log ε 2.7); their IR spectra indicated the presence of OH and/or NH (3405 cm−1) and carbonyl (1628 cm−1) groups.
Compound 3 was purified as a yellow solid. Its positive HRESIMS displayed a molecular ion peak at m/z 313.1301, indicating a molecular formula of C16H17N4O3 for the [M + H]+ and suggesting 11 degrees of unsaturation. The 1H NMR spectrum of 3 displayed five downfield multiplets between δ 7.7 and δ 6.7, and six aliphatic protons between δ 4.8 and δ 2.9. The 13C NMR spectrum showed resonances for 16 carbons including seven methines, two methylenes, and seven quaternary carbons (Table 2). Interpretation of the 1H, 13C, 1H-1H COSY, HMQC, and HMBC NMR spectra led to the assembly of three spin systems within 3. The first spin coupling system is an ABC system including signals at δ 6.82 (d, H-5), 6.68 (dd, H-7), and 7.17 (d, H-8). The second spin system comprised H-3β/4α (J = 13.6 Hz), H-3β/4β (J = 4.0 Hz), H-4α/4β (J = 15.3 Hz). The remaining coupling system included H-1 (δ 4.80, m), H2-10 [(δ 3.48, m) and (δ 3.18, m)], and two imidazole signals corresponding to H-13 (δ 7.68) and H-15 (δ 7.00). The placement of the methylimidazole and carboxylic acid moieties at C-1 and C-3, respectively, was supported by the HMBC experiments, which showed H2-10/C-9a (δ 131.6), C-11 (δ 136.0) and C-15 (δ 117.2) and H-3 (δ 3.86, m)/C-1′ (δ 174.9) connectivities. Similarly, the assignment of all quaternary carbons within 3 was secured from HMBC correlations (Figure 3). The NOE correlations of H-5/H-4 (δ 2.90), H-4/H-3, and H-3/H-1 clearly indicated that the carboxylic acid and the imidazole side chain were oriented on the same face. From the above evidences, the structure of compound 3 was determined as (−)-(1R*, 3R*)-6-hydroxy-1-(4-methylene-1H-imidazole)-2,3,4,9-tetrahydro-1H-β-carboline-3-carboxylic acid.
Table 2.
NMR Data of Compounds 3 and 4 a.
| Position | 3 | 3 b | 4 | |||
|---|---|---|---|---|---|---|
| δC Mult | δH (J in Hz) | δC Mult | δH (J in Hz) | δC Mult | δH (J in Hz) | |
| 1 | 55.8, CH | 4.80, m | 54.6 | 5.01, br, s | 55.2, CH | 4.72, m |
| 2 | - | - | - | - | - | - |
| 3 | 60.2, CH | 3.86, m | 59.7 | 4.26, br, d (7.3) | 60.6, CH | 3.70, m |
| 4 | 25.0, CH2 | 3.30, dd (15.3, 4.0) | 24.2 | 3.36, m | 25.7, CH2 | 3.28, dd (15.4, 3.8) |
| - | 2.90, dd (15.3, 13.6) | - | 3.04, dd (15.1, 12.4) | - | 2.96, dd (15.4, 13.8) | |
| 4a | 108.5, qC | - | 107.9 | - | 109.3, qC | - |
| 4b | 128.7, qC | - | 128 | - | 129.1, qC | - |
| 5 | 103.8, CH | 6.82, d (2.1) | 103.4 | 6.86, d (2.3) | 103.8, CH | 6.84, d (2.4) |
| 6 | 152.4, qC | - | 152.2 | - | 152.4, qH | - |
| 7 | 113.7, CH | 6.68, dd (8.7, 2.1) | 113.8 | 6.75, dd (8.7, 2.3) | 113.5, CH | 6.67, dd (8.7, 2.4) |
| 8 | 113.3, CH | 7.17, d (8.7) | 113.1 | 7.22, d (8.7) | 113.2, CH | 7.17, d (8.7) |
| 8a | 133.7, qC | - | 133.4 | - | 133.8, qC | - |
| 9 | - | - | - | - | - | - |
| 9a | 131.6, qC | - | 129.7 | - | 130.7, qC | - |
| 10 | 31.0, CH2 | 3.48, m | 28.8 | 3.73, br, d (14.2) | 28.4, CH2 | 3.64, m |
| - | 3.18, m | - | 3.36, m | - | 3.24, m | |
| 11 | 136.0, qC | - | 130.7 | - | 130.7, qC | - |
| 12 | - | - | - | - | - | - |
| 13 | 137.7, CH | 7.68, s | 136.2 | 8.73, s | 141.0, CH | 7.69, s |
