

Abstract
We previously demonstrated that RanBP9 overexpression increased Aβ generation and amyloid plaque burden, subsequently leading to robust reductions in the levels of several synaptic proteins as well as deficits in the learning and memory skills in a mouse model of Alzheimer's disease (AD). In the present study, we found striking reduction of spinophilin-immunoreactive puncta (52%, p<0.001) and spinophilin area (62.5%, p<0.001) in the primary cortical neurons derived from RanBP9 transgenic mice (RanBP9-Tg) compared to wild-type (WT) neurons. Similar results were confirmed in WT cortical neurons transfected with EGFP-RanBP9. At 6-months of age, the total spine density in the cortex of RanBP9 single transgenic, APΔE9 double transgenic and APΔE9/RanBP9 triple transgenic mice were similar to WT mice. However, in the hippocampus the spine density was significantly reduced (27%, p<0.05) in the triple transgenic mice compared to WT mice due to reduced number of thin spines (33%, p<0.05) and mushroom spines (22%, p<0.05). This suggests that RanBP9 overexpression in the APΔE9 mice accelerates loss of spines and that hippocampus is more vulnerable. At 12-months of age, cortex showed significant reductions in total spine density in the RanBP9 (22%, p<0.05), APΔE9 (19%, p<0.05) and APΔE9/RanBP9 (33%, p<0.01) mice compared to WT controls due to reductions in mushroom and thin spines. Similarly, in the hippocampus the total spine density was reduced in the RanBP9 (23%, p<0.05), APΔE9 (26%, p<0.05) and APΔE9/RanBP9 (39%, p<0.01) mice due to reductions in thin and mushroom spines. Most importantly, RanBP9 overexpression in the APΔE9 mice further exacerbated the reductions in spine density in both the cortex (14%, p<0.05) and the hippocampus (16%, p<0.05). Because dendritic spines are considered physical traces of memory, loss of spines due to RanBP9 provided the physical basis for the learning and memory deficits. Since RanBP9 protein levels are increased in AD brains, RanBP9 might play a crucial role in the loss of spines and synapses in AD.
Keywords: RanBP9, dendritic spines, transgenic mice, cortex, hippocampus, APΔE9 mice
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease of elderly characterized by two neuropathological hallmarks, extracellular amyloid plaques and intraneuronal neurofibrillary tangles (Goedert and Spillantini, 2006). The progression of disease pathology is further accompanied by a marked loss of synapses. Synapse loss which best correlates with cognitive impairment (Dekosky and Scheff, 1990; Scheff et al., 1990; Scheff et al., 2007; Terry et al., 1991) has been reported as an early event in the pathogenesis of AD. In fact, dendritic spines which are considered structural correlates of learning and memory (Alvarez and Sabatini, 2007; Nimchinsky et al., 2002) have been reported to be substantially reduced in AD brains (Merino-Serrais et al., 2013; Fiala et al., 2002; Knobloch and Mansuy, 2008). Since dendritic spines represent the major postsynaptic elements of excitatory synapses in the brain and are fundamental to long-term potentiation (LTP) and long-term depression (LTD), which are considered the predominant cellular mechanisms that underlie learning and memory (Cooke and Bliss, 1993), understanding the mechanisms by which spine loss occurs is important to unravel the pathogenesis of AD.
While there are obvious limitations in studying the spine loss directly in human brains, transgenic mouse models of AD provide great opportunity to study various aspects of spine loss including spatiotemporal correlations between the pattern of spine loss and learning and memory skills. Thus, several transgenic mouse models of AD overexpressing amyloid precursor protein (APP) and/or presenilin 1 (PS1), recapitulate loss of spines (Lanz et al., 2003; Moolman et al., 2004; Tsai et al., 2004; Spires et al., 2005). More recent studies have confirmed loss of spines in the APP/PS1 mice (Meng et al., 2013), J20 mice expressing both the Swedish (K670N/M671L) and the Indiana (V717F) mutations (Pozueta et al., 2013), Tg2576 mice expressing APP with Swedish mutation (Perez-Cruz et al., 2011), PS1 transgenic mice (Auffret et al., 2009) as well as the SAMP8 mouse model of aging (del Vallet et al., 2012). The overexpression of human tau in transgenic mouse model also reduces spine density (Rocher et al., 2010). Thus, rare familial AD (FAD)-associated gene mutations within APP, PS1 as well as tau hyper phosphorylation have been shown to induce spine loss and associated memory deficits. Interestingly, in addition to FAD associated genes, those genes that increase the risk of developing AD by their genetic association also contributes to loss of spines. The APOE4 allele is the strongest risk factor identified so far for developing late-onset AD (LOAD). A recent study provided compelling evidence that the APOE4 allele significantly reduced dendritic spine density which was well-correlated with learning and memory deficits (Rodriguez et al., 2013). However, it is not clear whether other genes that increase the risk of AD or the genes that are associated with progression of AD also adversely affect spine density.
The Ran-binding protein 9 (RanBP9) was first identified as a 55 kDa protein (Nakamura et al., 1998), but later studies by the same group revealed that the full-length RanBP9 is a 90 kDa protein (Nishitani et al., 2001). RanBP9 is ubiquitously expressed in different tissues and cell lines and is highly conserved in different organisms (Rao et al., 2002; Wang et al., 2002). RanBP9 is a multidomain protein that functions as a scaffolding protein by assembling multiprotein complexes in different subcellular regions, thereby mediates diverse cellular functions (Murrin and Talbot, 2007; Suresh et al., 2012). Recently, RanBP9 was found to be within the clusters of RNA transcript pairs associated with markers of AD progression (Arefin et al., 2012), suggesting that RanBP9 might contribute to the pathogenesis of AD. In fact, even before this discovery, we showed for the first time that RanBP9 increased Aβ generation by 4-fold in a variety of cell cultures (Lakshmana et al., 2010), primary neurons (Lakshmana et al., 2009) as well as mouse brains (Lakshmana et al., 2012), consequently leading to increased amyloid plaque burden (Lakshmana et al, 2012). Because RanBP9 protein levels are increased in J20 (Woo et al., 2012) and APΔE9 mice (Wang et al., 2013) as well as in the AD brains (Lakshmana et al., 2010; Palavicini et al., 2013a), RanBP9 is expected to positively contribute to the increased Aβ generation and to the associated synaptic and behavioral deficits seen in AD patients and in mouse models of AD. In line with these predictions, we recently demonstrated that RanBP9 overexpression in the APΔE9 mice led to learning and memory deficits in both the T maze (Palavicini et al., 2013a) and Barnes maze paradigms (Woo et al., 2012). These deficits appear to be due to RanBP9-mediated synaptic damage as reflected by reduced levels of synaptic proteins in the APΔE9 mouse brains (Lakshmana et al., 2012; Palavicini et al., 2013a; 2013b). Most importantly, we confirmed the inverse relationships between the protein levels of spinophilin, a marker of dendritic spines and RanBP9 levels in the synaptosomes derived from both mouse brains and AD brains (Palavicini et al., 2013a). These pieces of evidence taken together imply that RanBP9 might play a primary role in the loss of synapses in AD.
The primary objective of the present study was to examine whether RanBP9 overexpression in APΔE9 mice leads to alterations in dendritic spines. Remarkably, RanBP9 overexpression in APΔE9 mice accelerated loss of spines already at 6-months of age. Thus, the loss of spines observed in the present study provides the physical basis for the previously observed synaptic and behavioral deficits due to RanBP9 overexpression in the APΔE9 mouse model of AD.
Material and methods
Mice
All animal experiments were carried out based on ARRIVE guidelines and in strict accordance with the National Institute of Health's ‘Guide for the Care and Use of Animals’ and as approved by the Torrey Pines Institute's Animal Care and Use Committee (IACUC). Generation of RanBP9-Tg mice have been described previously (Lakshmana et al., 2012). The RanBP9 specific primers used in the polymerase chain reaction (PCR) is as follows. The forward primer is 5′ – gcc acg cat cca ata cca g -3′, and the reverse primer is 5 – tgc ctg gat ttt ggt tct c – 3′. Positive mice were then backcrossed with native C57Bl/6 mice and the colonies were expanded. RanaBP9-Tg line 629 was used to breed with B6.Cg-Tg, APPswe, PSEN1ΔE9 (APΔE9) mice for generating triple transgenic mice (APΔE9/RanBP9). We obtained APΔE9 mice from Jackson Labs (Bar Harbor, Maine, USA). These double transgenic mice express a chimeric mouse/human APP (Mo/HuAPP695swe) driven by prion promoter and a mutant human presenilin 1 (PS1-ΔE9) also driven by prion promoter for neuronal expression of transgenes.. These APΔE9 transgenic mice were generated by co-injection of APP695swe and PS1-ΔE9 encoding vectors controlled by their own mouse prion promoter element. These mice were backcrossed to maintain them in the C57Bl/6 background, expanded and genotyped to confirm the transgene using the following primers. The forward primer is 5′ – gac tga cca ctc gac cag gtt ctg – 3′ and the reverse primer is 5 - ctt gta agt tgg att ctc ata tcc g – 3′. The mice were fed with ad libitum food and water all the time. The food is the irradiated global rodent chow from Harlan. The mice were maintained in a 12-hour light/dark cycle at a temperature of 21-23°C and a humidity of 55±10. After weaning, mice were kept in home cages comprising single sex, single genotype and groups of only 5 mice per cage. All of the mice lived in an enriched environment with increased amounts of bedding and nesting materials.