| 14 | - | - | - | - | - | - |
| 15 | 117.2, CH | 7.00, s | 118.6 | 7.27, s | 134.6, qC | - |
| COOH | 174.9, qC | - | 170.6 | - | 176.0, qC | - |
| NCH3 | - | - | - | - | 33.3, CH3 | 3.71, s |
| SCH3 | - | - | - | - | 19.8, CH3 | 2.39, s |
a NMR data were measured in CD3OD on a Bruker DRX 400 MHz spectrometer; chemical shifts (ppm) are referenced to CD3OD (δ 3.33 ppm, 49.5 ppm). Proton coupling constants (J) in Hz are given in parentheses. The assignments were based on DEPT, 1H-1H COSY, HMQC, and HMBC experiments; b NMR data were reported in the literature [29]. Chemical shifts (ppm) are referenced to CD3OD (δ 3.30 ppm, 49.0 ppm).
Figure 3.

Key 2D NMR correlations of compound 3.
Literature survey revealed that the structure of alkaloid 3 was the same as that of hyrtioreticulin B, a metabolite isolated previously from the sponge H. reticulates by Tsukamoto et al. [29]. However, careful comparison of the overall 1H- and 13C-NMR dataof compound 3 with those of hyrtioreticulin B (Table 2) revealed significant differences between them. For instance, the ∆δH of chemical shift for H-13 in both compounds was more than 1.0 ppm and the differences of the 13C-NMR data were even more obvious, especially for the C-11 (∆δc = +4.8 ppm) and -COOH (∆δc = +3.8 ppm). From a natural product chemistry point of view, the differences are too big to be ignored. In order to secure the correctness of 3, the 1D and 2D NMR spectra were reanalyzed and the proposed structure was confirmed. It raises a necessity to clarify if the isolate 3 and hyrtioreticulin B are the same. Surprisingly, the 1H- and 13C-NMR spectra of hyrtioreticulin B are quite different from those of 3. We failed to obtain authentic sample of hyrtioreticulin B from Tsukamoto’s group for the direct comparison. At this stage, we can only ascribe these differences to the “alkaloidal nature” [30].
Compound 4 was obtained as a yellow solid. The ESIMS spectrum of 4 showed a pseudo-molecular ion peak at m/z 373 [M + H]+, and its molecular formula was established as C18H21N4O3S from the HRESIMS (m/z 373.1348 ([M + H]+, calcd 373.1334), indicating 11 degrees of unsaturation. The 1H and 13C NMR (Table 2) spectra of 4 showed great similarities to those of 3. The presence of a same 3- carboxylic acid, 6-hydroxyl functional groups on the tetrahydro-β-carboline ring as 3 was immediately recognized. In addition, NMR spectra displayed additional signals for one N-methyl group [δH 3.71 (3H, s), δC 33.3 (CH3)] and one thiomethyl group [δH 2.39 (3H, s), δC 19.8 (CH3)], that should be obviously substituted on the imidazole ring in agreement with the observed molecular weight difference of 60 mass units. Significant 1H-13C long-range correlation, as show in Figure 4, allowed to locate the methyl to N-12 and the thiomethyl group to C-15 and connected the imidazole system at C-10. Thus, the planar structure of 4 was unambiguously assigned. By analogy to Compound 3, the relative configuration of H-1 and H-3 in 4 was assigned as cis by the ROESY experiment (Figure 4). On the basis of above observations, the structure of 4 was determined to be 12-N-CH3, 15-SCH3 derivative of 3, named hainanerectamine C.