Primary neuronal cultures
To prepare cortical primary neuronal cultures, cortices from both the hemispheres were separated and freed from meninges under a dissection microscope from newborn (P0) pups of RanBP9 transgenic mice overexpressing flag-tagged RanBP9 (RanBP9-Tg) or from wild-type (WT) mice. The cortical tissue was washed 3X with Ca2+/Mg2+-free Hanks' balanced salt solution containing penicillin/streptomycin. The tissues were dissociated in 0.27% trypsin (in 10% Dulbecco's modified Eagle's medium/ Hanks' balanced salt solution) by incubating at 37 °C for 30 min. Neurons were collected by centrifugation and re-suspended in 10% Ham's F-12 medium (cat # 10-080-CV, Media Tech, Pittsburgh, PA, USA) containing penicillin/streptomycin. The neurons were further dissociated by triturating 20 times with a Pasteur pipette and passed through a cell strainer. After centrifugation, the neurons were re-suspended in neurobasal medium containing 2% B-27 supplement (cat# 1-7504-044, Life Technologies, Grand Island, NY, USA), glutamine (cat# 25030-081, Life Technologies), pyruvate (cat# 11360, Life Technologies), and penicillin/streptomycin (50 units/ml penicillin, 50 μg/ml streptomycin, cat # 30-002-C1, Media Tech) and plated on to a sterile coverslip in the 6-well plate. The coverslips (GG-18-PDL, Neuvitro, Germany) used were especially made for primary neurons, coated with Poly-D-Lysine (PDL). Half of the growth medium was changed twice weekly.
Sholl analysis
To understand the influence of RanBP9 on dendritic intersections, primary cortical neurons from WT and RanBP9-Tg mice were immunostained with MAP2 (1:150 dilution) and dendritic intersections were calculated by Sholl analysis. ImageJ software with Sholl analysis plugin was used for automated quantitation. This plugin automates the task of doing Sholl analysis on a neuron. Its algorithm is based on how Sholl analysis is done manually by creating a series of concentric circles around the soma of the neuron, and counts how many times the neuron intersects with the circumference of these circles. The images were first converted into 8-bit grayscale images. Thresholding was done to maintain similar background and noise on all neurons. The pixels were converted into microns using the set scale menu. Using the selection tool, a point at the center of soma was selected and the plugin was run. The starting radius, ending radius, radius step size and radius span were set. The number of intersections of dendrites was calculated with concentric spheres positioned at radial intervals of 2 μm. The results were saved as a picture file and also exported to text file. The dendritic morphology and spine quantification were done by a blinded analyzer. The dendritic intersections were quantified from a total of 30 neurons per genotype in 3 independent experiments.
Immunostaining of primary cortical neurons with spinophilin, MAP2 and GFP antibodies
Primary cortical neurons derived from RanBP9-Tg and age-matched WT control mice were immunostained for microtubule-associated protein, MAP2 (cat# M4403, Sigma-Aldrich, St Louis, MO, USA) at 1:150 dilution and spinophilin (cat # 9061S, Cell Signaling Danvers, MA, USA) at 1:150 dilution at 21 days in vitro (DIV) to detect endogenous proteins. In another set of experiments, primary neurons derived from WT mice were transiently transfected with either EGFP-N1 control vector or EGFP-N1-RanBP9 construct as follows. The pEGFP-N1 plasmid was purchased from Clontech (cat# 6085-1, Mountain View, CA, USA). EGFP-RanBP9-FL construct was received from Dr. Hideo Nishitani, Kyushu University, Japan. The primary cortical neurons derived from WT mice were transiently transfected with either EGFP-N1 control plasmid or EGFP-N1-RanBP9-FL construct on 15DIV and the transgene was allowed to be expressed until 21DIV. Transfections were carried out using nanoparticle based magnetofection called Neuromag following manufacturer's instructions. Briefly, the Neuromag reagent was vortexed and placed in a microfuge. A 2.0 μg of plasmid DNA was diluted in 400 μL of neurobasal medium without either B27 supplement or penicillin/streptomycin. The DNA solution was then added to the Neuromag solution and vortexed gently for about 5 seconds and incubated at room temperature for 30 minutes. The Neuromag/DNA complexes were then added on to the cultures drop by drop and the dish was shaken gently to ensure uniform distribution. The culture plate was placed on a magnetic plate in a CO2 incubator for 20 minutes. Following 20 minute incubation, the magnetic plate was removed and the neurons were grown in a CO2 incubator under standard conditions. We could reproducibly achieve up to 30% of transfection efficiency using Neuromag and the magnetic plate. At 21DIV, co-immunostaining was done at 1:150 dilutions for GFP (cat # G10362, Life Technologies, Grand Island, NY, USA) and at 1:150 dilutions for spinophilin (cat # 9061S, Cell Signaling, Danvers, MA, USA). following procedures as described previously (Lakshmana et al., 2010). The images were converted in to black and white, 8 pixels and the density of spinophilin was quantified from 36 neurons per genotype in 3 independent experiments using imageJ software.
Tissue extraction and immunoblotting
Mouse brain tissues from four different genotypes, viz., WT, RanBP9-Tg, APΔE9 and APΔE9/RanBP9 triple transgenic mice were dissected and cortex was removed on ice. Cortical lysates were prepared from 6-months-old mice from all four genotypes. In brief, we anesthetized the mice with isoflurane, decapitated immediately and rapidly removed the brain tissue in to 1% NP40 buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.02% sodium azide, 400 nM microcystine-LR, 0.5 mM sodium vanadate and 1% sodium Nonidet P-40) containing complete protease inhibitor cocktail for use with mammalian cell and tissue extracts (cat # P8340, Sigma, St. Louis, USA). Tissue was homogenized using Power Gen 125 (Fisher Scientific, Pittsburgh, USA) and centrifuged at 100,000 g for 1 hr. Protein concentrations from each sample were measured by bicinchoninic acid (BCA) method (cat # 23235, Pierce Biotechnology Inc., Rockford, USA). Equal amounts of proteins were loaded into each well and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes, blocked with 5% milk and incubated overnight with primary antibodies followed by one hour incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies. The primary antibodies used were the polyclonal antibody CT15 (against c-terminal 15 residues of APP) has been described previously (Lakshmana et al., 2009, 2010). CT15 antibody recognizes full-length APP and c-terminal fragments and was used at 1:10,000 dilutions. Monoclonal antibody against RanBP9 was produced by immunizing mice with a peptide corresponding to 146–729 amino acids of RanBP9 (mouse monoclonal used at 1:1000 dilutions). Anti-flag-tag antibody, M2 which recognizes N- or C-terminus flag fusion proteins was used at 1:1000 dilutions and was purchased from Sigma-Aldrich (cat # F3165, St. Louis, USA). Mouse monoclonal antibody against beta-actin (cat # A00702, used at 1:5000 dilution, recognizes mouse and human beta actin protein) was purchased from Genscript USA Inc. (Piscataway, NJ, USA). The secondary antibodies such as peroxidase-conjugated AffiniPure goat anti-mouse (Code # 115-035-146; 1:5000 dilutions) and anti-rabbit (code # 111-035-144; 1:5000 dilution) IgGs were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The protein signals were detected using Super Signal West Pico Chemiluminescent substrate (cat # 34080, Pierce, USA).
DiI labeling, confocal microscopic imaging and analysis of dendritic spines
Mice were perfused using 1.5% para formaldehyde (PFA) and 0.02% glutaraldehyde in 1X PBS. Brains were dissected, post fixed overnight and rinsed in 1X PBS overnight at 4 °C. A 200-300 μm coronal brain sections were taken using a Vibratome (Leica, VT1000S). The carbocyanin dye, DiI (1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindo-carbocyanine perchlorate) has been successfully used to label neuronal dendrites and spines by many investigators (Hongpaisan et al., 2007; Kim et al., 2007; Hongpaisan et al., 2013). Therefore, DiI (5% in dimethylbutane)-filled glass capillaries (Papa et al., 1995) were placed in the hippocampal and cortical regions and left overnight at 4°C in PBS. Glass pipettes (cat # TW100F-4, thickness, 1.0 mm and length, 4 inches) were purchased from World Precision Instruments (Sarasota, FL, USA) and pulled using an electrode-puller. The sharp edges were cut-off and the blunt end were inserted in to the bottle containing 5% DiI. It takes only few seconds for the dye to get in to the glass electrode by capillary action. Overnight was sufficient for the dye to diffuse throughout the neuron. We did not notice any non-specific staining and the stained neurons showed uniform distribution of the dye throughout the neuron. The brain sections were mounted on glass slides, using initially PBS as mounting medium and once the sections were properly aligned, PBS was replaced with another mounting medium. To avoid possible dehydration-induced shrinkage of dendritic structures and dye bleaching, we used glycerol-based mounting medium that also contained DAPI to stain nuclei (Vectashield), purchased from Vector Laboratories (Burlingame, CA, USA). Pyramidal neuronal dendrites stained with DiI (excitation, >510 nm and emission >568 nm) in the layer 6 of cortex and the CA1 region of the hippocampus were imaged by confocal laser scanning microscopy (Nikon C1Si laser scanning multispectral confocal microscope; Nikon, Melville, NY, USA) using 63x oil immersion objective (1.4NA). A stack of confocal images taken every 0.1 μm was collected to obtain all dendritic spines of an individual dendritic shaft.
A neuron injected with DiI is shown in Fig. 1A. To count spines, a region of interest (ROI) measuring 20 μm dendritic segment was randomly selected as shown in Fig. 1B. Further morphological reconstruction was performed using NeuronStudio, neuron morphology reconstruction software developed by Wearne et al (2005) and Rodriguez et al (2006). A typical example of 3D reconstruction of a 20 μm dendritic segment by NeuronStudio is shown in Fig. 1C. While quantitation of total spine density may provide some valuable information, quantifying different types of spines provides additional valuable information since spine morphology on which the spine classifications are based determines the strength, stability and function of excitatory synaptic connections. The most commonly used nomenclature divides spines in to 3 categories based on their relative length and head thickness (Peters et al., 1970). In our analysis, a spine was deemed thin if its head thickness was ≤ 0.65 μm and its neck length was ≥ 0.65 μm. A stubby spine has only a head ≤ 0.65 μm with no distinct neck. A spine with both its thickness and length ≥ 0.65 μm was considered as mushroom (Fig. 1E). Dendritic segment reconstructed by NeuronStudio (Fig. 1C) was used to classify the spines, which are labeled and color-coded as in Fig. 1D.
Fig.1.
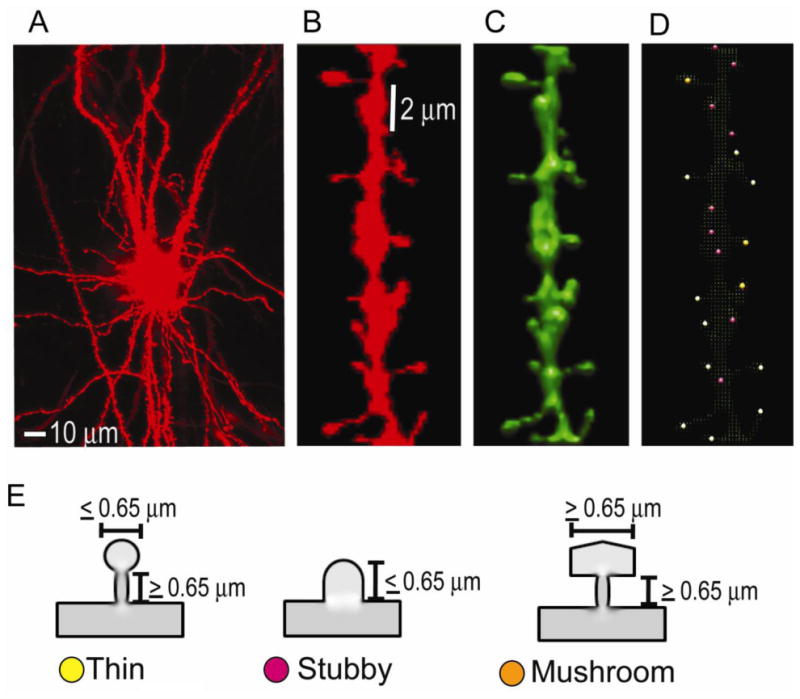
An example of diolistic dye, Dil-stained confocal image of a neuron, dendritic segment and dendritic spines used in the study. (A), A cortical neuron completely filled with DiI throughout the soma and dendritic segments.(B), A 20 μm segment of a dendrite with different types of spines. (C), The reconstructed 3D image of the dendritic segment in B by the NeuronStudio software. (D), The NeuronStudio image of the dendrite with color-coded spine types classified based on head to neck ratio. Thin, yellow; stubby, magenta; mushroom, orange. (E), Schematic diagrams illustrating thin (yellow), stubby (magenta) and mushroom (orange) spines with the indicated neck and head diameters.
Confocal images were processed using Image Pro Plus 3D Suite. Five 20 μm dendritic segments per mice, in a total of five mice, per genotype per age group in the cortical and hippocampal brain regions were analyzed. They were picked in an unbiased random fashion. Informative stack images were selected for each dendritic segment, and they were deconvoluted using the sharp stack 3D blind deconvolution feature. Image projections for each dendritic segment were generated using the extended depth of field feature (maximum intensity) and such projected images were modeled in 3D using the 3D constructor feature of the software suite.
Statistical analysis
The spinophilin-immunoreactive puncta was processed by imageJ and the data was analyzed by Student's t-test. We used two-tailed p value assuming populations may have different standard errors. To find the differences in the number of spines in the cortex and the hippocampus among the four genotypes of mice, one way analysis of variance (ANOVA) followed by Bonferroni multiple comparison post-hoc test was used using Instat3 software (GraphPad Software, San Diego, CA, USA). The data presented for spines are mean ± SEM per micron of the dendritic segment. The data were considered significant only if the p<0.05, * indicates p<0.05, **, p<0.01 and ***, p<0.001.
Results
RanBP9 overexpression robustly reduces dendritic intersections and spinophilin-immunoreactive puncta in primary cortical neurons
Based on the evidence that RanBP9 significantly reduced both pre and postsynaptic proteins in mouse brains (Lakshmana et al., 2012; Wang et al., 2014) which in turn led to adverse effects on the learning and memory skills (Palavicini et al., 2013a; 2013b), we were interested to quantify spinophilin-immunoreactive puncta in primary neurons overexpressing RanBP9. In all brain regions, spinophilin is localized specifically to the heads of dendritic spines and ∼93% of dendritic spines are thought to contain spinophilin (Hao et al., 2003). Therefore, spinophilin immunoreactivity makes it an excellent marker for quantitative assessment of spine density (Hao et al., 2003). First, we confirmed the expression of flag-RanBP9 in primary neurons derived from RanBP9-Tg mice using both flag antibody and RanBP9-specific monoclonal antibody (Fig. 2A). The generation and characterization of RanBP9 monoclonal antibody has been described previously (Denti et al., 2004), and has been used extensively in our laboratory (Lakshmana et al., 2009, 2010, 2012; Palavicini et al., 2013a, 2013b; Wang et al., 2013; Wang et al., 2014). Similarly, transient expression of EGFP-RanBP9 in primary neurons derived from WT mice was confirmed by both GFP antibody and RanBP9 antibodies (Fig. 2B). Initially, we quantified the number of dendritic intersections stained by MAP2 in a total of 30 neurons from three independent experiments using Sholl analysis plugin in the imageJ software. ANOVA followed by a post-hoc test revealed a highly significant decrease (p<0.001) in the number of intersections in the cortical neurons derived from RanBP9-Tg mice compared to those derived from WT mice (Fig. 3A & B). To quantify spinophilin puncta, primary cortical neurons derived from RanBP9-Tg or WT mice were double-labeled with MAP2 (red) and spinophilin (green) antibodies. Quantitation of spinophilin puncta per micron area on the dendrites from a total of 36 neurons per genotype from three independent experiments showed significant alterations. Thus, spinophilin puncta were reduced by 52% (p<0.001) in cortical neurons derived from RanBP9-Tg mice when compared to those derived from WT controls (Fig. 4A &B). Spinophilin area (calculated as the area occupied by spinophilin puncta to the total dendritic area) on the dendrite was also reduced by 62.5% (p<0.001) in RanBP9-Tg neurons compared to WT neurons (Fig. 4A &B).
Fig. 2.
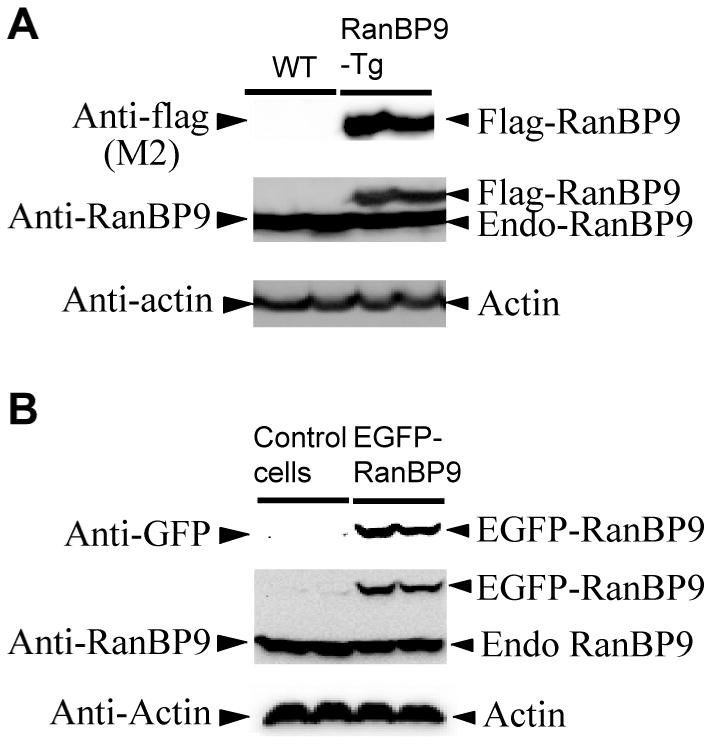
Confirmation of expression of flag-RanBP9 and EGFP-RanBP9 in primary cortical neurons. (A), Lysates prepared from cortical neurons derived from RanBP9-Tg mice at 21DIV were subjected to SDS-PAGE analysis and probed with flag antibody to detect flag-RanBP9 and RanBP9 monoclonal antibody to detect both flag-RanBP9 and endogenous RanBP9. (B), Primary cortical neurons derived from WT mice were transiently transfected on 19DIV with or without EGFP-RanBP9 and on 21DIV lysates were prepared and subjected to SDS-PAGE analysis. GFP antibody detected the expression of EGFP-RanBP9 and RanBP9 antibody detected both EGFP-RanBP9 and endogenous RanBP9. Actin was detected as a loading control.