Figure 4.

Key 2D NMR correlations of compound 4.
In conclusion, besides the sesterterpenoids that were already reported previously, six indole alkaloids and two β-carboline derivatives were also isolated and structurally characterized from the present collection (LS-103) of Hainan sponge H. erecta. It is interesting to note that although many indole alkaloids were reported from various natural resources [31], to the best of our knowledge, this is the first report of the isolation of indole alkaloid bearing a butenal moiety from a marine sponge. On the other hand, although many β-carboline alkaloids were reported from the genus Hyrtios and other marine sponges [32], only two tetrahydro-β-carboline containing both a methylimidazole and a carboxylic acid moiety at C-1 and C-3 have been reported from the animals of this genus [29]. The abundant production and accumulation of indole and β-carboline alkaloids in the title specimen is intriguing, as it seems unlikely that these compounds act solely as repellents against predators. Instead, they may play as an as yet unknown physiological role in these benthic animals.
Compound 1–8 were evaluated for their inhibitory activity against Aurora A [33,34,35]. Bioassay results showed that compounds 2–4 had moderate inhibitory effects with IC50 values of 24.5, 13.6, and 18.6 μg/mL, respectively. The cytotoxic activities of 1–8 against the growth of several tumor cell lines [A549 (human lung adenocarcinoma) and HT-29 (human promyelocytic leukemia)] were also evaluated. Unfortunately, the results indicated that all the tested compounds were inactive against the above cancer cells (50% effective dose of clonal inhibition ED50 > 10 μg/mL).
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241MC polarimeter. UV spectra were recorded on a Varian Cary 300 Bio spectrophotometer. IR spectra were recorded on a Nicolet-Magna FT-IR 750 spectrometer. NMR spectra were recorded on a Bruker DRX-400 spectrometer (Bruker, Germany) with the residual CH3OH (δH 3.30 ppm, δC 49.0 ppm) as an internal standard. EIMS and HREIMS data were obtained on a Finnigan-MAT-95 mass spectrometer. ESIMS and HRESIMS spectra were recorded on a Waters Q-TOF Micro LC-MS-MS mass spectrometer. Reversed-phase HPLC (Agilent 1100 series liquid chromatography using a VWD G1314A detector at 210 nm and a semi-preparative ODS-HG-5 (5 μm, 10 mm (i.d.) × 25 cm) column) was also employed. Commercial Si gel (Qing Dao Hai Yang Chemical Group Co., Qingdao, China, 200–300 and 400–600 mesh) was used for CC, and precoated Si gel plates (Yan Tai Zi Fu Chemical Group Co., Yantai, China, G60 F-254) were used for analytical TLC. The Aurora kinase inhibitor VX-680 was from Roche (purity = 99.88%). Z-LYTE Kinase Assay Kit (Cat No. PV3174, Invitrogen, Carlsbad, CA, USA).
3.2. Animal Material
The specimens of H. erecta were collected off Lingshui Bay, Hainan Province, China, in November 2003, and were identified by Prof. Rob W. M. van Soest (Zoologisch Museum, University of Amsterdam, Amsterdam, The Netherlands). A voucher sample (LS-103) is available for inspection at the Herbarium of Shanghai Institute of Materia Medica, CAS, Shanghai, China.