Fig. 3.
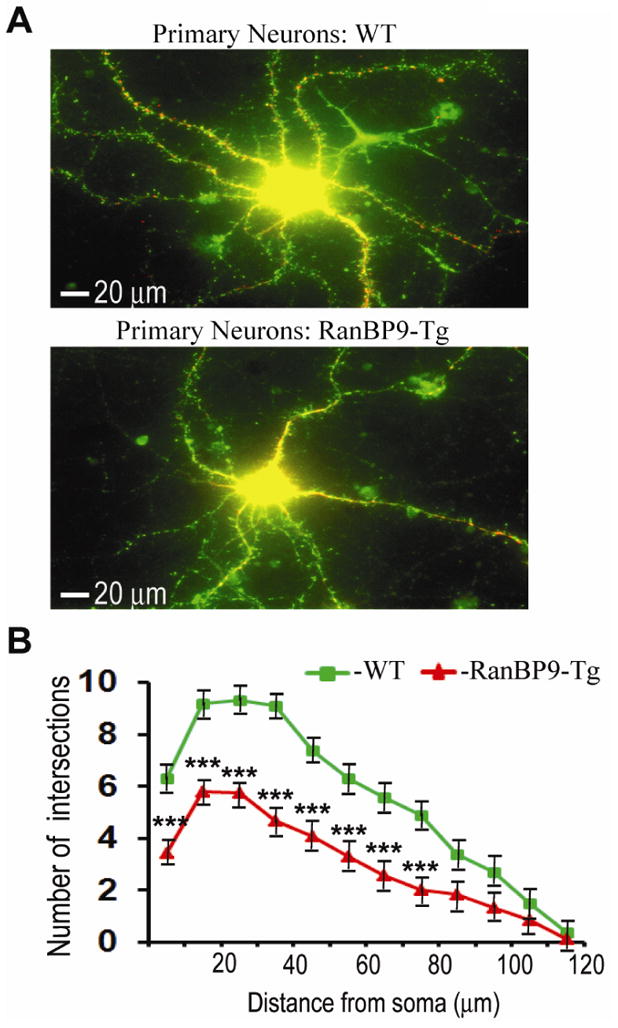
RanBP9 overexpression reduces dendritic outgrowth of primary cortical neurons at 21DIV. (A), Cortical neurons were derived from either WT or RanBP9-Tg mice and cultures were maintained until 21DIV. Neurons were co-stained with anti-MAP2 (red, 1:150 dilution) to stain the dendritic arbor and anti-spinophilin (green, 1:150 dilution) to stain dendritic spines and a representative merged image for each of WT and RanBP9-Tg neurons are shown. (B), Sholl analysis revealed the extent of reduction in the number of intersections. The ordinate represents the distance from soma in μm and the abscissa represents number of dendritic intersections that cross along the concentric circles at defined distance from soma. Significant differences in the dendritic arbor were revealed by ANOVA followed by post hoc test. ***, p<0.001 in RanBP9 cortical neurons versus WT. The data are mean + SEM of 30 neurons in three independent experiments.
Fig. 4.
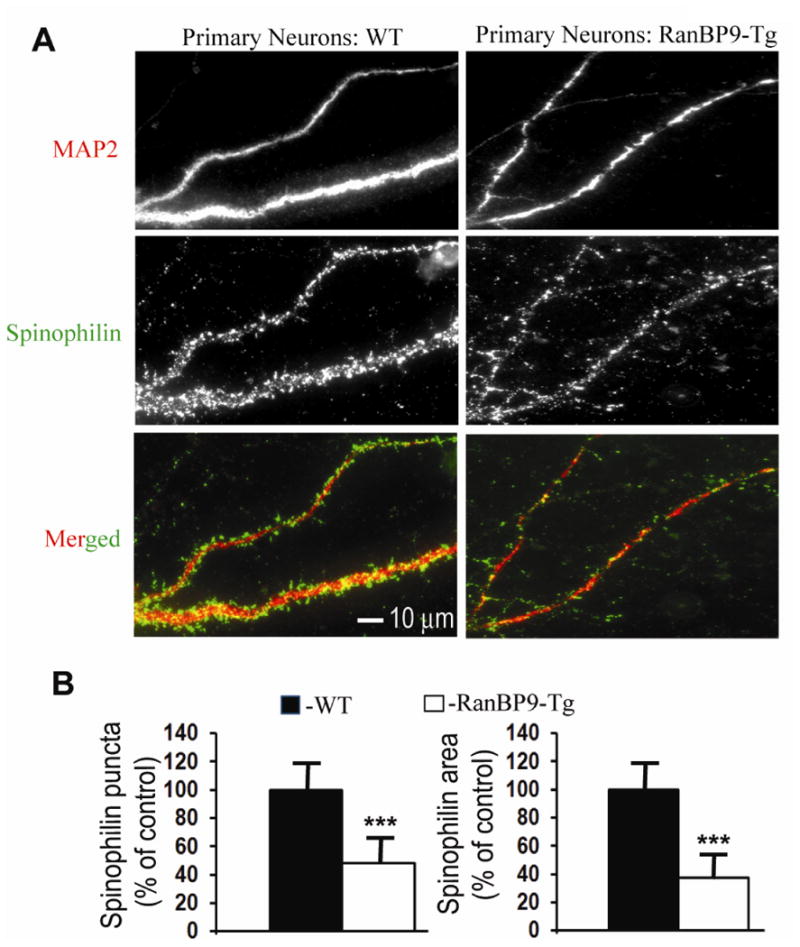
Spinophilin-immunoreactive puncta are robustly decreased in primary cortical neurons derived from RanBP9-Tg mice. (A), Primary cortical neurons derived from P0 pups of RanBP9-Tg mice or WT mice were cultured and maintained for 21DIV. At 21DIV, the neurons were co-immunostained for MAP2 to label the dendritic arbor (red) and spinophilin to label spines (green) and the images were acquired in their respective channels. Merged images show spinophilin-immunoreactive puncta on the dendrites. (B), Quantitation of the number of spinophilin-immunoreactive puncta and the spinophilin area by image J showed significantly reduced numbers in the cortical neurons derived from RanBP9-Tg mice compared to WT neurons. Student's t-test revealed significant differences. ***, p<0.001 in the RanBP9-Tg neurons compared to WT neurons. The data are mean ± SEM, n=3 independent experiments with 12 neurons analyzed per experiment.
To further confirm these findings, primary cortical neurons derived from WT mice were cultured until 15DIV and transfected with EGFP-RanBP9 or the control vector, EGFP-N1. At 21DIV neurons were immunostained for GFP (green) and spinophilin (red). The merged image in Fig 5A shows red colored spinophilin puncta on the green dendrites. Similar to neurons from transgenic mice, WT neurons transfected with EGFP-RanBP9 showed reduction of spinophilin puncta by 50.5% (p<0.001) and spinophilin area by 49% (p<0.001) when compared to EGFP-N1-transfected neurons (Fig. 5A &B). The data shown are an average of 36 neurons per genotype from three independent experiments. Thus, RanBP9 overexpression has adverse effects on the spinophilin immunoreactivity in the growing primary neurons.
Fig. 5.
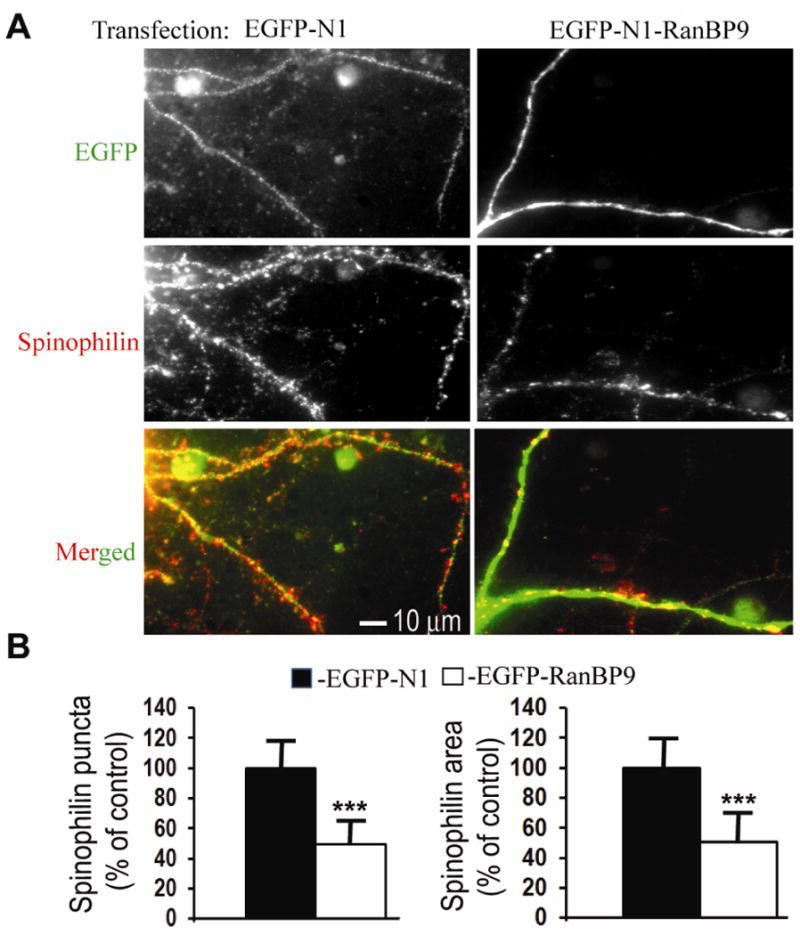
Spinophilin-immunoreactive puncta are robustly decreased in primary cortical neurons transiently overexpressing RanBP9. (A), Primary cortical neurons derived from P0 pups of WT mice were cultured and maintained until 15DIV. At 16DIV, the neurons were transiently transfected with either an EGFP-N1-RanBP9-FL construct or the control vector EGFP-N1 by a nanoparticle- based transfection reagent, Neuromag (magnetofection). At 21DIV, the neurons were co-immunostained for GFP to label the dendritic arbor (green) and spinophilin to label spines (red) and the images were acquired in their respective channels. Merged images show spinophilin-immunoreactive puncta on the dendrites. (B), Quantitation of the number of spinophilin-immunoreactive puncta and the spinophilin area by image J showed significantly reduced numbers in the cortical neurons transfected with EGFP-RanBP9 compared to neurons transfected with the EGFP-N1 control vector. Student's t-test revealed significant differences. ***, p<0.001 in the cortical neurons transfected with EGFP-RanBP9 compared to EGFP-N1 vector transfected neurons. The data are mean ± SEM, n=3 independent experiments with 12 neurons analyzed per experiment.
Confirmation of expression of transgenes in the RanBP9-Tg and APΔE9 mouse brains
To test whether RanBP9 overexpression indeed results in reduced spine density in mouse brains, we wanted to quantify spine density in four genotypes of mice; RanBP9-Tg, APΔE9, APΔE9/RanBP9 triple transgenic and age-matched WT controls. The generation and characterization of flag-tagged RanBP9-Tg mice has been described previously (Lakshmana et al., 2012). Among several of RanBP9-Tg lines, we selected line 629 for this study because of high levels of transgene expression in most brain regions studied including cortex and hippocampus. The reason for higher levels of transgene expression in line 629 is not known, but might depend on the integration site of the transgene. Expression of exogenous RanBP9 was determined using flag-specific antibody (M2) which detected flag-tagged RanBP9 only in the RanBP9-Tg and APΔE9/RanBP9 triple transgenic mice as shown in Fig. 6 (panel 1). Expression of both flag-RanBP9 and endogenous RanBP9 was confirmed using RanBP9-specific monoclonal antibody (Fig. 6, panel 2). Similarly high levels of expression of APP were detected only in the APΔE9 and APΔE9/RanBP9 genotypes, compared to either age-matched WT or RanBP9 single transgenic mice (Fig. 6, panel 3).
Fig. 6.
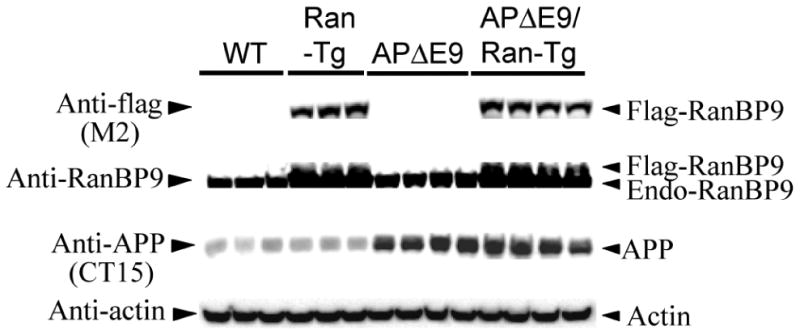
Confirmation of transgenes expression in the RanBP9 single transgenic, APΔE9 double transgenic and APΔE9/RanBP9 triple transgenic mice. Brain homogenates prepared from different genotypes of mice and age-matched WT controls at 6-months of age were subjected to SDS-PAGE electrophoresis and probed with anti-flag antibody to detect exogenously expressed flag-tagged RanBP9 in the RanbP9-Tg mice and triple transgenic mice only (panel 1). RanBP9-specific monoclonal antibody confirmed the expression of exogenously expressed flag-RanBP9 as well as endogenous RanBP9 (panel 2). The CT15 antibody (epitope last 15 residues of APP) detected exogenously expressed APP only in the APΔE9 and APΔE9/RanBP9 genotypes (panel 3). Actin was used as loading control (panel 4).
RanBP9 overexpression in APΔE9 mice exacerbates loss of spines in the apical dendrites of layer 6 of cortex at 12 months of age
To determine whether cognitive deficits by RanBP9 observed previously are due to a loss of spine density, we quantified the thin, stubby, mushroom and total spine numbers in the apical dendrites of pyramidal neurons in layer 6 of cortex in the APΔE9, APΔE9/RanBP9, RanBP9-Tg and age-matched WT controls at 6- and 12-months of age. Pyramidal neurons were selected because they are the most abundant and well-characterized cell type in the cortex (Mountcastle, 1998). Additionally, only pyramidal neurons project outside the cortex (Crick and Asanuma, 1986) and are the primary source of connections between different cortical areas (Braitenberg, 1978). Furthermore, pyramidal neurons are found primarily in those brain regions that are associated with advanced cognitive functions, suggesting that these neurons are likely to have a major role in information processing and cognitive function relevant to AD. In contrast to other cortical lamina, layer 6 contains multiple distinct classes of pyramidal neurons (Douglas and Martin, 2004), and regulate the activity in other layers such as layer 4 (Lee and Sherman, 2009), which all point to a pivotal role for layer 6 in controlling the flow of information in to and out of cortical networks. At 6-months of age, neither of the spine types nor the total spine numbers were altered (Fig. 7A & B). However, by 12 months of age, thin spine numbers were significantly reduced (30%, p<0.05) only in the APΔE9/RanBP9 triple transgenic mice compared to WT controls (Fig. 7A & B). While stubby spines remained unaltered in any of the genotypes studied, mushroom spine density was reduced in RanBP9 (43%, p<0.01), APΔE9 (28%, p<0.05) and APΔE9/RanBP9 (50%, p<0.01) mice compared to WT controls (Fig. 7A & B). Consistent with these reductions, the total spine density was decreased in RanBP9-Tg (22%, p<0.05), APΔE9 (19%, p<0.05) and APΔE9/RanBP9 (33%, p<0.01) mice compared to WT controls. Most importantly, APΔE9/RanBP9 triple transgenic mice had a significantly stronger reductions (14%, p<0.05) in the number of total spines when compared to APΔE9 double transgenic mice (Fig. 7A & B). Overall, these results suggest that at 12-months of age, RanBP9 exacerbates loss of spines in the cortex mostly due to reductions in the thin and mushroom type of spines.
Fig. 7.
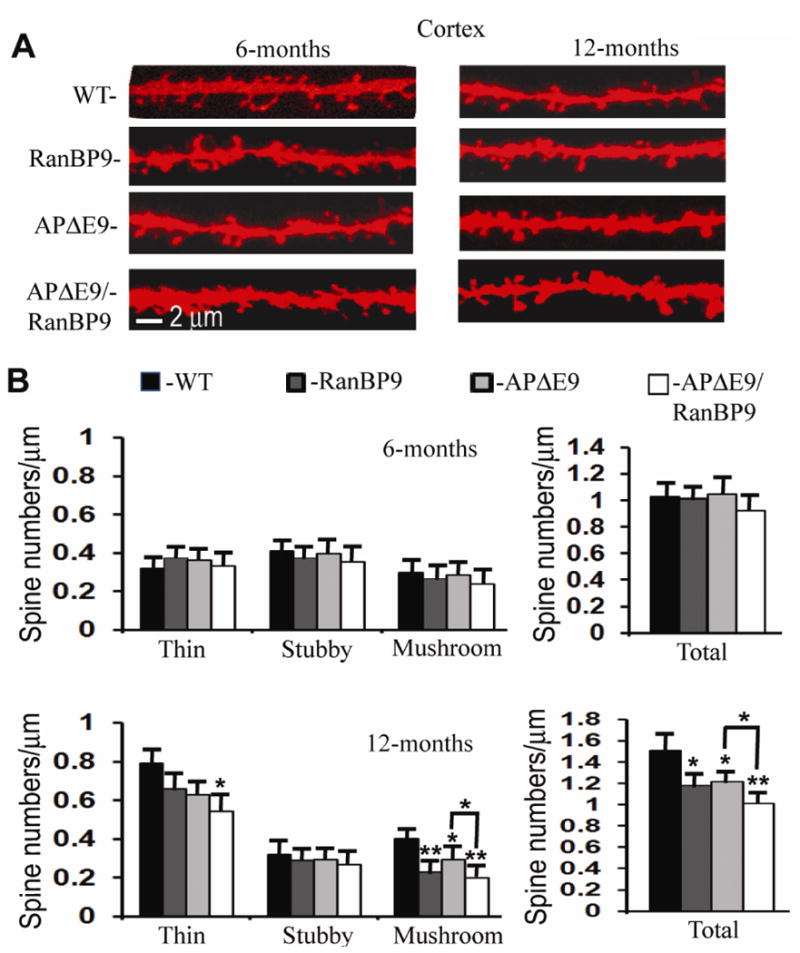
RanBP9 overexpression reduces total spine density in the cortex at 12-months of age but not at 6 months. (A), Representative examples of dendrite and spine morphologies of cortical neurons at 6- and 12-monthsof age used for quantitation of spines in different genotypes of mice. (B), Quantitation of Image-pro and NeuronStudio-processed images of 12-month old mice revealed significant reductions in total spines in the RanBP9-Tg (22%), APΔE9 (19%), and APΔE9/RanBP9 (33%) mice compared to WT mice. APΔE9/RanBP9 mice showed further reductions in total spines compared to APΔE9 (14%) mice. The reduction in total spines is due to reduced mushroom spines in the RanBP9-Tg (23%), and APΔE9 (28%) genotypes, while in the APΔE9/RanBP9-Tg genotype the reduction was due to both thin (32%) and mushroom spines (50%). ANOVA followed by post hoc Bonferroni multiple comparison test revealed significant differences. *, p<0.05, **, p<0.01 as indicated compared to WT mice or APΔE9 mice. The data are mean ± SEM, n=5 mice for each genotype.