3.3. Extraction and Separation
The frozen sponge (190 g, dry weight) was extracted with acetone (1 L × 4) exhaustively at room temperature. The acetone extract was concentrated in vacuo and the resulting residue (5.6 g) was partitioned between Et2O and H2O, n-BuOH and H2O, respectively. The Et2O-soluble extract (2.6 g) was chromatographed on a silica gel column (5 × 40 cm) using light petroleum ether with increasing amount of EtOAc (10:0, 9:1, 8:2, 7:3, 6:4, 5:5, 4:6, 3:7, 0:10, v/v, each 1L) as eluent. The fraction eluted with EtOAc/petroleum ether (4:6, 415.6 mg) was further purified by Silica gel CC (3 × 30 cm) eluting with CHCl3/MeOH (9:1) to give the pure compounds 1 (>95%, 5.7 mg) and 2 (>95%, 8.2 mg). The n-BuOH-soluble extract (>95%, 2.5 g) was subjected to gel filtration on Sephadex LH-20 (CH3OH, 4 × 110 cm), yielding a mixture that was further purified by RP-HPLC [semipreparative ODS-HG-5 (5 μm, 250 × 10 mm), CH3OH/H2O (4:6), 2.0 mL/min] to afford two pure compounds 3 (>95%, 6.2 mg) and 4 (>95%, 5.1 mg).
Compound 1: brown amorphous solid; + 4.0 (c 0.05, MeOH); UV (MeOH) λmax (log ε) 353 (0.7), 307 (3.4), 252 (3.7), 271 (3.6), 213 (4.0) nm; IR (KBr) νmax 3405, 2929, 1705, 1629, 1473, 1076 cm−1; EIMS m/z 247 [M]+ (10), 160 (100), 132 (22); HREIMS m/z 247.0835, calcd for C13H13NO4, 247.0844; 1H and 13C NMR see Table 1.
Compound 2: gray amorphous powder; UV (MeOH) λmax (log ε) 357 (3.1), 356 (3.0), 276 (3.8), 254 (3.6), 219 (4.5) nm; IR (KBr) νmax 3396, 2925, 2854, 1672, 1630, 1461, 1166 cm−1; EIMS m/z 201 [M]+ (100), 172 (68), 139 (56), 127 (24), 111 (28); HREIMS m/z 201.0787, calcd for C12H11NO2, 201.0784; 1H and 13C NMR data see Table 1.
Compound 3: yellow solid; −47 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 358 (2.7), 286 (3.3), 275 (3.4), 205 (3.9) nm; IR (KBr) νmax 3405, 2927, 1626, 1400, 1201, 1084, 625 cm−1; ESIMS m/z 313 [M + H]+; HRESIMS m/z 313.1301, calcd for C16H17N4O3, 313.1301; 1H and 13C NMR data see Table 2.
Compound 4: yellow solid; −40 (c 0.06, MeOH); UV (MeOH) λmax (log ε) 389 (2.8), 358 (2.9), 302 (3.3), 271 (3.5), 204 (4.0) nm; IR (KBr) νmax 3423, 1627, 1402, 1201, 1143, 1035, 621 cm−1; ESIMS m/z 373 [M + H]+; HRESIMS m/z 373.1348, calcd for C18H21N4O3S, 373.1334; 1H and 13C NMR see Table 2.
3.4. Biological Assay
Compounds 1–8 were evaluated for their inhibitory activity against Aurora A, a member of serine/threonine kinase family involving in the regulation of cell division and a new target in cancer treatment [33,34,35]. Compounds 1–8 were initially diluted to a 100× concentration in DMSO. The 100× concentrations were then diluted to a 4× working concentration in kinase buffer. 2.5 μL of the test compounds solution and the positive control at various concentrations were added to a barcoded corning, low volume NBS, 384-well plate, then 5 μL of the 2× peptide/kinase mixture and 2.5 μL of the 4× ATP solution were added to the plate. The assay plate was shaken on a plate shaker for 30 s, and then incubated for 60 min at room temperature. After the treatment 5 μL of the development reagent solution was added to the plate. The assay plate was shaken on a plate shaker for 30 s, and then incubated for 60 min at room temperature. The assay plate is read on fluorescence plate reader and the data is analyzed.