RanBP9 overexpression in the APΔE9 mice accelerates loss of apical dendritic spines already by 6-months of age in the CA1 region of the hippocampus
Since the hippocampus is central to spatial memory regulation, we monitored the spine density alterations in the hippocampus as well. In both AD brains (West et al., 1994; Padurariu et al., 2012) and tauopathy mouse model (Spires et al., 2006), the most prominent neuronal cell loss occurs in the CA1 region relative to other hippocampal sub regions. Moreover, CA1 region is the output of the hippocampus connecting directly with the subiculum and entorhinal cortex, which are pivotal for spatial memory. The apical dendrites in cortex and the hippocampus play crucial role in memory, learning and sensory associations by modulating the excitatory and inhibitory signals. Therefore, we quantified spine density in the apical dendrites of CA1 region. At 6-months of age, only APΔE9/RanBP9 triple transgenic mice showed significantly reduced number of thin spines (33%, p<0.05) as well as total spines (27%, p<0.01), while RanBP9-Tg and APΔE9 mice were similar to WT (Fig. 8A & B). Interestingly, a significant reduction (26%, p<0.05) in total spine numbers were also observed when compared between APΔE9 and APΔE9/RanBP9 mice. At 12-months of age, the alterations in spine density were even more severe. While stubby spines remained unaltered, thin spines were reduced in the APΔE9/RanBP9 mice (39%, p<0.05) and mushroom spines were reduced in RanBP9-Tg (40%, p<0.01), APΔE9 (46%, p<0.01) and APΔE9/RanBP9 (63%, p<0.01) genotypes (Fig. 8A & B). Total spine density was reduced by 23% (p<0.01) in RanBP9-Tg and 26% (p<0.01) in the APΔE9 mice, while the reduction in total spines in the APΔE9/RanBP9 triple transgenic mice was 39% (p<0.01) compared to WT controls. When APΔE9/RanBP9 triple transgenic mice were compared against APΔE9 mice, total spines were reduced by about 16% (p<0.05) (Fig. 8A & B). The data presented are an average of 25 dendritic segments from five mice per genotype per age group. Similar to cortex, spine numbers in the hippocampus also confirmed that RanBP9 overexpression consistently reduces spine density.
Fig. 8.
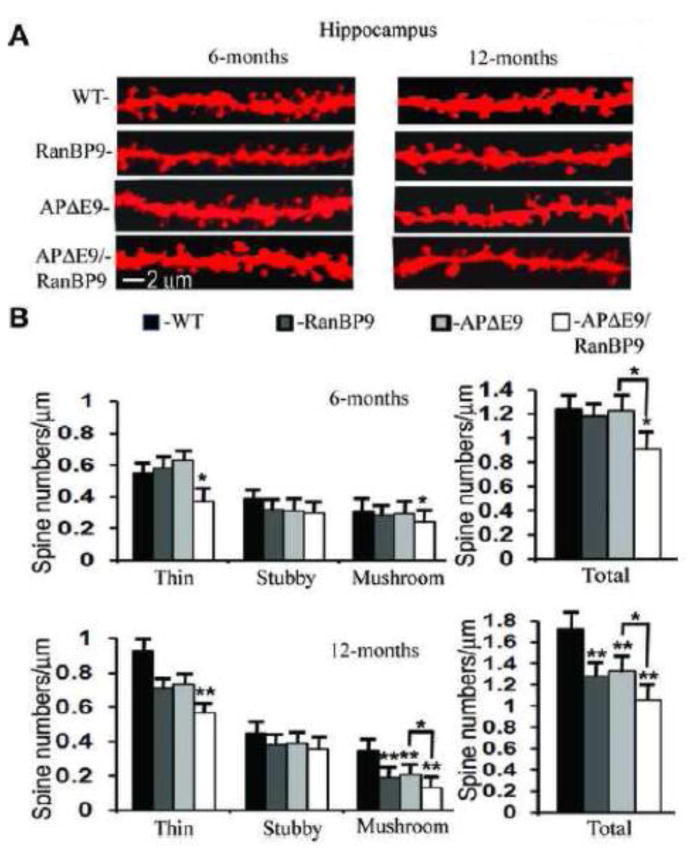
RanBP9 overexpression accelerates loss of spines in the hippocampus at both 6- and 12-months of age. (A), Representative examples of dendrite and spine morphologies of hippocampal neurons at 6- and 12-months of age used for quantitation of spines in different genotypes of mice. (B), Quantitation of Image-pro and NeuronStudio-processed images of 6- month old mice revealed a significant reduction in total spines only in the APΔE9/RanBP9 mice (27%), compared to WT mice which was due to reduced numbers of thin spines (33%) and mushroom spines (22%). At 12 months, reductions in total spines were evident in the RanBP9-Tg (23%), APΔE9 (26%) and APΔE9/RanBP9 mice (39%) compared to WT mice. The reduction in total spines in the APΔE9 mice (46%) and in ranBP9-Tg mice (40%) is due to reductions in the mushroom spines, while in the APΔE9/RanBP9 mice the reductions were due to reduced levels of both thin (39%) and mushroom (63%) spines. ANOVA followed by post hoc Bonferroni multiple comparison test revealed significant differences. *, p<0.05, **, p<0.01 as indicated compared to WT mice or APΔE9 mice. The data are mean ± SEM, n=5 mice for each genotype.
Discussion
Since RanBP9 overexpression led to robust increase in the levels of Aβ and amyloid plaque burden (Lakshmana et al., 2009; Lakshmana et al., 2012) which was accompanied by a significant decrease in the levels of synaptic proteins (Lakshmana et al., 2012, Wang et al., 2014) and by learning and memory deficits (Woo et al., 2012; Palavicini et al. 2013a; 2013b), we next wanted to investigate whether RanBP9 overexpression in a mouse model of AD has any influence on dendritic spines. It is well established that alterations in dendritic spines, especially the large spines, correlate well with changes in memory and cognitive function. For this reason, dendritic spines are considered physical traces of memory. Until recently, the lack of methods for high resolution imaging of dendritic spines has limited the studies on the alteration of spines in animal models of AD. The introduction of membrane-soluble fluorescent dyes has enabled us to examine dendritic spines in a rapid and reproducible manner.
The reduced spinophilin puncta observed in primary neuronal cultures derived from RanBP9-Tg mice as well as WT neurons transfected with EGFP-RanBP9 confirmed our earlier studies showing similar reductions in spinophilin immunoreactivity in APΔE9 mice overexpressing RanBP9 (Palavicini et al., 2013a). We also confirmed previously, by immunoblotting, that the extent of spinophilin reduction was dependent on the expression levels of RanBP9 (Palavicini et al., 2013a). The fact that spinophilin levels were not altered in those brain regions where exogenous RanBP9 was not expressed directly implicates RanBP9 in the reduced levels of spinophilin. Further, RanBP9 protein levels were inversely proportional to the spinophilin levels in the synaptosomes isolated directly from AD brains (Palavicini et al., 2013a). Therefore, the present results on spinophilin puncta in primary neuronal cultures complements our previous results as well as the actual spine numbers counted in the present study as described below. Thus the effect of RanBP9 on spinophilin are consistent in primary neurons, mouse brains and AD brains.
The most important observation made in the present study is the reductions in actual spine counts in the APΔE9 mouse brains under RanBP9 overexpression conditions. The spine density in the apical dendrites of APΔE9 mice was not altered in both the layer 6 of cortex and the CA1 region of hippocampus at 6-months of age, but by 12 months, significant reductions in spine density were seen in both brain regions. This reduction is mostly accounted for by decreased numbers of mushroom type spines in both the cortex and the hippocampus. A previous study also reported significant reductions in large spines in the hippocampus of APΔE9 mice at 12-months of age (Knafo et al, 2009). Another recent study also demonstrated significant loss of total spines in the hippocampus of 12 month old APΔE9 mice (Wang et al., 2012). Our result on spine density reductions at 12 months thus corroborates many other studies in APΔE9 mice. Similar to the APΔE9 mouse model, both J20 and APP-PS1 mouse models of AD also showed significant reductions in spine density at 11 months but not at 3- or 8-months of age (Moolman et al., 2004). However, in the PDAPP and Tg2576 mouse models, loss of spines has been shown as early as 4.5 months in both the hippocampus and somatosensory cortex, but surprisingly the authors did not find significant reductions in spine density at 11- or 20-months of age (Lanz et al., 2003). Interestingly, RanBP9 overexpression in the APΔE9 mice led to significant reduction in the number of dendritic spines already at 6-months of age in the hippocampus but not cortex, suggesting that RanBP9 accelerates loss of spines in the hippocampus. Thus, the hippocampus is more vulnerable to the effect of RanBP9 than cortex. By 12 months however, RanBP9 overexpression in APΔE9 mice further exacerbated loss of spines in both the cortex and hippocampus mostly due to loss of mushroom and thin spines. More extensive loss of spines in the APΔE9/RanBP9 triple transgenic mice compared to either RanBP9 single transgenic or APΔE9 double transgenic mice also suggests that RanBP9 potentiates synaptic damage in APΔE9 mice.
The majority of previous studies on alterations in spine density in the mouse models of AD reported only the number of total spines. A notable aspect of the present study is the identification of specific loss of mushroom and thin spines but not of stubby spines. Mushroom spines are more stable and are considered to be ‘memory spines’ in contrast to thin spines which are considered to be ‘learning spines’ because they can enlarge in response to synaptic activity. Loss of both types of spines under RanBP9 overexpression conditions suggests that RanBP9 can affect both the learning and memory aspects of cognition. By decreasing the proportion of learning spines, RanBP9 may decrease a neuron's ability to form new synapses and to respond to changes in activity. This is supported by our recent study (Palavicini et al., 2013a), in which we demonstrated that RanBP9 overexpression in the APΔE9 mice affected both the learning latency and the correct responses in a memory test after the task was learnt. This is consistent with the idea that small spines are preferential sites for LTP and large spines might represent physical traces of LTP (Matsuzaki et al., 2004; Kasai et al., 2003). Reports by others also confirmed deficits in both the cognition (Malm et al., 2007) and cortical plasticity (Battaglia et al., 2007) in 12-month-old APΔE9 mice. The role of stubby spines in memory remains unknown. These spines are hypothesized to play a role in neuron excitability.
Increased Aβ and amyloid plaques caused by RanBP9 through its tripartite interaction with APP, low-density lipoprotein receptor-related protein (LRP) and β-site APP cleaving enzyme 1 (BACE1) (Lakshmana et al., 2009; Lakshmana et al., 2012) may be one of the primary mechanisms by which RanBP9 could induce spine loss. There are numerous published reports directly linking Aβ, Aβ oligomers and amyloid plaques to the loss of spines in several mouse models of AD, as well as in actual AD. In the present study, we also observed significant reductions in spine density in the RanBP9 single transgenic mice suggesting that RanBP9 per se in the absence of APP, Aβ and amyloid plaques can reduce spine density. This is not surprising because a significant amount of RanBP9 is present throughout dendritic branches in the mouse brain (Lakshmana et al., 2012). Moreover, RanBP9 is a ligand for Rho-GEF (Bowman et al., 2008) and Rho GTPases have key roles in the actin cytoskeleton and therefore in the spine morphology (Nakayama et al., 2000; Tashiro et al., 2000), In addition, activation of Rho GTPases was directly determined in single spines in relation to structural changes induced by LTP (Murakoshi et al., 2011). Consistent with these results, cofilin, another important regulator of neuronal cytoskeleton was reduced in the primary neurons derived from RanBP9-Tg mice (Woo et al., 20012) as well as in the adult mouse brains (Palavicini et al., 2013b). These multiple pieces of evidences strongly suggest that RanBP9 overexpression alters actin polymerization/depolymerization through Rho GTPases or cofilin and subsequently causes the loss of spines. The loss of large mushroom spines in particular may also be due to RanBP9-mediated decrease in the levels of cofilin (Woo et al., 2012; Palavicini et al., 2013b). It is well known that cofilin regulates the dynamics of actin cytoskeleton through their filament-severing and monomer-binding activities (Bernstein et al., 2010; Van Troys et al., 2008), thereby intracellular cytoskeletal levels are controlled by cofilin. Since actin is highly enriched in spines which also provide structural foundation for spine shape and size, alterations in cofilin levels are expected to alter the number of different types of spines. Thus loss of large mushroom spines by RanBP9 may be due to cofilin-mediated altered actin cytoskeleton in the neurons. The reason for loss of predominantly mushroom spines is not known, but might depend on the fact that distinct spine categories have physiologically distinct functions and RanBP9 might affect only those spines that perform a specific function. As the protein levels of RanBP9 is increased in the J20 APP transgenic mice (Woo et al., 2012), APΔE9 mice (Wang et al., 2013) and in actual AD brains (Lakshmana et al., 2010; Palavicini et al., 2013a), reduced spine density may be directly attributed to RanBP9. This conclusion is also consistent with reduced spinophilin puncta in the primary neurons overexpressing RanBP9 observed in the present study. Direct support also comes from AD brains in which we recently demonstrated inverse correlations between RanBP9 and spinophilin levels in the synaptosomes (Palavicini et al., 2013a). As both mouse models of AD (Lanz et al., 2003; Moolman et al., 2004; Tsai et al., 2004; Spires et al., 2005) and actual AD (Merino-Serrais et al., 2013; Fiala et al., 2002; Knobloch and Mansuy, 2008) have deficits in dendritic spine density, the current results on the effect of RanBP9 on dendritic spines strongly implicates RanBP9 to play a crucial role in the loss of dendritic spines and therefore in the pathogenesis of AD. Since dendritic spines are the primary targets of excitatory synaptic inputs to pyramidal neurons, significant reduction in spine density is likely the reason for the observed hippocampal-dependent spatial learning and memory deficits in the APΔE9/RanBP9 triple transgenic mice reported recently from our laboratory (Palavicini et al., 2013a).
In conclusion, we suggest that loss of spines is the physical basis for the RanBP9-induced significant reductions in the levels of spinophilin and other synaptic proteins reported previously from our laboratory. Since spines are considered physical traces of memory, loss of spines is also responsible for the RanBP9-induced spatial learning and memory deficits. As loss of spines and synapses are now considered the likely basis for cognitive deficits in AD, RanBP9 is undoubtedly, a key molecular target to be considered for future development of AD therapeutics.
Highlights.
Primary neurons transfected with RanBP9 decreased spinophilin-immunoreactive puncta
Primary neurons derived from RanBP9 transgenic mice confirmed the reduced spinophilin
Mushroom and thin spines were robustly reduced in APΔE9 mice overexpressing RanBP9
Both cortex and hippocampus showed decreased spine density at 12-months of age
Reduced spine density provided the physical basis for the reduced synaptic proteins
Acknowledgments
This work was supported by National Institute of Aging (NIA)/NIH grant numbers (1R03AG032064-01, M.K. Lakshmana), 1R01AG036859-01, M.K. Lakshmana). We thank Dr. Hideo Nishitani for the EGFP-RanBP9 construct. We are grateful to the vivarium staff at TPIMS for their cooperation
Abbreviations
- AD
Alzheimer's disease
- APP
Amyloid precursor protein
- LRP
low-density lipoprotein receptor-related protein
- LTD
long-term depression
- LTP
long-term potentiation
- MAP2
Microtubule associated protein 2
- NP40
Nonidet-P40
- PFA
Para formaldehyde
- PS1
Presenelin 1
- PVDF
Polyvinylidene fluoride
- RanBP9
Ran-binding protein 9
Footnotes
Conflict of Interest: All the authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Ann Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Arefin AS, Mathieson L, Johnstone D, Berretta R, Moscato P. Unveiling clusters of RNA transcript pairs associated with markers of Alzheimer's disease progression. PLoS One. 2012;7:e45535. doi: 10.1371/journal.pone.0045535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffret A, Gautheron V, Repici M, Kraftsik R, Mount HT, Mariani J, et al. Age-dependent impairment of spine morphology and synaptic plasticity in hippocampal CA1 neurons of a presenilin1 transgenic mouse model of Alzheimer's disease. J Neurosci. 2009;29:10144–10152. doi: 10.1523/JNEUROSCI.1856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia F, Wang HY, Ghilardi MF, Gashi E, Quartarone A, Friedman E, et al. Cortical plasticity in Alzheimer's disease in humans and rodents. Biol Psychiatry. 2007;62:1405–1412. doi: 10.1016/j.biopsych.2007.02.027. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20:187–195. doi: 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bowman AL, Catino DH, Strong JC, Randall WR, Kontrogianni-Konstantopoulos A, Bloch RJ. The rho-guanine nucleotide exchange factor domain of obscurin regulates assembly of titin at the Z-disk through interactions with Ran binding protein 9. Mol Biol Cell. 2008;19:3782–3792. doi: 10.1091/mbc.E08-03-0237. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braitenberg V. Cortical architectonics: general and areal. In: Brazier MAB, Petsche H, editors. Architectonics of the Cerebral Cortex. Raven Press; New York, NY: 1978. pp. 443–65. [Google Scholar]
- Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129:1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- Crick F, Asanuma C. The PDP Research Group. Certain aspects of the anatomy and physiology of the cerebral cortex. In: Rumelhart DE, McClelland JL, editors. Parallel Distributed Processing: Explorations in the Microstructures of Cognition. Volume 2: Psychological and Biological Models. MIT Press; Cambridge, MA: 1986. pp. 333–71. [Google Scholar]
- DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- del Valle J, Bayod S, Camins A, Beas-Zarate C, Velazquez-Zamora DA, Gonzalez-Burgos I, et al. Dendritic spine abnormalities in hippocampal CA1 pyramidal neurons underlying memory deficits in the SAMP8 mouse model of Alzheimer's disease. J Alzheimer's Dis. 2012;32:233–240. doi: 10.3233/JAD-2012-120718. [DOI] [PubMed] [Google Scholar]
- Denti S, Sirri A, Cheli A, Rogge L, Innamorati G, Putignano S, Fabbri M, Pardi R, Bianchi E. RanBPM is a phosphoprotein that associates with the plasma membrane and interacts with the integrin LFA-1. J Biol Chem. 2004;279:13027–34. doi: 10.1074/jbc.M313515200. [DOI] [PubMed] [Google Scholar]
- Douglas RJ, Martin KAC. Neuronal circuits of the neocortex. Annu Rev Neurosci. 2004;27:419–451. doi: 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Hao J, Janssen WG, Tang Y, Roberts JA, McKay H, Lasley B, et al. Estrogen increases the number of spinophilin-immunoreactive spines in the hippocampus of young and aged female rhesus monkeys. J Comp Neurol. 2003;465:540–550. doi: 10.1002/cne.10837. [DOI] [PubMed] [Google Scholar]
- Hongpaisan J, Sun MK, Alkon DL. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer's disease transgenic mice. J Neurosci. 2011;31:630–643. doi: 10.1523/JNEUROSCI.5209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Xu C, Sen A, Nelson TJ, Alkon DL. PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol Dis. 2013;55:44–62. doi: 10.1016/j.nbd.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kim BG, Dai HN, McAtee M, Vicini S, Bregman BS. Labeling of dendritic spines with the carbocyanine dye DiI for confocal microscopic imaging in lightly fixed cortical slices. J Neurosci Methods. 2007;162:237–243. doi: 10.1016/j.jneumeth.2007.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knafo S, Alonso-Nanclares L, Gonzalez-Soriano J, Merino-Serrais P, Fernaud-Espinosa I, Ferrer I, et al. Widespread changes in dendritic spines in a model of Alzheimer's disease. Cerebral Cortex. 2009;19:586–592. doi: 10.1093/cercor/bhn111. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Lakshmana MK, Yoon IS, Chen E, Bianchi E, Koo EH, Kang DE. Novel role of RanBP9 in BACE1processing of amyloid precursor protein and amyloid beta peptide generation. J Biol Chem. 2009;284:11863–11872. doi: 10.1074/jbc.M807345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmana MK, Chung JY, Wickramarachchi S, Tak E, Bianchi E, Koo EH, et al. A fragment of the scaffolding protein RanBP9 is increased in Alzheimer's disease brains and strongly potentiates amyloid-beta peptide generation. FASEB J. 2010;24:119–127. doi: 10.1096/fj.09-136457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmana MK, Hayes CD, Bennett SP, Bianchi E, Reddy KM, Koo EH, et al. Role of RanBP9 on amyloidogenic processing of APP and synaptic protein levels in the mouse brain. FASEB J. 2012;26:2072–2083. doi: 10.1096/fj.11-196709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003;13:246–253. doi: 10.1016/s0969-9961(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Lee CC, Sherman SM. Modulator property of the intrinsic cortical projection from layer 6 to layer 4. Front Syst Neurosci. 2009;3:3. doi: 10.3389/neuro.06.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci. 2007;27:3712–3721. doi: 10.1523/JNEUROSCI.0059-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng C, He Z, Xing D. Low-Level Laser Therapy Rescues Dendrite Atrophy via Upregulating BDNF Expression: Implications for Alzheimer's Disease. J Neurosci. 2013;33:13505–13517. doi: 10.1523/JNEUROSCI.0918-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rabano A, Avila J, et al. The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain. 2013;136:1913–1928. doi: 10.1093/brain/awt088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Mountcastle VB. Perceptual Neuroscience: The Cerebral Cortex. Harvard University Press; Cambridge, MA: 1998. [Google Scholar]
- Murrin LC, Talbot JN. RanBPM, a scaffolding protein in the immune and nervous system. J Neuroimmune Pharmacol. 2007;2:290–295. doi: 10.1007/s11481-007-9079-x. [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472:100–104. doi: 10.1038/nature09823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Masuda H, Horii J, Kuma KI, Yokoyama N, Ohba T, Nishitani H, Miyata T, Tanaka M, Nishimoto T. When overexpressed, a novel centrosomal protein,RanBPM, causes ectopic microtubule nucleation similar to gamma-tubulin. J Cell Biol. 1998;143:1041–1052. doi: 10.1083/jcb.143.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Ann Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Nishitani H, Hirose E, Uchimura Y, Nakamura M, Umeda M, Nishii K, Mori N, Nishimoto T. Full-sized RanBPM cDNA encodes a protein possessing a long stretch of proline and glutamine within the N-terminal region, comprising a large protein complex. Gene. 2001;272:25–33. doi: 10.1016/s0378-1119(01)00553-4. [DOI] [PubMed] [Google Scholar]
- Padurariu M, Ciobica A, Mavroudis I, Fotiou D, Baloyannis S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer's disease patients. Psychiatr Danub. 2012;24:152–8. [PubMed] [Google Scholar]
- Palavicini JP, Wang H, Bianchi E, Xu S, Rao JS, Kang DE, et al. RanBP9 aggravates synaptic damage in the mouse brain and is inversely correlated to spinophilin levels in Alzheimer's brain synaptosomes. Cell Death & Dis. 2013a;4:e667. doi: 10.1038/cddis.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palavicini JP, Wang H, Monond D, Bianchi E, Xu S, et al. RanBP9 overexpression down-regulates phospho-cofilin, causes early synaptic deficits and impaired learning, and accelerates accumulation of amyloid plaques in the mouse brain. J Alzheimer's Dis. 2013b;39:727–740. doi: 10.3233/JAD-131550. [DOI] [PubMed] [Google Scholar]
- Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cutures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cruz C, Nolte MW, van Gaalen MM, Rustay NR, Termont A, Tanghe A, et al. Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer's disease. J Neurosci. 2011;31:3926–3934. doi: 10.1523/JNEUROSCI.6142-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nature Commun. 2013;4:1939. doi: 10.1038/ncomms2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MA, Cheng H, Quayle AN, Nishitani H, Nelson CC, Rennie PS. RanBPM, a nuclear protein that interacts with and regulates transcriptional activity of androgen receptor and glucocorticoid receptor. J Biol Chem. 2002;277:48020–48027. doi: 10.1074/jbc.M209741200. [DOI] [PubMed] [Google Scholar]
- Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, et al. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 2013;223:385–393. doi: 10.1016/j.expneurol.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Hof PR, Wearne SL. Rayburst sampling, an algorithm for automated three-dimensional shape analysis from laser scanning microscopy images. Nature Protocols. 2006;1:2152–2161. doi: 10.1038/nprot.2006.313. [DOI] [PubMed] [Google Scholar]
- Rodriguez GA, Burns MP, Weeber EJ, Rebeck GW. Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learning & Memory. 2013;20:256–266. doi: 10.1101/lm.030031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh B, Ramakrishna S, Baek KH. Diverse roles of the scffolding protein RanBPM. Drug Discov Today. 2012;17:379–387. doi: 10.1016/j.drudis.2011.10.030. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cerebral Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nature Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–667. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tang XC, Zhang HY. Huperzine A alleviates synaptic deficits and modulates amyloidogenic and nonamyloidogenic pathways in APPswe/PS1dE9 transgenic mice. J Nneurosci Re. 2012;90:508–517. doi: 10.1002/jnr.22775. [DOI] [PubMed] [Google Scholar]
- Wang D, Li Z, Messing EM, Wu G. Activation of Ras/Erk pathway by a novel MET-interacting protein RanBPM. J Biol Chem. 2002;277:36216–36222. doi: 10.1074/jbc.M205111200. [DOI] [PubMed] [Google Scholar]
- Wang H, Dey D, Carrera I, Minond D, Bianchi E, Xu S, et al. COPS5 (Jab1) Protein Increases beta Site Processing of Amyloid Precursor Protein and Amyloid beta Peptide Generation by Stabilizing RanBP9 Protein Levels. J Biol Chem. 2013;288:26668–26677. doi: 10.1074/jbc.M113.476689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang R, Xu S, Lakshmana MK. RanBP9 Overexpression Accelerates Loss of Pre and Postsynaptic Proteins in the APΔE9 Transgenic Mouse Brain. PLoS One. 2014;9:e85484. doi: 10.1371/journal.pone.0085484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wearne SL, Rodriguez A, Ehlenberger DB, Rocher AB, Henderson SC, Hof PR. New techniques for imaging, digitization and analysis of three-dimensional neural morphology on multiple scales. Neuroscience. 2005;136:661–680. doi: 10.1016/j.neuroscience.2005.05.053. [DOI] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–72. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- Woo JA, Jung AR, Lakshmana MK, Bedrossian A, Lim Y, Bu JH, et al. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death and Differ. 2012;19:1413–1423. doi: 10.1038/cdd.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]