4. Conclusions
Our investigation demonstrated that the sponge H. erecta is a good source of bioactive substances. Compounds 2–4 exhibited moderate inhibitory activity against Aurora A, a member of serine/threonine kinase family involving in the regulation of cell division and a new target in cancer treatment, with IC50 values of 24.5, 13.6, and 18.6 μg/mL, respectively. These results suggest that continuing investigation of novel secondary metabolites together with the potentially useful bioactivities from this marine organism are worthwhile for future drug development. Moreover, further study should be conducted to unambiguously confirm the identity of structures of compound 3 and hyrtioreticulin B by total synthesis in future.
Acknowledgments
This research work was financially supported by the National Marine 863 Projects (Nos. 2011AA09070102 and SS2013AA090202), the Natural Science Foundation of China (Nos. 41306130, 21072204, 81273430, 20721003). We are indebted to Ernesto Mollo of ICB-CNR, Italy, for the collection of the animal material. We are also grateful to Jia Li of SIMM-CAS, for the biological assays. W.-F. He thanks State Key Laboratory of Drug Research-SIMM (SIMM1203KF-12), China Postdotoral Science Foundation (2012M520956) for the financial supports. We are also grateful to Rob W. M. van Soest (Zoologisch Museum, University of Amsterdam, Amsterdam 94766, The Netherlands) for identification of the sponge.
Supplementary Files
Supplementary Information (PDF, 4409 KB)
Supplementary Information
NMR and HR-ESI-MS spectra of the new compounds are available in Supplementary Information.
Author Contributions
Conceived and designed the experiments: Yue-Wei Guo. Extraction and isolation: Wen-Fei He, Duo-Qing Xue. Sample collection: Li-Gong Yao. Biological assay: Jing-Ya Li, Jia Li. Wrote the paper: Wen-Fei He, Duo-Qing Xue.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Youssef D.T.A., Yamaki R.K., Kelly M., Scheuer P.J. Salmahyrtisol A, a novel cytotoxic sesterterpene from the red sea sponge Hyrtios erecta. J. Nat. Prod. 2002;65:2–6. doi: 10.1021/np0101853. [DOI] [PubMed] [Google Scholar]
- 2.Ryu G., Matsunaga S., Fusetani N. Three new cytotoxic sesterterpenes from the marine sponge Hyrtios cf. erectus. J. Nat. Prod. 1996;59:515–517. doi: 10.1021/np960130e. [DOI] [PubMed] [Google Scholar]
- 3.Williams D.E., Tahir A., Andersen R.J. A new acyclic diketotriterpenoid isolated from the Indonesian marine sponge Hyrtios erectus. J. Nat. Prod. 1999;62:653–654. doi: 10.1021/np980526l. [DOI] [PubMed] [Google Scholar]
- 4.Nasu S.S., Yeung B.K.S., Hamann M.T., Scheuer P.J., Kelly-Borges M., Goins K. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios spp. J. Org. Chem. 1995;60:7290–7292. [Google Scholar]
- 5.Salmoun M., Devijver C., Daloze D., Braekman J.C., Gomez R., de Kluijver M., van Soest R.W.M. New sesquiterpene/quinones from two sponges of the genus Hyrtios. J. Nat. Prod. 2000;63:452–456. doi: 10.1021/np9903346. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi M., Aoki S., Sakai H., Kawazoe N., Kihara N., Sasaki T., Kitagawa I. Altohyrtin A, a potent anti-tumor macrolide from the Okinawan marine sponge Hyrtios altum. Tetrahedron Lett. 1993;34:2795–2798. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi M., Aoki S., Sakai H., Hihara N., Sasaki T., Kitagawa I. Altohyrtins B and C and 5-desacetylaltohyrtin A, potent cytotoxic macrolide congeners of altohyrtin A, from the Okinawan marine sponge Hyrtios altum. Chem. Pharm. Bull. 1993;41:989–991. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]
- 8.Pettit G.R., Cichacz Z.A., Gao F., Herald C.L., Boyd M.R. Isolation and structure of the remarkable human cancer cell growth inhibitors spongistatins 2 and 3 from an eastern Indian ocean Spongia sp. J. Chem. Soc. Chem. Commun. 1993:1166–1168. [Google Scholar]
- 9.Pettit G.R., Cichacz Z.A., Gao F., Herald C.L., Boyd M.R., Schmidt J.M., Hooper J.N.A. Antineoplastic agents. 257. Isolation and structure of spongistatin 1. J. Org. Chem. 1993;58:1302–1304. [Google Scholar]
- 10.Pettit G.R. Progress in the discovery of biosynthetic anticancer drugs. J. Nat. Prod. 1996;59:812–821. doi: 10.1021/np9604386. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi J., Murayama T., Ishibashi M., Kosuge S., Takamatsu M., Ohizumi Y., Kobayashi H., Ohta T., Nozoe S., Sasaki T. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron. 1990;46:7699–7702. doi: 10.1016/S0040-4020(01)90065-1. [DOI] [Google Scholar]
- 12.Bourguet-Kondracki M.L., Martin M.T., Guyot M. A new β-carboline alkaloid isolated from the marine sponge Hyrtios erecta. Tetrahedron Lett. 1996;37:3457–3460. doi: 10.1016/0040-4039(96)00573-4. [DOI] [Google Scholar]
- 13.Salmoun M., Devijver C., Daloze D., Braekman J.C., van Soest R.W.M. 5-hydroxytryptamine-derived alkaloids from two marine sponges of the genus Hyrtios. J. Nat. Prod. 2002;65:1173–1176. doi: 10.1021/np020009+. [DOI] [PubMed] [Google Scholar]
- 14.Yu Z.-G., Bi K.-S., Guo Y.-W. Hyrtiosins A–E, five new scalarane sesterterpenes from the South China Sea sponge Hyrtios erecta. Helv. Chim. Acta. 2005;88:1004–1009. doi: 10.1002/hlca.200590070. [DOI] [Google Scholar]
- 15.Iguchi K., Shimada Y., Yamada Y. Hyrtiosal, a new sesterterpenoid with a novel carbon skeleton from the Okinawan marine sponge Hyrtios erectus. J. Org. Chem. 1992;57:522–524. doi: 10.1021/jo00028a023. [DOI] [Google Scholar]
- 16.Yu Z.-G., Bi K.-S., Li Z.-Y., Guo Y.-W. Crystal structure of hyrtiosal, C25H38O3. New Cryst. Struct. 2004;219:415–416. [Google Scholar]
- 17.Cimino G., de Stefano S., Minale L. Scalaradial, a third sesterterpene with the tetracarbocyclic skeleton of scalarin, from the sponge Cacospongia mollior. Experientia. 1974;30:846–847. [Google Scholar]
- 18.Jacobs R.S., Bober M.A., Pinto J., Williams A.B., Jacobson P.B., de Carvalho M.S. In: Marine Biotechnology. Attaway D.H., Zaborsky O.R., editors. Volume 1. Plenum Press; New York, NY, USA: 1993. pp. 77–99. [Google Scholar]
- 19.Crews P., Bescansa P. Sesterterpenes from a common marine sponge, Hyrtios erecta. J. Nat. Prod. 1986;49:1041–1052. doi: 10.1021/np50048a012. [DOI] [PubMed] [Google Scholar]
- 20.Sun T., Wang Q., Yu Z.-G., Zhang Y., Guo Y.-W., Chen K.-X., Shen X., Jiang H.-L. Hyrtiosal, a PTP1B inhibitor from the marine sponge Hyrtios erectus, shows extensive cellular effects on PI3K/AKT activation, glucose transport, and TGFβ/Smad2 signaling. ChemBioChem. 2007;8:187–193. doi: 10.1002/cbic.200600349. [DOI] [PubMed] [Google Scholar]
- 21.Byon J.C., Kusari A.B., Kusari J. Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol. Cell. Biochem. 1998;182:101–108. doi: 10.1023/A:1006868409841. [DOI] [PubMed] [Google Scholar]
- 22.Du L., Shen L.-L., Yu Z.-G., Chen J., Guo Y.-W., Tang Y., Shen X., Jiang H.-L. Hyrtiosal, from the marine sponge Hyrtios erectus, inhibits HIV-1 integrase binding to viral DNA by a new inhibitor binding site. ChemMedChem. 2008;3:173–180. doi: 10.1002/cmdc.200700223. [DOI] [PubMed] [Google Scholar]
- 23.Xie Y.-L., Yu Z.-G., Liu L.-H., Guo Y.-W., Lou L.-G. Biphasic regulation of extracellular signal-regulated kinases by scalaradial, a secretory phospholipase A2 inhibitor. Cancer Biol. Ther. 2006;5:988–992. doi: 10.4161/cbt.5.8.2884. [DOI] [PubMed] [Google Scholar]
- 24.Xie Y.-L., Liu L.-H., Huang X.-C., Guo Y.-W., Lou L.-G. Scalaradial inhibition of epidermal growth factor receptor-mediated akt phosphorylation is independent of secretory phospholipase A2. J. Pharmacol. Exp. Ther. 2005;314:1210–1217. doi: 10.1124/jpet.105.086520. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino T., Yamamoto M., Uchiyama T. Formations of (5-hydroxy) indole S-(−)-lactic acid, N-acetyl-5-hydroxy-l-tryptophan, and (5-hydroxy) indole carboxylic acid in the metabolism of tryptophan and 5-hydroxy-tryptophan by chromobacterium violaceum. Biosci. Biotechnol. Biochem. 1993;57:1609–1610. doi: 10.1271/bbb.57.1609. [DOI] [Google Scholar]
- 26.Cardellina J.H., II, Nigh D., VanWagenen B.C. Plant growth regulatory indoles from the sponges Dysidea etheria and Ulosa ruetzleri. J. Nat. Prod. 1986;49:1065–1067. doi: 10.1021/np50048a015. [DOI] [Google Scholar]
- 27.Dale J.A., Mosher H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and alpha-methoxy-alpha-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973;95:512–519. doi: 10.1021/ja00783a034. [DOI] [Google Scholar]
- 28.Ohtani I., Kushmi T., Kashman Y., Kakisawa H. A new aspect of the high-field NMR application of Mosher’s method. The absolute configuration of marine triterpene sipholenol A. J. Org. Chem. 1991;56:1296–1298. [Google Scholar]
- 29.Yamanokuchi R., Imada K., Miyazaki M., Kato H., Watanabe T., Fujimuro M., Saeki Y., Yoshinaga S., Terasawa H., Iwasaki N., et al. Hyrtioreticulins A–E, indole alkaloids inhibiting the ubiquitin-activating enzyme, from the marine sponge Hyrtios reticulates. Bioorg. Med. Chem. 2012;20:4437–4442. doi: 10.1016/j.bmc.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 30.Tsukamoto S. (Kumamoto University, Kumamoto, Japan). Personal communication. 2014.
- 31.Bialonska D., Zjawiony J. Aplysinopsins-marine indole alkaloids: Chemistry, bioactivity and ecological significance. Mar. Drugs. 2009;7:166–183. doi: 10.3390/md7020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao R.-H., Peng W.-L., Wang Z.-H., Xu A.-L. β-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007;14:479–500. doi: 10.2174/092986707779940998. [DOI] [PubMed] [Google Scholar]
- 33.Glover D.M., Leibowitz M.H., McLean D.A., Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 34.Naruganahalli K.S., Lakshmanan M., Dastidar S.G., Ray A. Therapeutic potential of Aurora kinase inhibitors in cancer. Curr. Opin. Investig. Drugs. 2006;7:1044–1051. [PubMed] [Google Scholar]
- 35.Gautschi O., Mack P.C., Davies A.M., Lara P.N., Jr., Gandara D.R. Aurora kinase inhibitors: A new class of targeted drugs in cancer. Clin. Lung Cancer. 2006;8:93–98. doi: 10.3816/CLC.2006.n.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information (PDF, 4409 KB)